
Abstract
Bcl-w is a newly described cell death suppressor member of the Bcl-2 gene family. As these genes may have a role in the outcome of ischemic brain injury, the regional expression of Bcl-w protein in rat brain was examined at 6 to 72 hours after 90 minutes of transient middle cerebral artery occlusion. Bcl-w protein, although constitutively expressed at low levels in nonischemic brain, was found to be overexpressed in ischemic brain at all time points studied. Up-regulation of Bcl-w protein was particularly abundant in the penumbral region of the cortex and mainly in cells lacking DNA fragmentation. In the cortical penumbra, Bcl-w protein was detected predominantly in neurons and showed mitochondrial localization, as determined using double-label immunohistochemistry. Bcl-w expression was also detectable, to a lesser extent, in reactive astrocytes in the infarct border zone and in microvessel walls in the infarct regions. At the mechanistic level, incubation of isolated brain mitochondria with the addition of recombinant Bax or high concentration of calcium resulted in release of cytochrome c from the mitochondria. In the presence of recombinant Bcl-w protein, however, the release of cytochrome c induced by Bax or calcium was largely inhibited. Further, recombinant Bcl-w protein inhibited calcium-induced loss of mitochondrial transmembrane potential, indicative of permeability transition, in a dose-dependent manner. These results suggest that Bcl-w may be an endogenous neuroprotectant against ischemic neuronal death and that, like its analogues such as Bcl-2 and Bcl-x-long, Bcl-w may achieve this protection via the mitochondrial death-regulatory pathway.
Brain injury resulting from cerebral ischemia is a complex process involving mixed types of cell death. Whereas ischemic neurons often die from necrosis, a significant number of neurons exhibit a type of death reminiscent of apoptosis, in which the genetic program of apoptosis, or programmed cell death, is activated (for reviews, see Choi, 1996; Schulz et al., 1999). In strong support of a role for apoptosis in ischemic brain injury, several apoptosis-regulatory genes have recently been found to be activated in neurons after ischemia (Krajewski et al., 1995; Chen et al., 1995, 1996, 1998a; Ni et al., 1997; Namura et al., 1998).
Recent studies on basic mechanisms of apoptosis have established a central paradigm in which a group of cysteine proteases, known as caspases, are responsible for the execution of the death program (for review, see Thornberry and Lazebnik, 1998). A number of death regulators play important roles in the initiation and regulation of caspase activation. Of these, the Bcl-2 family of death-modulatory genes, which now has at least 16 identified members, is prominent (for review, see Adams and Cory, 1998). This gene family contains both pro- and antiapoptotic members, with the antiapoptotic Bcl-2 and the proapoptotic Bax the best-studied examples. Bcl-2 family proteins share structural homology in the Bcl-2 homology-binding domains (BH 1 to 4), which are important for their functions as well as for protein interactions among the family members (Yin et al., 1994; Cheng et al., 1996; Wang et al., 1996; Chittenden et al., 1995; Hunter et al., 1996). Functionally, Bcl-2 family proteins such as Bcl-2, Bcl-xL, Bax, and Bid regulate apoptosis through the mitochondria by controlling mitochondrial membrane permeability and the release of caspase-activating factors (for review, see Green and Reed, 1998). Among these gene products, Bcl-2 and Bax have been shown to be regulated by ischemia (Krajewski et al., 1995; Chen et al. 1995, 1996), and modulation of Bcl-2 gene expression affects the outcome of ischemic injury (Martinou et al., 1994; Linnik et al., 1995; Lawrence et al., 1996). Thus, certain Bcl-2 family proteins may be important modulators of neuronal death in ischemia.
The closest homologues of Bcl-2 are Bcl-xL and the recently identified antiapoptotic gene Bcl-w (Gibson et al., 1996). Bcl-w is highly conserved from mouse to human, with 99% homology at the amino acid level. Further, there is a 46% homology of Bcl-w to Bcl-2 (a comparable homology is that of Bcl-xL and Bcl-2 at 42%) (Gibson et al., 1996). Structurally, Bcl-w contains each of the four BH domains and the hydrophobic C-terminal transmembrane domain. These properties determine that Bcl-w is a membrane-bound protein and is able to heterodimerize with a number of proapoptotic Bcl-2 family proteins (O'Connor et al., 1998; Holmgreen et al., 1999). A death suppressor action of Bcl-w has been demonstrated by its inhibition of apoptosis in hematopoietic cell lines induced by various apoptosis inducers (O'Connor et al., 1998; Holmgreen et al., 1999; Hsu et al., 1998).
Bcl-w is widely expressed in mammalian tissues including the central nervous system (Print et al., 1998). Expression of Bcl-w is increased during brain development, with the highest levels in the mature brain (Hamner et al., 1999). However, inactivation of Bcl-w by homologous recombination in mouse embryonic stem cells produces animals that are normal in brain development presumably due to the redundant function of other anti-apoptotic genes (Print et al., 1998). These observations suggest that Bcl-w may play an important role in mature brain rather than during brain development. Nevertheless, the functional role of Bcl-w in the brain, especially in settings of stress, is unexplored. Accordingly, in the present study, we examined the expression of Bcl-w protein in the rat model of transient cerebral ischemia and investigated the functions of Bcl-w in isolated brain mitochondria.
METHODS
Surgical procedures
All experiments were performed on male Sprague—Dawley rats, each weighing 275 to 300 g. Focal cerebral ischemia was induced in rats by reversible occlusion of the middle cerebral artery (MCA), as previously described (Chen et al., 1996, 1997). In brief, anesthesia was induced with 4% isoflurane through a facemask; rats were then intubated and ventilated with 1.5% isoflurane in an air/oxygen mixture (90:8.5%). Blood pressure, blood gases, and blood glucose concentration were maintained in the normal range throughout the experiments. Temporalis muscle and rectal temperatures were maintained in the range of 36.5 to 37.5°C using a heating pad and a temperature-regulated heating lamp. The bifurcation of the common carotid artery was exposed under the operating microscope, the external carotid artery was dissected and coagulated distally, and the internal carotid artery was isolated and separated from the vagus nerve. A 3-0 monofilament nylon suture (Ethicon, Somerville, NJ, U.S.A.) was introduced into the internal carotid artery through the stump of the external carotid artery and gently advanced for a distance of 20 mm from the common carotid artery bifurcation to block the origin of the MCA for 90 minutes. The MCA blood flow was restored by withdrawing the intraluminal suture from the internal carotid artery. Six rats served as sham-operated controls in which animals underwent the same surgical procedures but no suture was inserted. Three normal untreated rats served as naive controls.
