
Abstract
Recent findings in animals emphasize that experimental ischemic brain damage can be strikingly reduced by estrogen; however, the neuroprotective mechanisms are not well understood. It was hypothesized that estrogen signaling via cognate estrogen receptors (ERs) within the vasculature is an important aspect of cerebral ischemic protection in the female brain, in part by amplifying intraischemic cerebral blood flow (CBF). In the present study, the hypothesis that chronic treatment with the pure ER antagonist ICI182,780 (ICI) would increase ischemic brain damage by a blood flow-mediated mechanism was investigated. Adult C57Bl/6J mice were pretreated with either subcutaneous ICI (100 μg/day) or oil/ ethanol vehicle for 1 week before 2 hours of middle cerebral artery occlusion (MCAO) and 22 hours of reperfusion. End-ischemic regional CBF was evaluated in additional cohorts using [14C]iodoantipyrine autoradiography. Infarction volume as measured by cresyl violet histology was greater in the striatum of ICI-treated females (70 ± 3% of contralateral striatum vs. 40 ± 12% in vehicle-treated females). Cortical injury was not enhanced relative to control animals (39 ± 6% of contralateral cortex in ICI group vs. 27 ± 8% in vehicle-treated group). Physiologic variables and ischemic reduction of the ipsilateral cortical laser-Doppler flow signal were similar between groups. Further, ICI treatment did not alter end-ischemic cortical or striatal CBF. The deleterious effect of ICI was limited to females, as there were no differences in stroke damage or CBF between male treatment groups. These data suggest that estrogen inhibits ischemic brain injury in striatum of the female by receptor-mediated mechanisms that are not linked to preservation of intraischemic CBF.
Estrogen is a natural neuroprotectant and potential therapeutic agent in many forms of cardiovascular and cerebrovascular disease. Exogenous 17β-estradiol treatment reduces injury after middle cerebral artery occlusion (MCAO) in ovariectomized female (Simpkins et al., 1997; Dubal et al., 1998; Shi et al., 1998; Zhang et al., 1998; Rusa et al., 1999) and male (Toung et al., 1998) rodents as well as in aged, reproductively senescent female animals (Alkayed et al., 1999). The steroid exhibits diverse actions in blood vessels, neurons, and glia, many of which may be relevant to estrogen's antiischemic properties. Endogenous estrogens are critical to protection from stroke ordinarily observed in female animals relative to their age-matched male or ovariectomized counterparts (Hall et al., 1991; Alkayed et al., 1998; Zhang et al., 1998; Wang et al., 1999). In female rats with native estrogens, intraischemic cerebral blood flow (CBF) is partially preserved in brain regions such as striatum, and these animals have smaller infarcts (Alkayed et al., 1998). Exogenous estrogen treatment also increases intraischemic CBF during global cerebral ischemia produced by four-vessel occlusion (Hurn et al., 1995).
17β-Estradiol is known to be vasoactive in the cerebral circulation, under both physiological (Goldman et al., 1976; Magness et al., 1998) and ischemic (Hurn et al., 1995; Pelligrino et al., 1998) conditions. Furthermore, estrogen receptors (ERs) are present in vascular smooth muscle (Harder and Coulson, 1975) and in endothelial cells (Colburn and Buonassisi, 1978). We have recently observed abnormal CBF responses to MCAO in transgenic female mice deficient in ERs, subtype α, as compared with wild-type females (Hurn et al., 1998). Therefore, we hypothesized that natural estrogens signal via cognate receptors within the vasculature to alter cerebral ischemic mechanisms, in part by up-regulating hormone-responsive vasodilators and amplifying intraischemic CBF. In the present study, we tested the hypotheses that treatment with the pure ER antagonist ICI182,780 in the wild-type female mouse (1) reduces intraischemic CBF during MCAO and (2) consequently increases infarction volume.
MATERIALS AND METHODS
This study was approved by the Johns Hopkins Institutional Animal Care and Use Committee and is in compliance with the guidelines of the National Institutes of Health for care and handling of animals. Adult female C57Bl/6J mice (3 months old, 18 to 29 g, n = 9 to 10 per group) were randomized to treatment group and received daily subcutaneous injections of either ICI (100 μg/day in 0.1 mL of sesame oil with 2.5% ethanol) or oil/ethanol vehicle for 7 days prior to MCAO. Age-matched males were also treated with ICI or vehicle as additional control cohorts (n = 10 per group). ICI is designated as a “pure” antiestrogen without agonist activity; chronic administration directly decreases tissue ER protein (MacGregor and Jordan, 1998; Parker et al., 1993; Gibson et al., 1991). The choice of the 100-μg/day treatment was based on previous work indicating that ICI at this dose antagonizes estrogen-induced effects on estrous and ingestive behavior (Wade et al., 1993a) and was modified for body weight in mouse. Pre-MCAO somatosensory and motor behavior was evaluated before and after the week of drug treatment to determine any adverse effects of ICI treatment. These tests assessed balance (time to fall from narrow pole up to a maximum of 120 seconds), agility (turning in a blind alley or on an inclined screen), forelimb strength (hanging from suspended wire), and autogrooming time (Nelson et al., 1995; Walensky et al., 1998).
