
Abstract
Clinical studies demonstrate that estrogen replacement therapy in postmenopausal women may enhance cognitive function and reduce neurodegeneration associated with Alzheimer's disease and stroke, This study assesses whether physiologic levels of estradiol prevent brain injury in an in vivo model of permanent focal ischemia. Sprague-Dawley rats were ovariectomized; they then were implanted, immediately or at the onset of ischemia, with capsules that produced physiologically low or physiologically high 17β-estradiol levels in serum (10 or 60 pg/mL, respectively), One week after ovariectomy, ischemia was induced. Estradiol pretreatment significantly reduced overall infarct volume compared with oil-pretreated controls (mean ± SD: oil = 241 ± 88; low = 139 ± 91; high = 132 ±88 mm3); this protective effect was regionally specific to the cortex, since no protection was observed in the striatum. Baseline and ischemic regional CBF did not differ between oil and estradiol pretreated rats, as measured by laser Doppler flowmetry. Acute estradiol treatment did not protect against ischemic injury. Our finding that estradiol pretreatment reduces injury demonstrates that physiologic levels of estradiol can protect against neurodegeneration.
Until recently, estradiol, the primary hormone secreted by the ovary, has been thought to predominantly influence reproductive functions. It is now known that this pleiotropic hormone has profound effects on plasticity in the developing and adult brain (McEwen et al., 1995). Recent clinical studies demonstrate that estradiol replacement in postmenopausal women ameliorates cognitive dysfunction (Sherwin, 1994; Robinson et al., 1994; Henderson et al., 1996) and decreases the incidence and rate of neurodegeneration associated with diseases such as Alzheimer's disease (Henderson et al., 1996; Paganini-Hill and Henderson, 1994; Brenner et al., 1994).
Although in vitro studies clearly demonstrate that estradiol is a neurotrophic factor that increases neuronal survival (Goodman et al., 1996; Singer et al., 1996; Green et al., 1997; Weaver et al., 1997, Brooke et al., 1997), induces neurite outgrowth (Toran-Allerand, 1991), and increases spine densities (Murphy and Segal, 1996), few studies have examined this role in in vivo models of brain injury. Reports have been contradictory. Sex differences in postischemic necrosis revealed less neuronal pathology in female gerbils compared with male gerbils (Hall et al., 1991). Pharmacologic doses of estradiol have been reported to both exacerbate brain injury (Emerson et al., 1993) and reduce mortality and brain damage (Simpkins et al., 1997) in female rats. Hum and colleagues (1995) report that postischemic hyperemia was greater in female than in male rabbits and that pharmacologic levels of estradiol resulted in greater CBF during ischemia. Studies that administer precise, physiologically relevant treatments to test for neuroprotection are lacking.
This study determined whether physiologically relevant levels of estradiol protect against brain injury and if pretreatment is necessary to exert these effects. Our findings demonstrate that pretreatment with two biologically relevant doses of estradiol significantly protects against cerebral ischemia.
MATERIALS AND METHODS
Animals and experimental treatments
Female Sprague-Dawley rats (225 to 250 g) were maintained in a 14:10 hour light—dark cycle with unrestricted access to food and water. Under methoxyflurane anesthesia, rats were bilaterally ovariectomized to eliminate endogenous estradiol production. Immediately after ovariectomy or at the onset of ischemia, silastic capsules (3.8 mm diameter, 30 mm length, 0.07 volume) were implanted subcutaneously. Animals received a capsule filled with vehicle (sesame oil), a capsule filled with a low dose of 17β-estradiol (180 µg/mL), or two capsules filled with a high dose of 17β-estradiol (1 mg/mL). Within 1 hour of implantation, the Silastic capsules produce a transient rise of estradiol in blood that stabilizes by 24 hours. After this transient rise, the capsules consistently release hormone over time, producing stable levels of estradiol in serum (Wise et al., 1981).