Western blot analysis
The rats were killed at 6, 24, or 72 hours after the completion of ischemia (n = 3-4/experimental condition) or 24 hours after sham operation by lethal overdose with intraperitoneal injection of 8% chloral hydrate. The brains were rapidly removed; the cortex and caudate putamen ipsilateral and contralateral to the ischemic hemisphere were separately dissected, homogenized, and lysed (Chen et al., 1996). The lysates were cleared by centrifugation at 14, 000 g for 30 minutes at 4°C. The protein was denatured in sodium dodecyl sulfate gel-loading buffer (100 mmol/L Tris-HCl, 200 mmol/L dithiothreitol, 4% sodium dodecyl sulfate, 0.2% bromophenol blue, and 20% glycerol) at 100°C for 6 minutes and then separated on 12% sodium dodecyl sulfate polyacrylamide gels (40 μg of protein/sample). Immunoblotting was performed as previously described (Chen et al., 1996) using a chemiluminescent detection system (Clontech, Palo Alto, CA, U.S.A.). The antibody used to detect rat Bcl-w is a rabbit polyclonal antibody raised against the Bcl-w recombinant protein (StressGene, Victoria, British Columbia, Canada). The working dilution for Bcl-w in the present study was 1:500. Immunoreactivity for Bcl-w on each individual lane of the blots was semiquantified using a gel densitometric scanning program (Chen et al., 1998b).
Immunohistochemistry
Rats were anesthetized with 8% chloral hydrate at 0, 1.5, 3, 6, 16, 24, or 72 hours after 90 minutes of ischemia or 24 hours after sham operation (n = 4/time point) and then perfused with 200 mL of heparinized 0.9% saline followed by 500 mL of 4% paraformaldehyde in 0.1 mol/L phosphate-buffered saline (PBS; pH 7.4). The brains were removed, immersed in 4% paraformaldehyde for 4 hours, and then placed in 30% sucrose at 4°C for 3 days before they were snap-frozen in 2-methylbutane at −30°C. Brains were cut into 15-|xm-thick coronal sections using a cryostat, mounted, and kept at −80°C until use. Sections at the levels of midcaudate (anteroposterior = +0.2 mm from bregma) were processed for immunohistochemical staining. The sections were air-dried and then washed three times with PBS. After the sections were permeabilized with 2.0% Triton X-100 for 30 minutes followed by three PBS washes, they were incubated in PBS containing 0.5% bovine serum albumin and 2% normal goat serum for 20 minutes to reduce nonspecific activity. Sections were then incubated for 2 hours at room temperature at 1:200 dilution in the same Bcl-w antibody used for western blot analysis. Sections were washed in PBS three times for 10 minutes each and then incubated for 2 hours at room temperature at a 1:3, 000 dilution with goat anti-rabbit Cy3.18 immunoconjugate (Jackson Immunochemicals, West Grove, PA, U.S.A.). This was followed by incubation of the sections in the nuclear DNA dye Hoechst 33258 at 10 μg/mL for 5 minutes. Sections were then washed in PBS four times for 15 minutes each on an orbital shaker, mounted in Gelvatol, and coverslipped. A Zeiss light microscope equipped for epifluorescent illumination was used for observation. For the assessment of nonspecific immunostaining, alternating sections from selective experimental conditions (24 hours after ischemia) were incubated without the primary antibody or with the Bcl-w antibody that was reabsorbed with the rat Bcl-w recombinant protein at the concentration of 10 μg/mL.
To confirm ischemic infarction and neuronal death in this animal model, brain sections obtained at 24 and 72 hours after ischemia (n = 4/group) were stained with cresyl violet.
Double-label immunohistochemistry
To determine if Bcl-w protein is associated with the mitochondrial membrane in ischemic brains, double-label immunohistochemistry for Bcl-w and the mitochondrial membrane-bound protein cytochrome c oxidase IV was performed in sections obtained 6 and 24 hours after ischemia (n = 3/time point). The sections were first processed for Bcl-w immunohistochemistry as described above and then incubated for 2 hours at room temperature with a monoclonal antibody against cytochrome c oxidase IV (Molecular Probes, Eugene, OR, U.S.A.) at a dilution of 1:3, 000. After three PBS washes, the sections were incubated at room temperature for 1 hour with a horse anti-mouse fluorescein isothiocyanate/fluorescein immunoconjugate (Vector Laboratories, Burlingame, CA, U.S.A.).
To identify the cell types in which Bcl-w protein is expressed after ischemia, double-label immunohistochemistry was performed for Bcl-w and either the neuronal nuclear marker NeuN (Chemicon International, Temecula, CA, U.S.A.) (Liu and Ferenci, 1998) or the glial marker glial fibrillary acidic protein (Sigma, St. Louis, MO, U.S.A.) (Chen et al., 1997) was used. The procedures were essentially the same as described above.
Detection of DNA fragmentation and double staining
In situ detection of DNA fragmentation (as a marker for cell death) in ischemic brains was performed using the large fragment of DNA polymerase I, as previously described (Chen et al., 1998b). The reaction mixture contained 5 mmol/L MgCl2, 5 mmol/L 2-mercaptoethanol, 20 μg/mL bovine serum albumin, 10 μmol/L each of dGTP, dCTP, dTTP, and biotinylated dATP, and 30 U/mL large fragment of DNA polymerase I (Sigma) in PBS (pH 7.4). The reaction was terminated by washing the sections three times in PBS. The incorporated biotinylated dATP in damaged DNA was detected using fluorescein isothiocyanate/avidin D (cell-sorting grade; Vector Laboratories), which selectively binds to biotin and produces a green-yellow color under fluorescent microscopy.