Cerebral ischemia was induced by reversible MCAO as previously published (Eliason et al., 1997; Hurn et al., 1998). In brief, mice were anesthetized with 1 to 1.2% halothane in oxygen-enriched air by facemask, and rectal and temporalis muscle temperatures were controlled at 37 ± 0.5°C throughout the experiment with heating lamps and water pads. Unilateral MCAO was performed by inserting a 6-0 nylon monofilament into the internal carotid artery via an external carotid artery stump and then positioning the filament tip for occlusion at a distance of 6 mm beyond the internal carotid/pterygopalatine artery bifurcation. The animal was rapidly awakened and scored for neurological damage as follows: 0 = no deficit, 1 = failure to extend forelimb, 2 = circling, 3 = unilateral weakness, 4 = no spontaneous motor activity. Mice with clear neurological deficits were reanesthetized for removal of the suture at the end of the 2-hour occlusion period. At 22 hours of reperfusion, each animal was perfusion-fixed with 10% formalin in phosphate buffer. Brains were then postfixed in formalin and 30% sucrose in phosphate buffer, cut as serial coronal sections (40 μm) on a freezing microtome, and stained with cresyl violet. A set of 12 evenly spaced sections through forebrain was mounted for determination of infarction volume by image analysis (Inquiry; Loats, Westminster, MD, U.S.A.). The following areas were measured in each section: cortical infarct, total ipsilateral cortex, total contralateral cortex, striatal infarct, total ipsilateral striatum, and total contralateral striatum. The area of cortical infarction in each section was corrected for edema and expressed as a percentage of the contralateral cortex as follows: [1 – (total ipsilateral cortex – cortical infarct)/total contralateral cortex)] × 100%. Corresponding volumes were then calculated for the total set of slices. The analogous procedure was carried out for striatum. All measurements were carried out by an investigator blinded to treatment assignment.
In separate cohorts of mice, arterial blood pressure and cortical laser—Doppler flowmetry (LDF; model MBF3D; Moor Instruments, Wilmington, DE, U.S.A.) were determined during MCAO and the first 30 minutes of reperfusion. A shallow indentation was made in the parietal skull (2 mm posterior, 3 mm lateral to bregma) with a low-speed drill for placement of the LDF probe (DP3 optical, 1 -mm diameter). A thin oil interface and the probe were applied with a hood to block ambient light. The LDF signal was recorded semicontinuously and then averaged over 15-minute intervals for comparison among treatment groups. Arterial blood samples via femoral catheter (100-μL sample volume) were analyzed for hemoglobin concentration, pH, Po2, Pco2, and plasma glucose at baseline and at end-ischemia. Plasma estradiol was measured in the final arterial sample in all animals by radioimmunoassay (Diagnostic Products Corp., Los Angeles, CA, U.S.A.), as previously described (Hurn et al., 1995). Plasma progesterone was measured by a similar technique in females, as previously described (Hurn et al., 1995).
Regional CBF was measured at end-ischemia in a final set of animals, using [14C]iodoantipyrine ([14C]IAP) autoradiography, as previously described in rat (Alkayed et al., 1998) and modified for mouse. The MCAO was initiated, and then each animal was assessed for neurological deficit. Mice with clear deficit were reanesthetized after ~60 minutes of MCAO and instrumented with an arterial (Clay-Adams PE 10 [Sparks, MD, U.S.A.]; 0.28-mm inner diameter, 0.61-mm outer diameter, 15 cm long) and venous (PE 10, 10 cm long) femoral catheters. At 120 minutes of MCAO, arterial blood pressure, pH, Pco2, and Po2 were measured; then intravenous infusion and arterial sampling started. A total of 4 μCi of [14C]IAP in 81 μL of isotonic saline (1 mCi/10 mL stock diluted in equal volume of saline to obtain 5 μCi/100 μL) was infused intravenously over 45 seconds at a constant infusion rate of 6.48 ml/h (Harvard 22 pump; 2-mL glass Popper syringe with 8.92-mm diameter). Simultaneously, the arterial catheter was opened, and blood was allowed to flow freely into heparinized saline drops of known volume placed in paraffin wells. Nine blood samples were collected at 5-second intervals and promptly covered. The mouse was decapitated 45 seconds after the infusion start, and the brain was quickly removed (<60 seconds), frozen at −50°C in 2-methylbutane on dry ice, and stored at −80°C. Each brain was sectioned on a cryostat (20-μm-thick coronal sections at −18°C) and thaw-mounted onto glass coverslips. Sections were apposed for 1 week to film (Kodak SB-5, Rochester, New York, U.S.A.) with 14C standards. Sample volume was measured using a pipette and calculated by subtracting the volume of a saline drop from the total volume of blood sample plus saline. Parallel time control saline drops were used to account for change in volume due to evaporation. Concentration of [14C]IAP was determined by liquid scintillation spectroscopy (Beckman, Somerset, NJ, U.S.A.; model 3801) after decolorization with 0.2 mL of tissue solubilizer (Soluene-350; Packard Instruments Co., Downers Grove, IL, U.S.A.). Autoradiographic images representing seven coronal levels (+4, +3, +2, +1, 0, −1, and −2 mm from bregma, three images each) were digitized and regional CBF determined by image analysis software (Inquiry; Loats Associates, Westminster, MD, U.S.A.). Rates of regional CBF were calculated by Kety—Schmidt modification of the Fick principle, as previously described (Jay et al., 1988). Two methods of analysis were used. First, CBF was sampled at discrete 0.08-mm2 regions within the cortical and striatal regions most vulnerable to MCAO, averaged over three to seven consecutive coronal slices (Alkayed et al., 1998). Second, the images of each coronal slice were scanned and pixels stratified according to corresponding blood flow rates. Pixels with flow rates falling within a range of blood flow were summed and converted to volume units (Rusa et al., 1999).