Model of focal cerebral ischemia
One week after ovariectomy, rats were anesthetized with a mixture of ketamine/acepromazine (80.0/0.52 mg/kg intraperitoneally). In all animals, body temperature was monitored with a rectal probe and maintained at 36.5° to 37.5°C with a heating pad throughout surgery and recovery. The right femoral artery was cannulated for monitoring physiologic variables (pH, Po2, P
Histologic preparation
Rats were killed 24 hours after the onset of ischemia. Brains were removed and sectioned into 1-mm slices. Alternate slices were stained for 15 minutes in 2% triphenyltetrazolium chloride (Bederson et al., 1986), fixed in 10% buffered formalin, and quantified for infarct size. Total, cortical, and striatal ischemic injury was quantified using a computer-assisted imaging system (NIH Image, v. 1.60), and the size of the infarct is expressed as volume (mm3).
Quantification of serum estradiol levels
Trunk blood, collected when rats were killed, was centrifuged, and sera were frozen until the time of assay. Samples (0.5 mL) were analyzed for estradiol by radioimmunoassay after ether extraction.
Measurement of regional cerebral blood flow
In a separate experiment, ovariectomized and estradiol-primed animals were anesthetized with a mixture of ketamine/acepromazine (80.0/0.05 mg/kg intraperitoneally). Temperature was monitored with a rectal probe and maintained at 36.5° to 37.5°C with a heating pad. Through a craniotomy, a noninvasive laser Doppler probe was positioned (approximately 3 to 5 mm posterior to bregma, 4 mm lateral to midline) over the right