To determine if Bcl-w protein is expressed in the same cells that contain DNA fragmentation after ischemia, double staining for Bcl-w immunohistochemistry and DNA damage labeling was performed in brain sections obtained 24 and 72 hours after ischemia (n = 3/time point). Brain sections were first processed for Bcl-w immunohistochemistry, as described above. After washes in PBS for 1 hour, the sections were then subjected to labeling for DNA fragmentation using the method described above. Sections were examined by fluorescence microscopy using excitation/emission wavelengths of 550/565 (red) and 495/515 (green-yellow) nm for Bcl-w and DNA damage labeling, respectively.
cDNA cloning of rat Bcl-w and expression of fusion proteins
To study the in vitro effect on mitochondria by Bcl-w, a cDNA encoding the whole reading frame of Bcl-w was isolated from rat brain using polymerase chain reaction. Polyadenylated mRNAs were isolated from the rat cerebral cortex using a fast track mRNA isolation kit (Invitrogen, San Diego, CA, U.S.A.) and were used as templates for cDNA synthesis, as described (Chen et al., 1998a). Polymerase chain reaction was performed using primers designed according to the Bcl-w sequence previously reported (Hamner et al., 1999): 5′-TCACTTGCTAGCAAAAAAGGCCCC-3′ (antisense) and 5′-ATGGCGACCCC-AGCCTCAACCCC-3′ (sense). The products generated from polymerase chain reaction were subcloned into pSPORT1 vector (GibcoBRL, Grand Island, NY, U.S.A.), amplified, purified, and subjected to sequencing analysis (University of Pittsburgh Service Core Facility).
To generate Bcl-w fusion protein, the Bcl-w cDNA was amplified using the primers 5′-GCCGCCACCATGGCGAC-CCCAGC-3′ (sense) and 5′-TCACTTGCTAGCAAAAAAGGCCCC-3′ (antisense). The cDNA was fused into the glutathione S-transferase (GST) gene in PGEX-2T vector according to the manufacturer's instructions (Amersham Pharmacia Biotech, Little Chalfont, Buckinghamshire, England). The GST-Bcl-w fusion protein was expressed in Escherichia coli BL21 cells and absorbed to a glutathione-Sepharose 4B column. The fusion protein was further cleaved by incubation in thrombin protease for 16 hours at room temperature. The eluate was collected by centrifugation at 500 g for 5 minutes at 4°C. The purified Bcl-w protein was verified by western blot analysis.
A GST—Bax fusion protein was also generated and purified using the method described above. The cDNA for expressing Bax fusion protein is a truncated human Bax that lacks the 21-amino acid hydrophobic C-terminus but retains the function (Yin et al., 1994).
In vitro effect of Bcl-w on isolated mitochondria
It has been proposed that the mitochondrion is the central site for the regulation of apoptosis by Bcl-2 family proteins. Whereas proapoptotic proteins such as Bax and Bak induce mitochondrial permeability transition and cytochrome c release, antiapoptotic proteins such as Bcl-2 and Bcl-xL inhibit these changes (Narita et al., 1998; Jurgensmeier et al., 1998; Eskes et al., 1998). To determine if Bcl-w possesses the ability to protect mitochondria as Bcl-2 and Bcl-xL do, the in vitro effect of Bcl-w on isolated brain mitochondria was studied.
Mitochondria were purified from normal nonischemic rat brain, as previously described (Berman and Hastings, 1999). Mitochondria were suspended at 10 mg of protein/mL in the MSB buffer containing 400 mmol/L mannitol, 50 mmol/L Tris-HCl (pH 7.2), 5 mg/ml bovine serum albumin, and 10 mmol/L KH2PO4 and kept on ice for up to 4 hours (Jurgensmeier et al., 1998).
For the cytochrome c release assay, mitochondria (1 mg of protein/mL) were incubated with recombinant Bax (50 μg/mL) or 100 μmol/L Ca2+ in the presence or absence of recombinant Bcl-w (50 or 250 μg/mL) in the MSB buffer for 1 hour. Mitochondria were pelleted by centrifugation and subjected to immunoblotting with antibodies to Bax (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) or Bcl-w (StressGene). The resulting supernatants were analyzed by immunoblotting with a monoclonal antibody against cytochrome c (Pharmingen, San Diego, CA, U.S.A.).
For the measurement of mitochondrial permeability transition, mitochondria (1 mg of protein/mL) were incubated with 100 μmol/L Ca2+ in the presence or absence of recombinant Bcl-w (50 to 500 μg/mL) or cyclosporin A (1 to 10 μmol/L) for 30 minutes. Mitochondrial transmembrane potential (ΔΨ) was analyzed by measuring the ΔΨ-dependent uptake of rhodamine-123, as previously described (Narita et al., 1998; Marzo et al., 1998). Fluorescence was determined in a Millipore (Bedford, MA, U.S.A.) CytoFluor 2300 automated plate-reading fluorometer with excitation at 505 nm and emission at 534 nm.