All data are expressed as means ± SD. Statistical evaluation was performed by Student's t-test to compare infarction volumes between drug- and vehicle-treated groups. Physiological and LDF values and behavioral variables were assessed by two-way analysis of variance and post-hoc Newman—Keuls test to determine differences between groups over time. Regional CBF was assessed by two-way analysis of variance and post-hoc Newman—Keuls test to evaluate differences between groups in striatum and cortex at the end-ischemic timepoint. Postischemic neurological scores were analyzed by the Mann-Whitney U test. The criterion for statistical significance was set at P < 0.05.
RESULTS
Animals were matched for age and sex. Although body weight was consistently greater in male than female mice, there were no weight differences between ICI- and vehicle-treated cohorts for either sex. Table 1 summarizes physiological variables during MCAO among ICI-and vehicle-treated groups. There were no differences in these parameters or in glucose, oxygen saturation, or rectal temperature (data not shown). There were no differences among groups in neurological score as assessed during MCAO or at end-reperfusion. Plasma estrogen and progesterone levels ranged from 1 to 18 pg/mL and 1 to 22 ng/mL in females. Estrogen was below the level of detection in males. Daily ICI treatment did not alter generalized activity, baseline performance of behavioral tests, or time in autogrooming.
Physiologic measurements
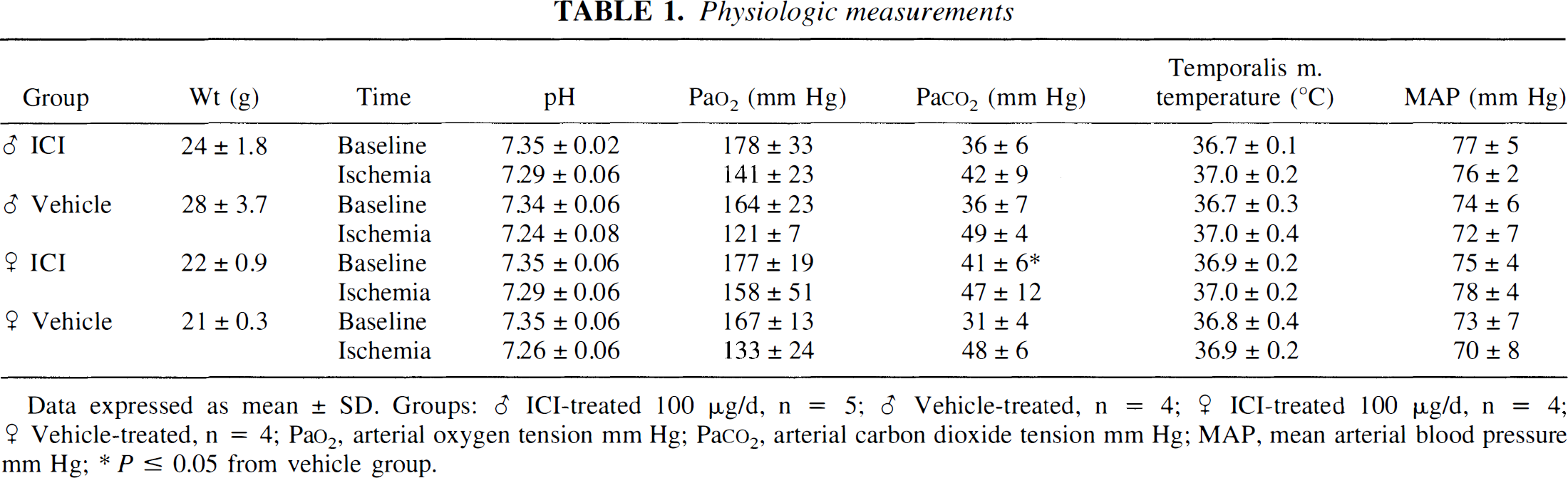
Data expressed as mean ± SD. Groups: ♂ ICI-treated 100 μg/d, n = 5; ♂ Vehicle-treated, n = 4; ♀ ICI-treated 100 μg/d, n = 4; ♀ Vehicle-treated, n = 4; Pao2, arterial oxygen tension mm Hg; Paco2, arterial carbon dioxide tension mm Hg; MAP, mean arterial blood pressure mm Hg;
P ≤ 0.05 from vehicle group.