parietal cortex over an area away from large pial vessels to obtain low, stable readings (Cholet et al., 1997). This region corresponds to an area affected by MCA occlusion, since ischemic injury spans the brain from approximately 4.2 mm anterior to bregma to 5.8 mm posterior to bregma. Baseline measurements were taken once a minute for 10 minutes; ischemia then was induced. Ten minutes after induction of ischemia, measurements were taken once a minute for 60 minutes. Data are expressed as 5-minute averages.
Data analysis
All data are expressed as mean ± SD. Infarct volumes in three group comparisons and serum estradiol levels were analyzed with one-way analysis of variance (ANOVA). Post hoc analyses were carried out with the Newman-Keuls test. Comparison between infarct volumes for acute treatment was analyzed with a Student's t test. Physiologic variables and laser Doppler measurements were analyzed using repeated measures ANOVA. All differences were considered significant at P < 0.05.
RESULTS
There were no significant differences in pH, blood gases, glucose, or MABP measurements among the treatment groups of rats (Table 1). Low and high physiologic levels of estradiol pretreatment significantly reduced overall infarct volume compared with oil-treated controls [F(2,31) = 5.14; P = 0.012] (Fig. 1A). Low and high physiologic estradiol pretreatment exerted an equivalent degree of neuroprotection.
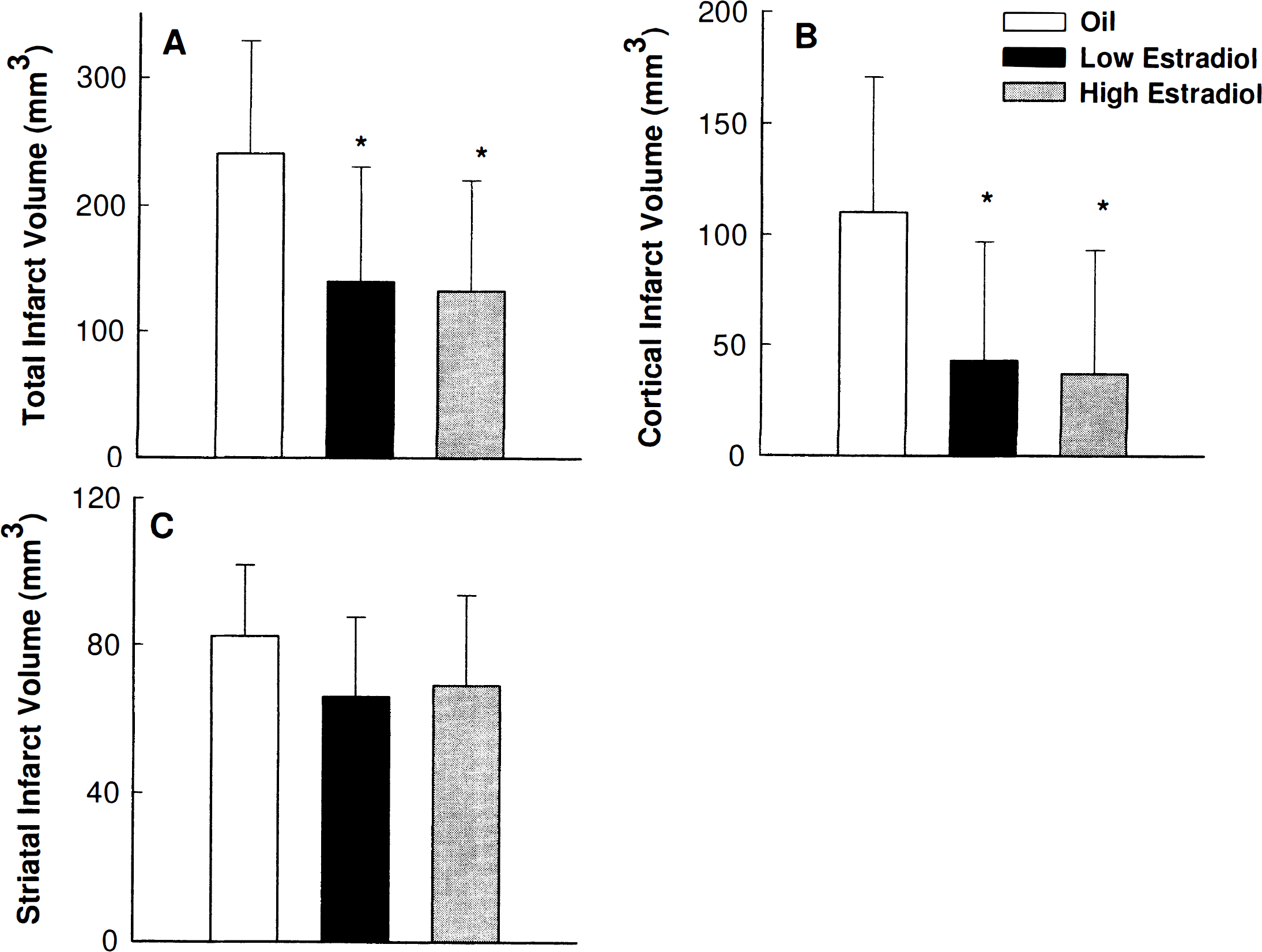
Estradiol pretreatment significantly reduced the total and cortical infarcts produced by permanent middle cerebral artery (MCA) occlusion.