RESULTS
Western blot analysis
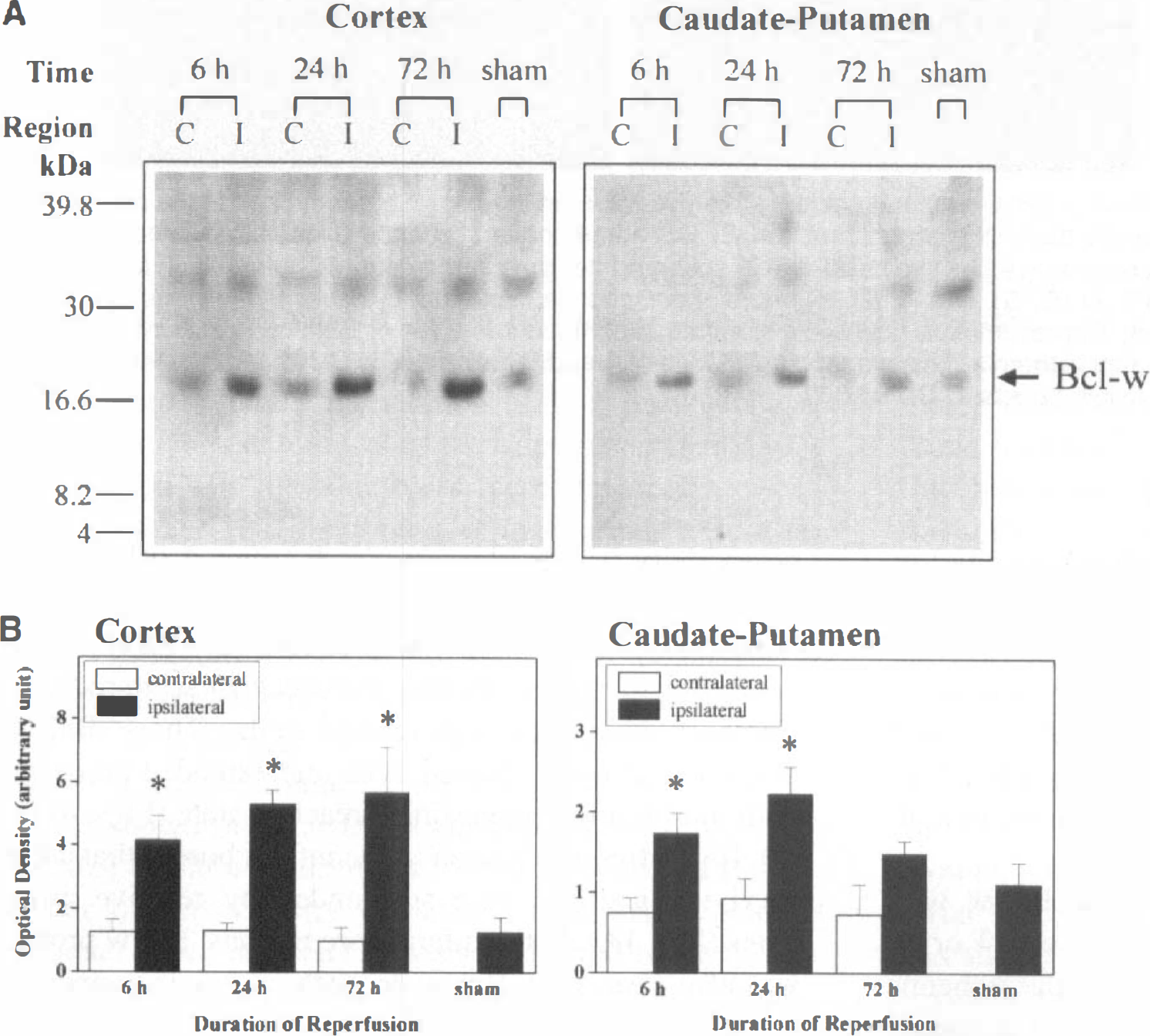
Western blotting revealed the presence of Bcl-w immunoreactivity in both cortex and caudate putamen samples from sham-operated brains. The levels of Bcl-w protein expression were increased four- to sixfold in the cortex from 6 to 72 hours after 90 minutes of MCA occlusion with a maximum concentration seen in the 72-hour time point (Fig. 1). In the caudate putamen, the increase was approximately twofold, with the maximum concentration occurring at 24 hours.
Immunohistochemistry
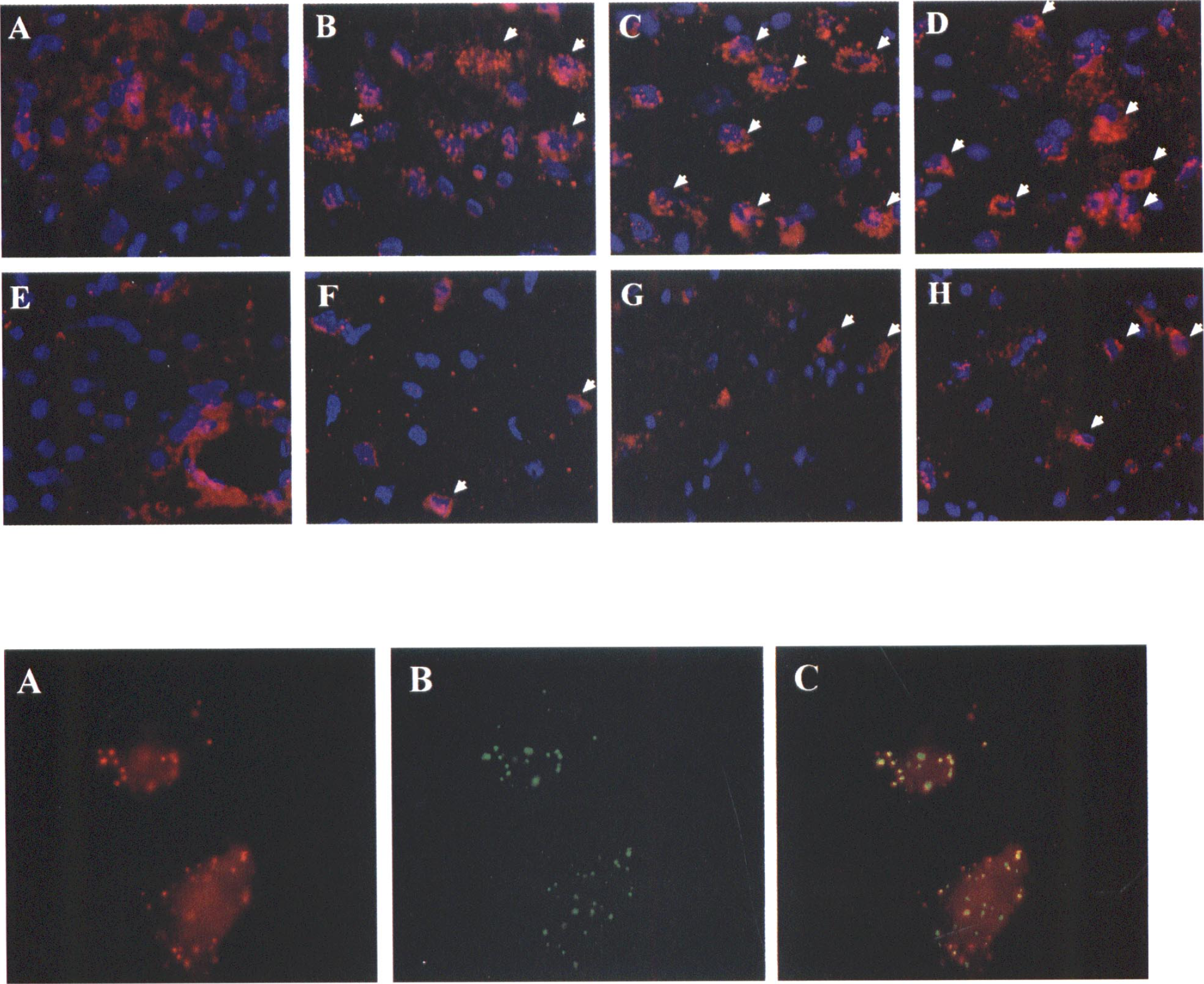
Immunohistochemical identification of Bcl-w protein was performed on brain sections using indirect immunofluorescence with double labeling with the DNA dye Hoechst 33258. In normal nonischemic brains, Bcl-w immunoreactivity was detectable at low levels in neurons throughout most forebrain regions, including cortex, caudate putamen, thalamus, and hippocampus. As shown in the top of Fig. 2A, the localization of Bcl-w immunoreactivity was distinct from the Hoechst 33258 staining and exclusively in the cytoplasm. Bcl-w immunoreactivity was increased in both cortex and caudate putamen beginning at 6 hours and peaking at 24 hours after transient focal ischemia (Fig. 2 bottom, B and F). The increased immunoreactivity of Bcl-w had a punctate appearance, suggesting a membrane-bound rather than a soluble protein. Although not all neurons showed prominent Bcl-w up-regulation with ischemia, in many cells the expression was robust, especially in the cortical penumbra 24 and 72 hours after ischemia (Fig. 2 top, C and D). Penumbral expression