Infarction volume was augmented in the striatum (P < 0.05) of ICI- versus vehicle-treated females but not in cortex (Fig. 1). During MCAO, the LDF signal was sharply reduced in each animal and then stabilized at ~30% of baseline. The percent reduction of LDF signal was not different in drug- and vehicle-treated groups (Fig. 2). The effect of ICI on infarction volume was limited to females, as there were no differences in stroke damage or LDF reduction during ischemia between male treatment groups. Striatal infarction was not different in ICI- and vehicle-treated males (48 ± 25 % of contralateral striatum vs. 46 ± 39% in vehicle-treated males). Cortical injury was also unchanged by drug treatment (23 ± 25% of contralateral cortex vs. 24 ± 25 in vehicle-treated males).
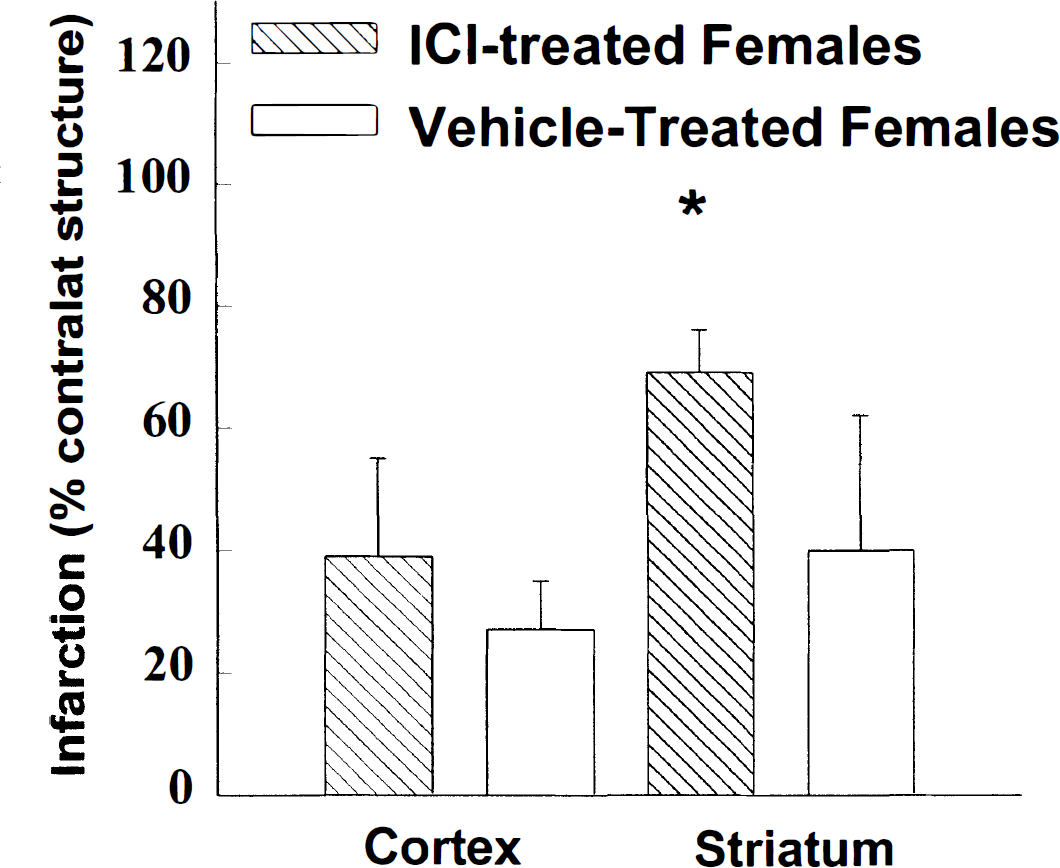
Infarction in female mice pretreated for 7 days with ICI182,780 (ICI) or oil vehicle. Infarct volumes are corrected for edema and expressed as a percentage of the contralateral structure as described in Materials and Methods. Contralateral cortical volumes were 55 ± 4 mm3 in ICI-treated (n = 9) and 57 ± 4 mm3 in vehicle-treated (n = 9) groups. Contralateral striatal volumes were 15 ± 2 mm3 in both groups. All measurements were carried out by an investigator blinded to treatment assignment. *P < 0.05 from vehicle-treated group.