Physiologic measurements among treated groups
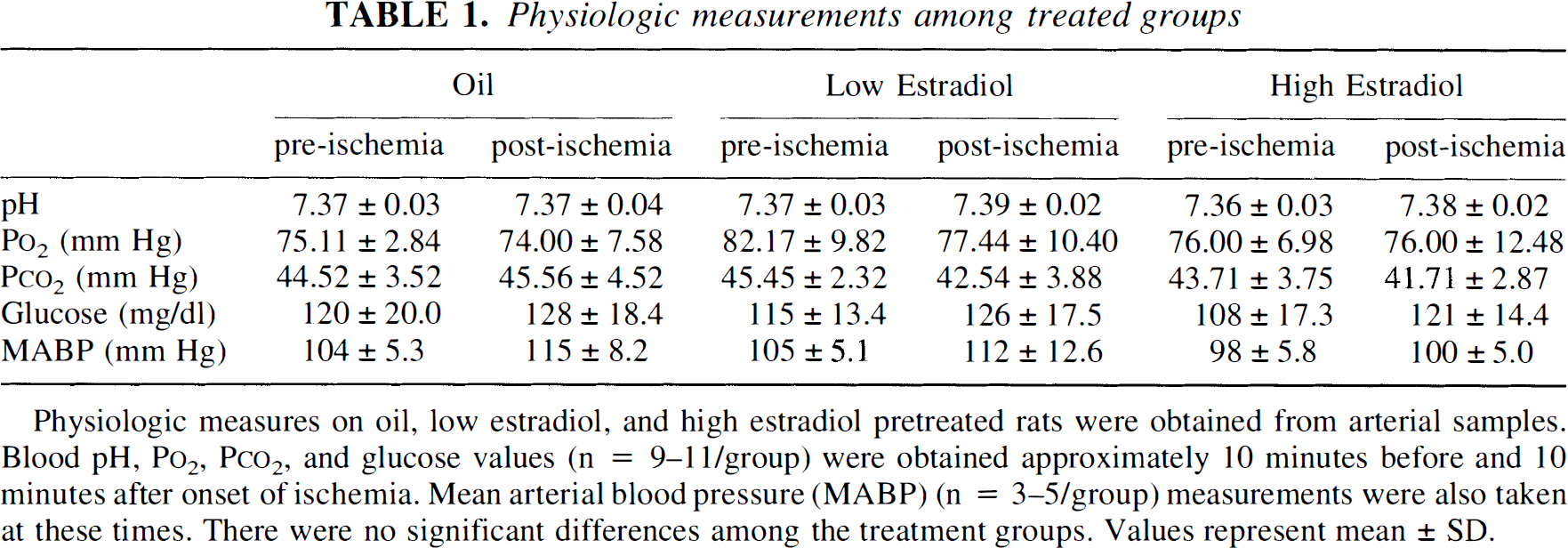
Physiologic measures on oil, low estradiol, and high estradiol pretreated rats were obtained from arterial samples. Blood pH, P
The total infarcts were further analyzed by region to compare treatment effect on cortical and striatal infarcts. Estradiol pretreatment reduced the cortical infarct by over 60% in low- and high-estradiol—primed animals when compared with oil-treated controls [F(2,31) = 5.68; P < 0.008] (Fig. 1B). In contrast, estradiol did not exert a significant effect in the striatum of low- and high-estradiol—primed rats compared with oil-treated controls [F(2,31) = 1.78; P = 0.18] (Fig. 1C). When hormone was administered at the time of the MCA occlusion, we did not detect any significant protective effect [F(1,16) = 1.04; p = 0.18] (Fig. 2).
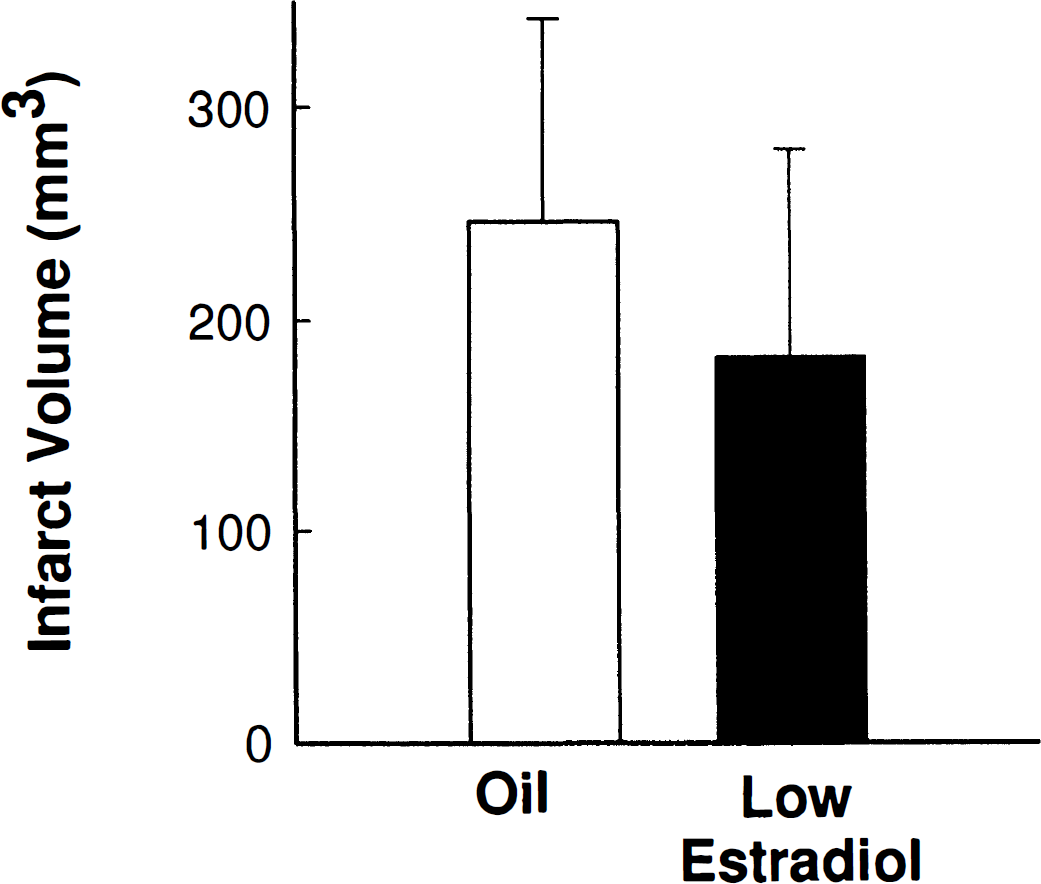
Estradiol treatment (n = 9) at the time of MCA occlusion did not significantly protect against ischemic injury compared with oil-treated controls (n = 9). Values represent mean ± SD.