of Bcl-w was at all time points more prominent than that in the ischemic core region of caudate putamen (Fig. 2 top, F to H). Sections that were incubated without the primary antibody or with the primary antibody reabsorbed with the recombinant Bcl-w protein showed background fluorescence only (data not shown).
Double-label immunohistochemistry
Subcellular localization of Bcl-w immunoreactivity in neurons was studied using double-label immunohistochemistry with the mitochondrial membrane-bound enzyme cytochrome c oxidase. The punctate membrane-bound features of Bcl-w immunoreactivity in ischemic neurons, seen in the bottom panels of Fig. 2, are now shown to be those of mitochondrial membrane localization.
To confirm the mitochondrial localization of Bcl-w, mitochondrial protein extracts were prepared from cortices of sham-operated brains and brains subjected to 90 minutes of MCA occlusion followed by 24 or 72 hours of reperfusion (n = 3/condition) and subjected to western blot analysis for Bcl-w. The western blot showed increased Bcl-w contents in the mitochondrial fraction after ischemia (Fig. 3).
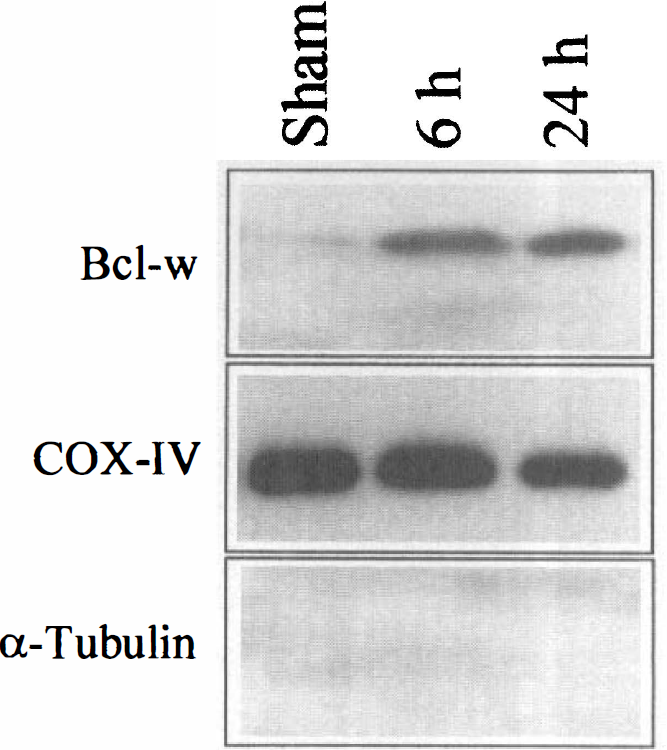
Representative western blots show increases of Bcl-w immunoreactivity in the mitochondrial fraction after ischemia. Mitochondrial protein was purified from the cortex at 6 or 24 hours after ischemia or 24 hours after sham operation and subjected to western blot analysis. Control western blots show that the purified protein fraction is enriched in the mitochondrial membrane-bound protein cytochrome c oxidase IV (COX-IV) but does not contain the cytosolic protein α-tubulin.