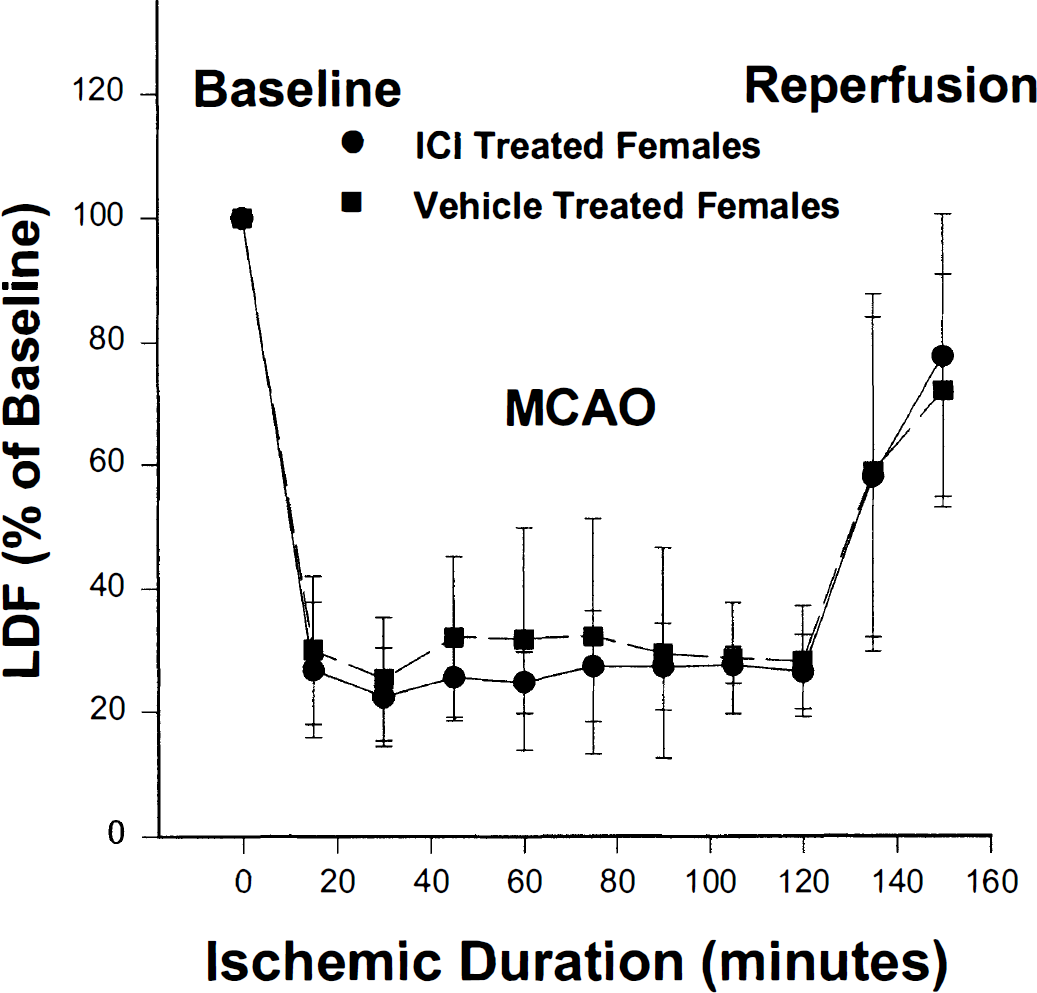
Reduction in laser—Doppler flowmetry signal during middle cerebral artery occlusion and early reperfusion in ICI182,780- and vehicle-treated female mice (n = 4 per treatment group).
In females, ICI treatment did not alter regional CBF in the contralateral nonischemic tissue (Fig. 3). Whereas striatal injury was exacerbated in ICI-treated females, end-ischemic striatal CBF was reduced equivalently in both female treatment groups (Fig. 3). To further examine differences in end-ischemic CBF distribution in these same animals, we quantified brain tissue volume that experienced near-zero CBF as well as tissue volumes experiencing less severely reduced CBF. Figure 4 shows the results of this partitioning of brain volumes into incremental levels of absolute regional CBF in ICI- versus vehicle-treated animals. There were no differences between groups at any flow increment.