Figure 3 shows serum 17β-estradiol concentrations in ovariectomized rats pretreated with Silastic capsules. The low and high estradiol serum concentrations resulting from pretreatment were significantly different from each other and from the oil-treated controls [F(2,15) = 84.48, P < 0.00001]. In our paradigm of hormone replacement, low-dose estradiol pretreatment produces serum levels equivalent to endogenous, circulating levels in adult female Sprague-Dawley rats. High-dose estradiol replacement produces serum levels equivalent to the transient, preovulatory estradiol surge in the rats (DePaolo et al., 1979). When low-dose estradiol is administered at the time of MCA occlusion (acute treatment), estradiol levels in blood immediately rise to proestrous values. These values stabilize, by 24 hours, to endogenous levels of estradiol, which mimic other days of the estrous cycle (Wise et al., 1981).
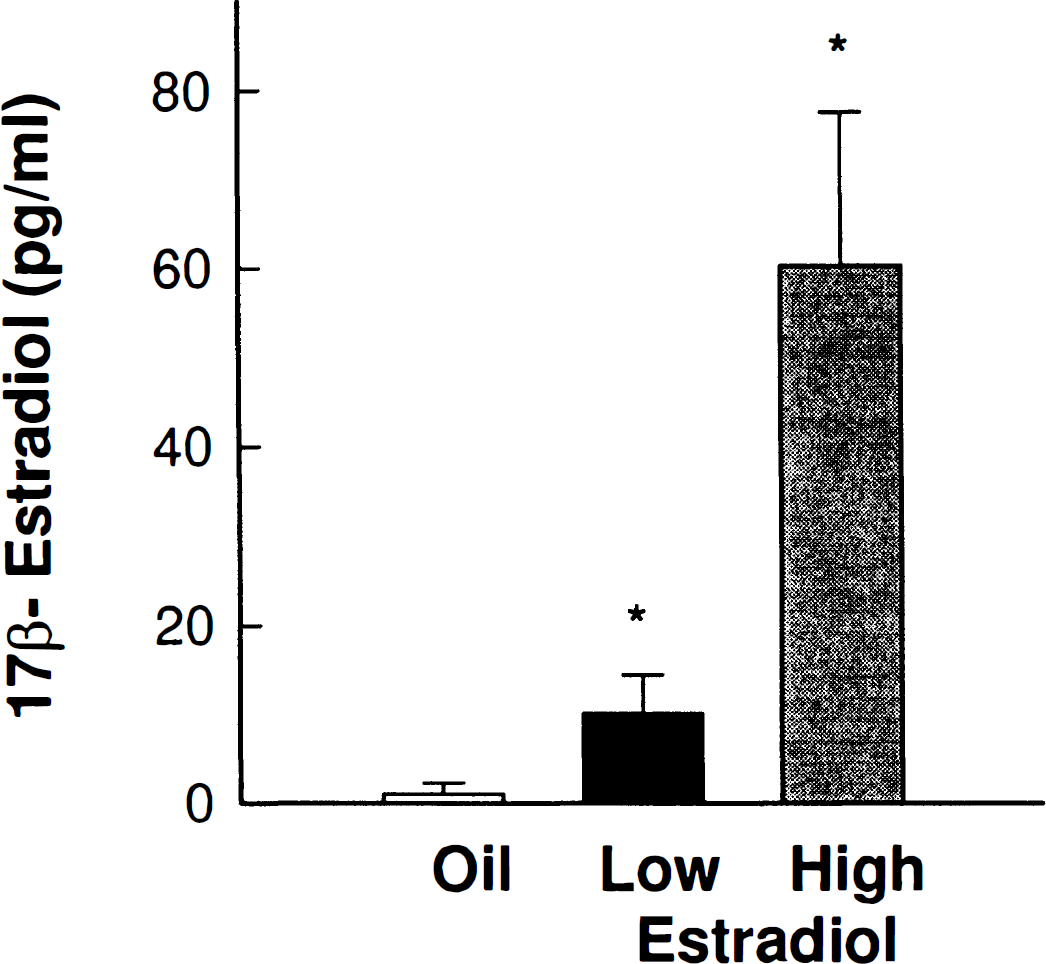
Serum 17β-estradiol concentrations in ovariectomized rats pretreated with Silastic capsules show that low (P < 0.0002, n = 5) and high (P < 0.0002, n = 8) physiologic estradiol levels were significantly different from the hormone levels of oil-pretreated controls (n = 5). Low and high estradiol serum concentrations also were significantly different from each other (P < 0.0002). Values represent mean ± SD.
In a separate experiment, regional CBF was measured by laser Doppler flowmetry through a craniotomy over an area affected by MCA occlusion on the right parietal cortex. Estradiol pretreatment did not alter the normal, or nonischemic, perfusion of blood to the cortex (oil: n = 5, 48.8 ± 6.7; low-dose estradiol: n = 5, 45.8 ± 5.3; high-dose estradiol: n = 4, 46.6 ± 6.2) [F(2,11) = 0.34; P = 0.72]. Furthermore, regional CBF after onset of ischemia did not differ between oil-pretreated and low-dose estradiol-pretreated rats [F(1,4) = 0.01; P = 0.92] (Fig. 4). The reduction of cortical blood flow in response to ischemia in oil- and estradiol-pretreated rats is within the range of reductions observed in ischemic penumbra, or secondarily compromised tissue (Nedergaard, 1996).