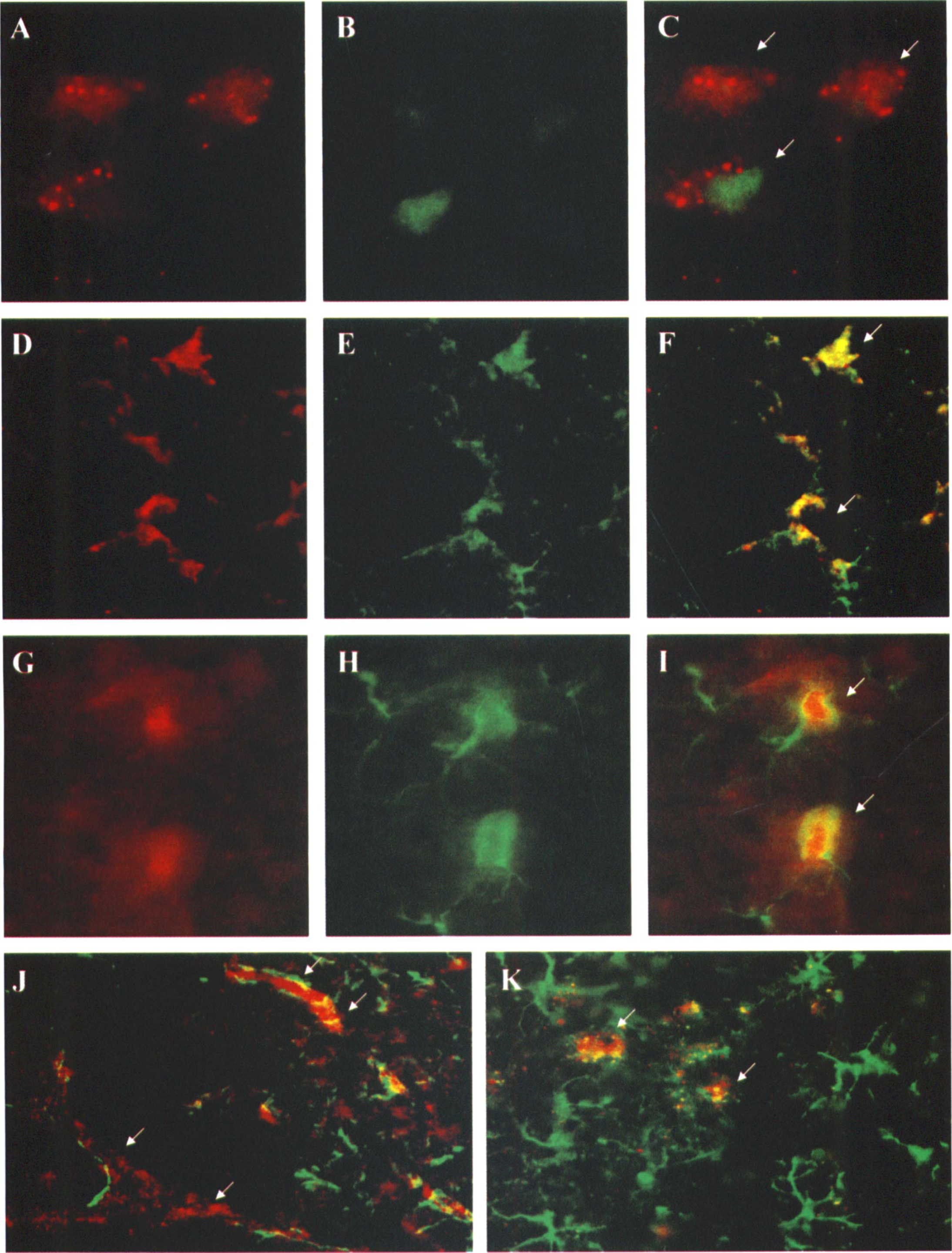
Double-label immunohistochemistry of Bcl-w with neuronal and glial markers in brains obtained 24 or 72 hours after ischemia is shown in Fig. 4. In the ischemic cortex, especially the penumbral regions, Bcl-w immunoreactivity was predominantly distributed in cells that were stained positively with the neuronal nuclear marker NeuN (Figs. 4A to 4C). In the cortical infarct border zone, Bcl-w expression was also seen in scattered glial fibrillary acidic protein-stained cells. These Bcl-w-positive astrocytes showed thick and extended processes with morphology suggesting a reactive state (Figs. 4D to 4I). It was frequently seen in the infarct border that a few surviving neurons were surrounded by reactive astrocytes (Fig. 4K). In the infarct core regions, Bcl-w protein was also expressed. However, such expression appeared to be limited to vessel walls (Fig. 4J).
Double-label immunofluorescent images of Bcl-w (
Discordance between Bcl-w expression and DNA fragmentation
Representative double-label of Bcl-w immunohistochemistry and DNA damage is shown in Fig. 5. Positive staining for DNA fragmentation and Bcl-w immunoreactivity was frequently observed in the same microscopic fields in the cortical infarct border zone 24 and 72 hours after ischemia. However, increased Bcl-w immunoreactivity was found in cells that did not show DNA damage. Cells that contained DNA damage showed low levels or absence of Bcl-w immunoreactivity.
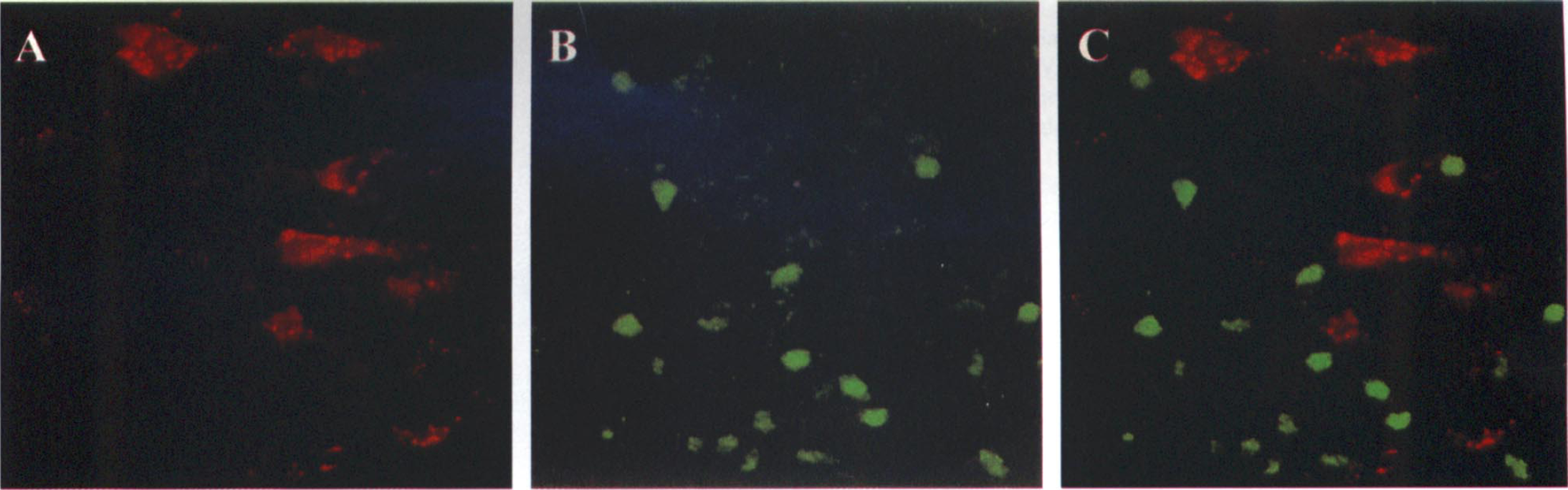
Double-label of Bcl-w immunofluorescence (A) and DNA fragmentation (B) in the cortical infarct inner border at 72 hours after ischemia. The overlapping image (C) shows that there is no colocalization of Bcl-w expression and DNA fragmentation in ischemic cells.