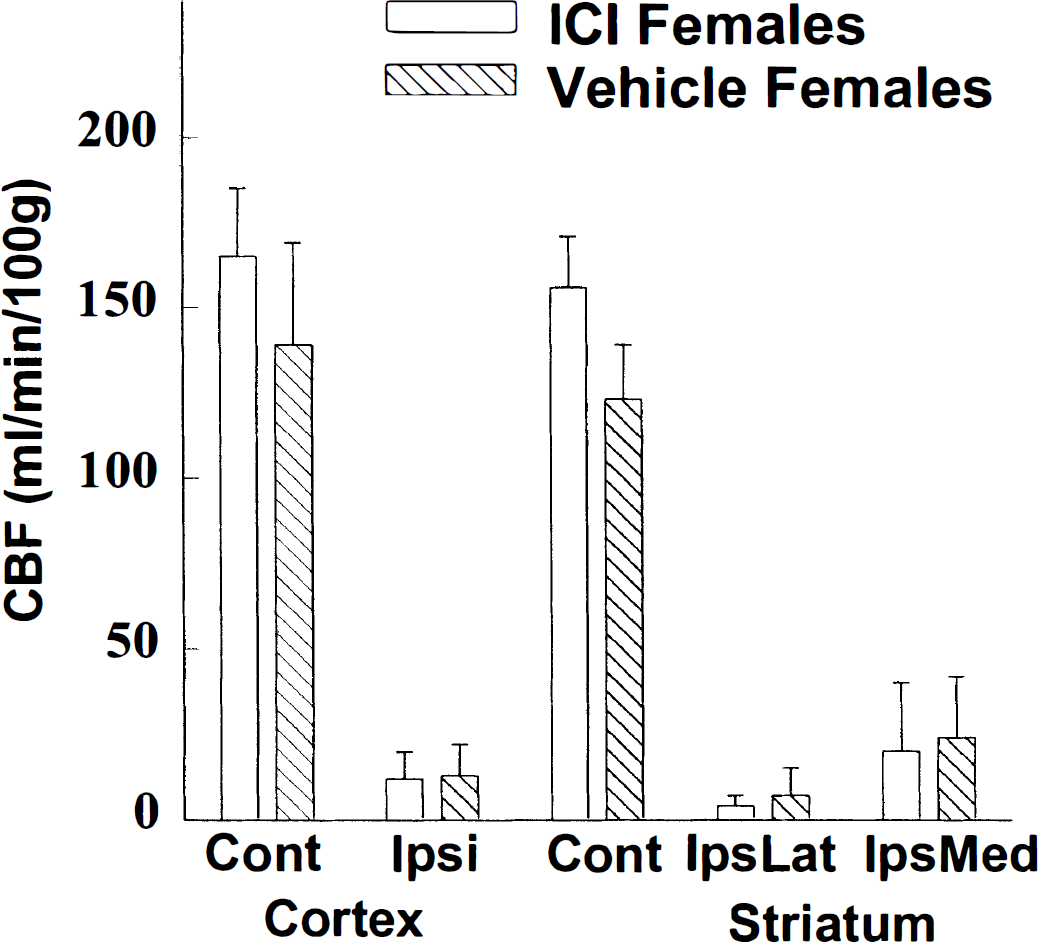
Regional cerebral blood flow (CBF) within (1) contralateral nonischemic cortex, (2) ipsilateral ischemic cortex, and (3 and 4) striatum, subdivided into lateral and medial areas, at 120 minutes of middle cerebral artery occlusion, as measured by [14C]iodoantipyrine autoradiography. Groups are ICI182,780-and oil vehicle-treated female mice (n = 4 per group).
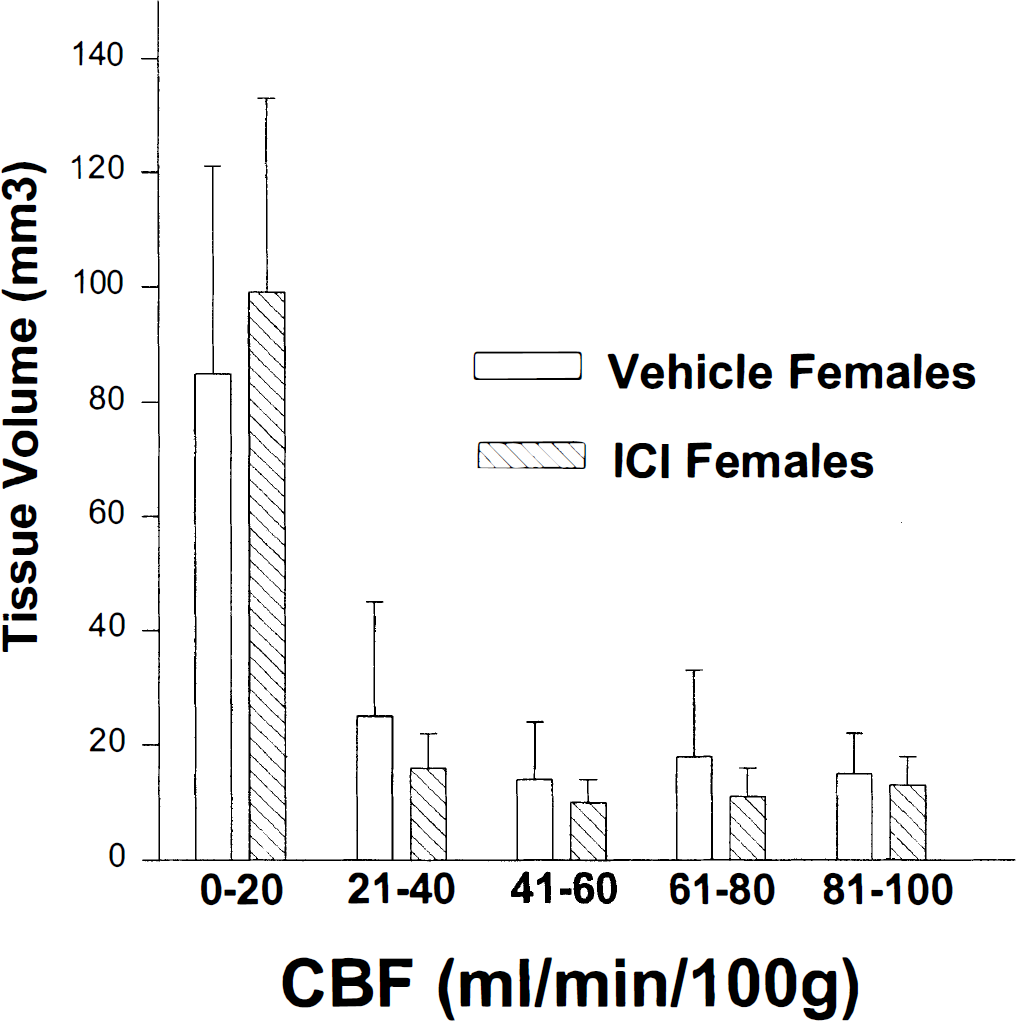
Brain volumes at incremental levels of absolute regional cerebral blood flow in ICI182,780 (ICI)- versus vehicle-treated female mice. There were no differences between groups at any flow increment, suggesting that ICI treatment did not alter the distribution of tissue area with low to near-zero perfusion during middle cerebral artery occlusion.
DISCUSSION
This study demonstrates two important findings. First, ICI treatment exacerbated histological injury in striatum after MCAO in the adult female mouse but had no effect on the male. The enhanced damage was regionally specific in that striatum, but not cortex, was vulnerable to an ER antagonist. Second, ICI treatment did not reduce end-ischemic regional CBF within the MCA territory. Further, there were no differences in CBF distribution across the ischemic hemisphere in drug-treated animals, suggesting that ICI does not recruit tissue into the ischemic core from better-perfused penumbral zones. These data suggest that endogenous estrogen inhibits experimental stroke injury in selected brain regions by receptor-mediated mechanisms that do not involve preservation of intraischemic CBF.
Endogenous reproductive steroids are important in reducing the risk of stroke in women. The risk of cerebrovascular events in women rises after menopause (Wenger et al., 1993), but the mechanism(s) by which native estrogens protect against cerebrovascular disease is unclear. Whether pharmacological replacement therapy also alters stroke pathophysiology remains controversial, with reports of increased (Wilson et al., 1985), decreased (Finucane et al., 1993; Paganini-Hill et al., 1988), and unchanged (Petitti et al., 1998) stroke risks in postmenopausal hormone users. There are few clinical data evaluating estrogen's neuroprotective properties in ongoing ischemic injury, but the steroid is clearly a significant endogenous neuroprotectant in experimental stroke. Our previous work, and that of others, suggests that endogenous estrogen acts by a mixture of blood flow-mediated and nonvascular mechanisms to reduce tissue injury during vascular occlusion (Hall et al., 1991; Alkayed et al., 1998; Zhang et al., 1998; Wang et