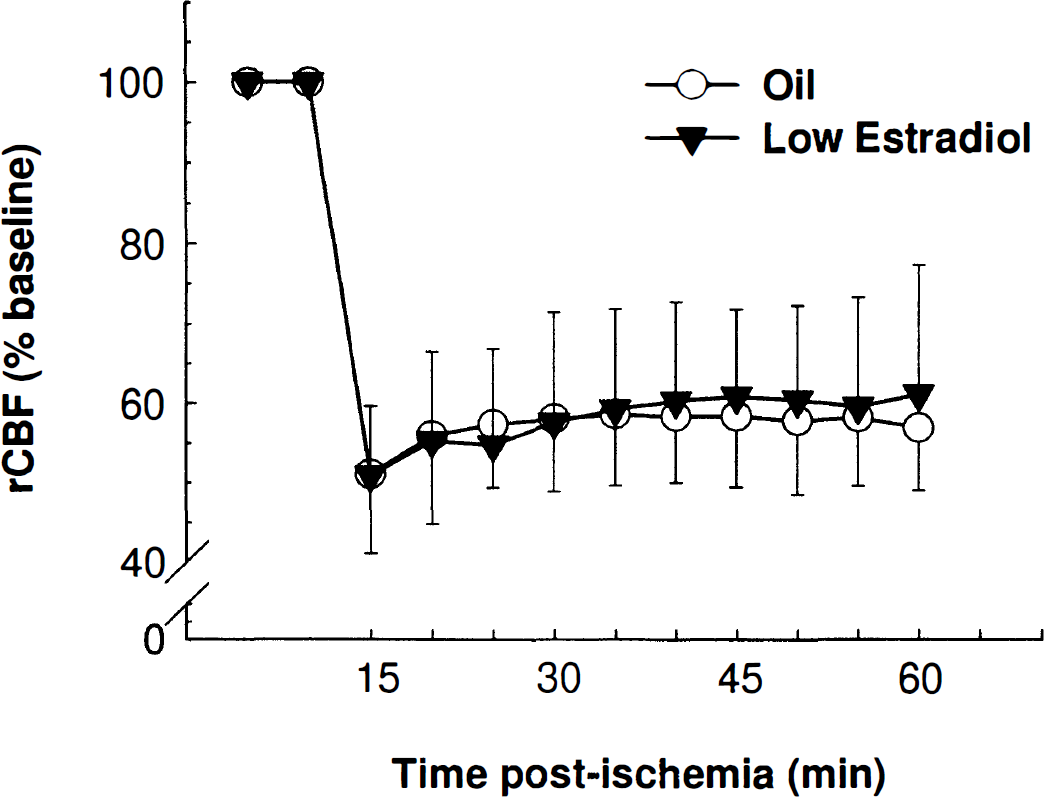
Regional cerebral blood flow (rCBF) did not differ between oil-pretreated (n = 3) and low-dose estradiol-pretreated (n = 3) rats, either before or after onset of ischemia. Laser Doppler flow measurements were taken over an area affected by MCA occlusion on the right parietal cortex. Baseline values did not differ between oil- and estradiol-pretreated rats. Ischemic laser Doppler flow measures began 10 minutes after onset of ischemia and are represented as a percentage of baseline. Each point represents a 5-minute average. Values represent mean ± SD.
DISCUSSION
Our data clearly demonstrate, for the first time, that physiologic concentrations of estradiol greatly attenuate the extent of brain damage by permanent cerebral ischemia. Pretreatment with estradiol reduces infarct size by almost 50%. In contrast, we observed no protective effect when estradiol was administered immediately after the MCA was occluded. Since estradiol priming of the brain is necessary for neuroprotection, our findings suggest that the presence of estradiol exerts its protective effect by influencing the expression of genes that enhance neuronal survival.
A study (Simpkins et al., 1997) reports the effects of pharmacologic levels of estradiol in a reperfusion model of ischemia. Although both studies demonstrate that 17β-estradiol exerts protective effects against cerebral ischemia, several important differences in the results should be emphasized. First, in the previous study, there was substantial incidence of mortality in ovariectomized rats, which was reduced by estradiol treatment. In contrast, in the current study, only five animals died, and they were equally distributed among the treatment groups. This difference in mortality may result from the more invasive surgical method or the strain of rats used in the former study, or both. Second, treatment with pharmacologic levels of estradiol initiated after surgery protected against injury. In contrast, using our experimental paradigm, physiologic estradiol treatment before ischemia was essential to demonstrate a protective effect.