In vitro effect of Bcl-w
Both recombinant rat Bcl-w protein and truncated Bax protein were expressed in E. coli BL21 cells (see Methods) and purified (Fig. 6A). Incubation of mitochondria for 1 hour without the addition of Bax resulted in no release of cytochrome c to the supernatant (Fig. 6B). Incubation with the addition of recombinant Bax or calcium resulted in redistribution of cytochrome c from the mitochondria to the supernatant. In the presence of Bcl-w protein, however, the release of cytochrome c induced by Bax or calcium was inhibited. Western blot analysis detected increased amounts of Bax and Bcl-w in the mitochondrial pellet (Fig. 6C), suggesting that Bax and Bcl-w were incorporated into the mitochondrial membrane.
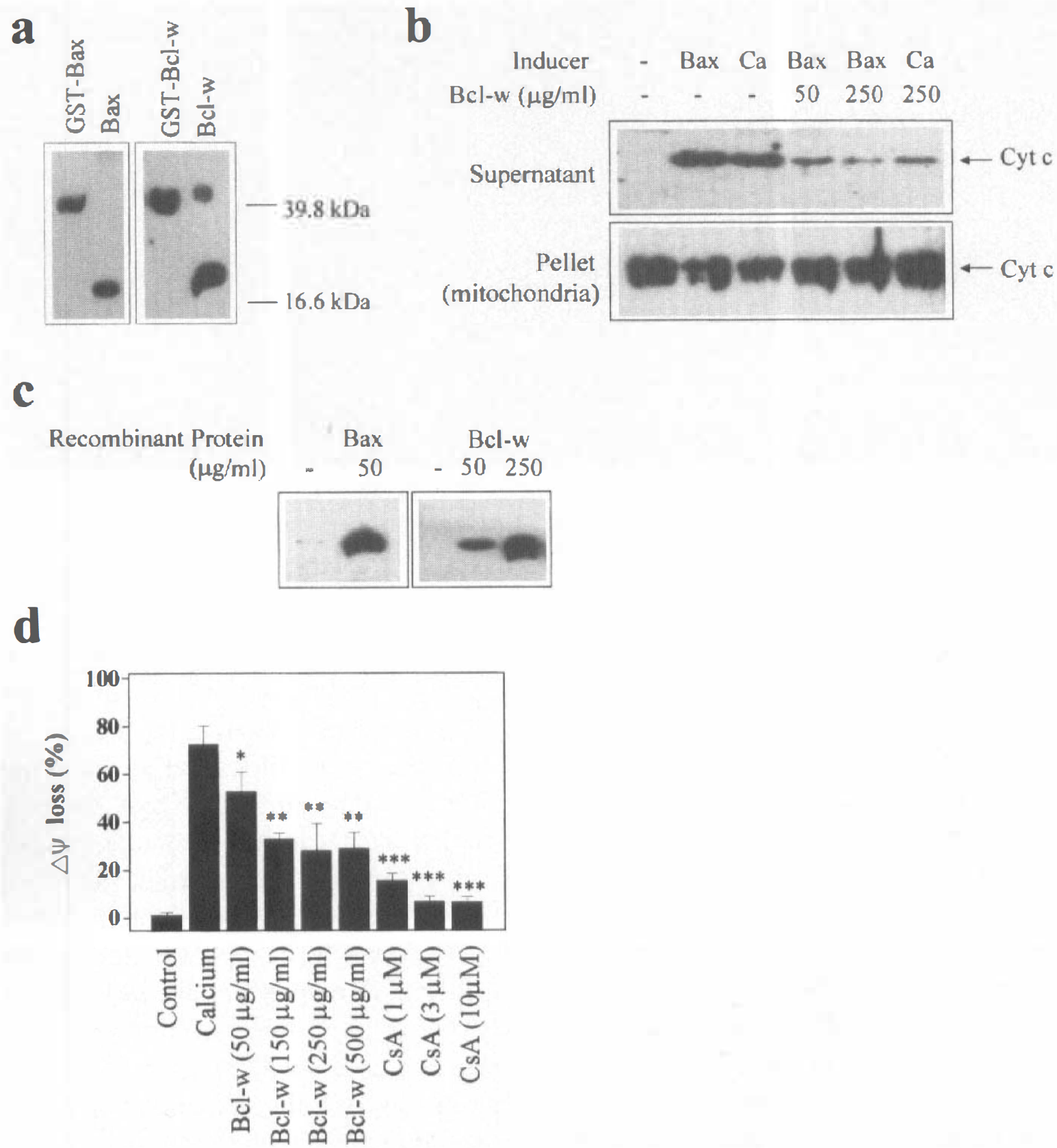
As shown in Fig. 6D, recombinant Bcl-w protein inhibited calcium-induced loss of mitochondrial ΔΨ, indicator of permeability transition, in a dose-dependent manner. Such an effect was also achieved using the mitochondrial permeability transition pore inhibitor cyclosporin A, as previously reported (Narita et al., 1998).