al., 1999). Further, exogenous estradiol treatment reduces ischemic brain injury, potentially by mechanisms that are independent of preservation of intraischemic CBF (Rusa et al., 1999; Wang et al., 1999). When LDF is assessed as an indicator of cortical perfusion, others have reported that estrogen treatment reduces ischemic damage without enhancing intraocclusion LDF values (Dubal et al., 1998; Shi et al., 1998).
One potential link among these multiple mechanisms is the hypothesis that the steroid's protective activity is initiated via cognate ERs. Classic estrogen signaling occurs via a nuclear receptor that acts as a ligand-activated transcription factor, binding target DNA and altering expression of the appropriate RNA species. Estrogen receptor modulation of gene transcription efficiency occurs as the ER protein binding domain interacts with a hormone-responsive element, a consensus palindromic DNA sequence. For example, an estrogen response element is present on the gene for endothelial nitric oxide synthase; consequently, it has been hypothesized that estrogen amplifies vasodilator release (Venema et al., 1994; Mendelsohn and Karas, 1994). The steroidal ICI family of ER antagonists are pure antiestrogens, devoid of estrogenic activity, and interact with both ERs and ER subtypes (Kuiper et al., 1997; MacGregor and Jordan, 1998). In this study, we utilized ICI in a pharmacological paradigm to evaluate the importance of ER-mediated steroid signaling to stroke outcome in female animals. We have previously observed that infarction volume after MCAO is paradoxically smaller in transgenic female mice deficient in ER, subtype α, than in wild-type females (Hurn et al., 1998). Further, intraischemic reduction in CBF is less in ER-deficient than wild-type females, perhaps accounting for their unexpectedly small amount of tissue injury after 2 hours of MCAO. Therefore, the present study was designed to evaluate the effect of generalized, pharmacological antagonism of ERs on intraischemic CBF and tissue outcome in wild-type female mice. The data indicate that loss of estrogen signaling via nuclear estrogen receptors exacerbates stroke injury in striatum, but not by depressing intraischemic regional CBF. ICI182,780-induced perfusion defects during reperfusion cannot be excluded, however, and it is feasible that vascular mechanisms were operant later within the 22-hour recovery time frame. Estrogen has known vasoprotective actions in the noncerebral circulation, which involve both receptor-dependent and -independent means (for review, see Duckles et al., 1996; Mendelsohn and Karas, 1999).