The differences between our results suggest that estradiol may exert neuroprotective effects through different mechanisms. Previous studies suggest that estradiol may act through receptor-mediated genomic (McEwen et al., 1995) or membrane-activated nongenomic pathways (Watters et al., 1997; Zhou et al., 1996; Weaver et al., 1997; Goodman et al., 1996). For example, pharmacologic doses of estradiol can act nongenomically to reduce oxidative injury, possibly by scavenging reactive oxygen species (Goodman et al., 1996). It is likely that the type of injury (reperfusion or permanent ischemia) or the treatment paradigm (pharmacologic or physiologic) may determine which mechanisms of estradiol action predominate.
Our findings that physiologic concentrations of estradiol exert a profound neuroprotective effect highlight the vulnerability of a hypoestrogenic brain to injury or abnormal function. The data suggest that postmenopausal women may be at greater risk for brain injury or may suffer greater neural dysfunction during normal aging than women who receive estradiol replacement therapy. In our paradigm of hormone replacement, the high dose of estradiol pretreatment achieved serum concentrations of hormone comparable with those that we observe on proestrus, the preovulatory phase of the reproductive cycle. The low dose of estradiol pretreatment resulted in basal concentrations of estradiol that circulate on other days of the estrous cycle (DePaolo et al., 1979). In our paradigm of acute, low-dose estradiol administration, no protection was observed, despite the transient proestrous-like values of estradiol produced immediately upon implantation (Wise et al., 1981).
Our data show that pretreatment with both physiologic low and physiologic high doses were equally effective at reducing the size of the ischemic injury. Maximal protection is achieved with the low level of estradiol pretreatment. This equivalent protection by low- and high-dose estradiol pretreatment may reflect the substantial occupancy of nuclear estrogen receptors that is already present, even with the low level of estradiol treatment (Camp and Barraclough, 1985).
Estradiol exerted a striking protective effect in the cortex compared with the striatum. This may reflect differential blood perfusion to cortex and striatum after permanent occlusion of the MCA; however, it is unlikely that this explains the extent of the difference (Tsuchidate et al., 1997). Furthermore, estradiol exerted a protective effect in the cortex, despite an equivalent reduction in regional cortical blood flow between oil- and estradiol-pretreated rats. Alkayed and colleagues (1998) report no differences in ischemic cortical blood flow in gonadally intact compared with ovariectomized rats using quantitative autoradiography. Taken together, these results suggest that estradiol acts as a neuroprotective factor in the cortex through blood flow—independent mechanisms.
Several lines of evidence suggest that the cortex is an important target for estrogen action. Both the alpha isoform of estradiol receptor and the newly discovered beta isoform have been identified with abundance in cortex but not in the striatum (Shughrue et al., 1997), suggesting that the cortex is directly modified by receptor-mediated, estradiol-induced plasticity. Various cellular and molecular mechanisms may explain estradiol's neuroprotective abilities. Estradiol enhances neurite outgrowth in cortical explant cultures (Toran-Allerand, 1991) and prevents cell death in dispersed cortical cultures (Singer et al., 1996; Brooke et al., 1997). In addition, it increases the expression of neurotrophins and their receptors in several areas of the brain, including cortex (Singh et al., 1995; Toran-Allerand, 1996), possibly through direct actions at the level of transcriptional activation of the genes.
Alternatively, estradiol may modulate the effects of glutamate release by altering NMDA receptor function (Woolley et al., 1997; Weaver et al., 1997) and reducing calcium mobilization (Brooke et al., 1997). It may alter the balance of proapoptotic and antiapoptotic genes (Huang et al., 1997; Spyridopoulos et al., 1997). Finally, estradiol may influence the expression of proteins involved in synaptic plasticity (Shughrue and Dorsa, 1993) or microtubule stability (Ferreira and Caceres, 1991).
In summary, our results clearly establish that physiologic concentrations of estradiol play a neuroprotective role in the injured brain. Furthermore, our findings suggest that estradiol may act by altering factors that favor neuronal viability and attenuate cell death through blood flow—independent mechanisms.
Footnotes
Acknowledgment
The authors thank Sue Craddock for technical assistance.