Alternatively, the deleterious effect of ER inhibition may be mediated at the parenchymal level. Such an observation is not entirely surprisingly, given that estrogen has a widespread group of cellular targets within brain, including regions not associated with sexual or reproductive function. Early autoradiographic studies using [3H]estradiol injections clearly demonstrated numerous sites of nuclear accumulation and retention of radioactivity in brain and spinal cord of both sexes (Stumpf, 1968). Estrogen has direct and rapid effects on neuronal tissue; for example, synaptic architecture within areas such as hippocampus changes with the estrous cycle and can be altered in <24 hours by exogenous estradiol (Woolley and McEwen, 1992). The effects of ICI in the present study were restricted to the caudate-putamen, and this finding could reflect a relatively high density of ER distribution in striatal versus cortical regions. However, the distribution of ER, subtype α or β, within striatum in female animals or humans is unclear and is under active investigation (Couse et al., 1997; Shughrue et al., 1997; Osterlund et al., 1997). Last, although ICI is a steroid and highly lipophillic, its ability to penetrate the blood-brain barrier has not been demonstrated under ischemic conditions. The agent has been shown to antagonize estrogen's effects on reproductive and ingestive neurobehavior (Wade et al., 1993a) but did not block [3H]estradiol uptake in hypothalamus in a subsequent study (Wade et al., 1993b). Effects on estradiol uptake in other brain regions have not been studied. Nevertheless, ICI treatment in the present study resulted in a regionally distinct, and gender-specific, deleterious effect in this stroke model. Conversely, chronic ER blockade had no effect on cortical injury in either male or female mice, emphasizing that estrogen's signaling in ischemic brain is not uniformly receptor mediated. Multiple mechanisms that do not act through classic ER pathways should also be considered: for example, the highly rapid induction of early immediate genes (Zhou and Dorsa, 1994), nongenomic membrane-associated activity in neurons (Mermelstein et al., 1996; Ross and Gu, 1999), and antioxidant activity in many cell types (Green et al., 1997; Ayres et al., 1998).
These data indicate that infarction volume was equivalent in vehicle-treated male and female C57Bl mice, unlike previous findings in gerbil (Hall et al., 1991) and rat (Li et al., 1996; Alkayed et al., 1998; Zhang et al., 1998). The present study is limited in that it was not designed to study primary differences between sexes in mice, other than to determine if any pro-injury effects of ICI were gender dependent. Males were selected so as to be of the same reproductive age and genetic strain as females. However, the 3- to 4-month-old female mice were uniformly smaller and of lower body weight than their age-matched male counterparts. These variables are known to influence the severity of intravascular occlusion and stroke outcome unless the size of the intraluminal suture is varied accordingly (Hata et al., 1998). Therefore, size and weight differences in our study confound true comparisons of outcome between male and female treatment groups.
Female mice were not stratified by timing within a 4-to 5-day estrous cycle, and consequently, plasma estrogen and progesterone at the time of ischemia ranged over normal physiological levels. ICI182,780 treatment was carried out for a week, or at least one cycle of normal hormonal variation, before MCAO and companion blood flow studies. Therefore, chronic ER blockade increased striatal damage and failed to alter local ischemic CBF under conditions of high and low circulating estrogen. Effects of ovarian cyclicity on baseline or intraischemic CBF are unclear. However, timing of ischemia within the reproductive cycle has recently been shown to correlate with the amount of tissue damage sustained after MCAO in female rats (Carswell et al., 1999). Further studies are needed to confirm this observation in mouse and to determine if antiestrogen treatment produces equally deleterious effects at each phase of the estrous cycle.
In conclusion, we find that ER antagonism selectively increases striatal injury after MCAO in females, suggesting that receptor-mediated mechanisms are important to estrogen's protective properties within this brain region. The key effectors of estrogen's activity in brain downstream from the receptor and the relative importance of receptor subtype α versus β in this hormonal signaling mechanism remain to be determined.
Footnotes
Acknowledgment
The authors thank Dr. David Warner (Department of Anesthesiology, Duke University School of Medicine) for his kind advice and assistance in applying the [14C]IAP autoradiographic technique to the mouse brain.