DISCUSSION
Bcl-w is a newly identified antiapoptotic member of the Bcl-2 gene family that is constitutively expressed in adult brain. The data presented here demonstrate that the expression of Bcl-w protein is up-regulated in the brain after transient focal ischemia. Bcl-w protein is overexpressed predominantly in cells that survive the ischemic insult. This pattern of expression resembles that of Bcl-2 (Chen et al., 1995) but is opposite from that of the proapoptotic protein Bax in ischemic brain (Krajewski et al., 1995; Chen et al., 1996; Gillardon et al., 1996). These results thus support a role for Bcl-w and other antiapoptotic Bcl-2 members in promoting cell survival after cerebral ischemia.
The presence of Bcl-w protein in the cerebral cortex and caudate putamen of sham-operated and ischemic brains was confirmed using western blot analysis. A 21-kDa protein of the expected molecular mass for Bcl-w was found in sham and ischemic cortical and caudate putamen samples. Optical density semiquantification showed a four- to sixfold increase of the protein in the ischemic cortex, with maximum expression at 72 hours. In the caudate putamen, the up-regulation was only twofold, with a maximum at 24 hours. This pattern of Bcl-w expression in this model is consistent with a survival protein as the caudate putamen infarcts, but the cortex constitutes a penumbral region that largely survives.
The cellular localization of Bcl-w protein was determined immunocytochemically in brain sections counterstained with the nuclear DNA dye Hoechst 33258. In sham-operated brains or in the contralateral hemisphere of ischemic brains, Bcl-w immunoreactivity was detectable at low levels in a nonnuclear localization. The intensity and the number of cells showing this immunoreactivity were increased in both caudate putamen and cortex in the ischemic brain beginning at 6 hours after ischemia. The expression was particularly abundant in the cortical penumbra where the immunoreactivity nearly filled the cytoplasmic compartment in some ischemic neurons (Fig. 2 top, B to D). The expression was much less robust in the ischemic core region of caudate putamen where infarct occurred (Fig. 2 top, F to H).
The appearance of Bcl-w immunoreactivity was that of a punctate pattern in the cytoplasm of neurons. This pattern of staining suggests that the protein is membrane bound, as the presence of the hydrophobic C-terminal transmembrane domain in the Bcl-w protein would predict (Gibson et al., 1996). In an attempt to identify the specific cytoplasmic membrane to which Bcl-w was bound, sections were double-stained with the mitochondrial membrane-bound enzyme cytochrome c oxidase. As shown in the bottom panels of Fig. 2, cytochrome c oxidase immunoreactivity and that of Bcl-w overlapped, strongly suggesting mitochondrial localization for this gene product. The mitochondrial localization of Bcl-w was further confirmed using subcellular fractionation followed by western blot analysis (Fig. 3).
The cell types that exhibited increased Bcl-w immunoreactivity were assessed using double-label immunohistochemistry (Fig. 4). Bcl-w immunoreactivity was seen mainly in neurons in the cortical penumbra as most Bcl-w-expressing cells were also stained positively for the neuronal marker NeuN. In the cortical infarct border zone, Bcl-w expression was also detectable in reactive astrocytes. Within the cortical infarct, however, Bcl-w immunoreactivity was seen in the microvessel walls but detectable in few neurons. This is a pattern highly reminiscent of that seen with Bcl-2, which is also expressed in focal ischemia in all cell types that survive various degrees of ischemia (Chen et al., 1995). In contrast to neurons, Bcl-w expression in astrocytes showed a diffuse instead of a punctate pattern (Fig. 4). The different subcellular localization in these two different cell types cannot be readily explained by the available data. One possibility is that the sensitivity of the immunofluorescent method used in this study was insufficient to distinguish cytosolic from mitochondrial immunoreactivity in astrocytes where the cell bodies and processes are much narrower than neurons. Alternatively, as expression of Bcl-w was up-regulated in reactive astrocytes and reactive astrocytes were maximally detected at 72 hours in this model, translocation of Bcl-w from cytosol to the mitochondria may occur at later time points. Future studies employing longer duration of reperfusion may address this issue.
The preferential expression of Bcl-w in cells that survive ischemia was demonstrated in DNA fragmentation/Bcl-w immunohistochemistry double-label experiments in which DNA damage labeling served as a marker of cell death (Chen et al., 1998a). Whereas a large number of DNA-fragmented cells were seen in the cortical and caudate putamen infarct zones, very few cells in these areas exhibited Bcl-w immunoreactivity. Both DNA damage labeling and increased Bcl-w immunoreactivity were frequently seen in the infarct border zone. However, there was little overlap between these two labels (Fig. 5), indicating that DNA fragmentation developed predominantly in cells that expressed no or a low level of Bcl-w protein. This pattern of staining was opposite from that of the proapoptotic protein Bax, which was previously found to be up-regulated in dying neurons after ischemia (Krajewski et al., 1995; Chen et al., 1996; Gillardon et al., 1996). Accordingly, such differential expression of Bcl-w (similarly, Bcl-2) and Bax in ischemic brain may shift the ratio of antiapoptotic versus proapoptotic proteins in surviving and dying cells in opposite fashions. This ratio has been considered to be an important determinant of the fate of cell death in developmental as well as in pathological apoptosis (Oltvai et al., 1993; Korsmeyer et al., 1993; Yang and Korsmeyer, 1996). The up-regulation of Bcl-w expression in survival neurons seen in this study is thus compatible with, but, however, does not prove, a neuroprotective role of Bcl-w in the ischemic brain.
The mechanism of presumed neuroprotection by Bcl-w cannot be stated with certainty based on the available data. However, like several other Bcl-2 family members, Bcl-w contains the transmembrane C-terminal domain, which allows it to localize to mitochondrial membranes (Gibson et al., 1996). Moreover, Bcl-w possesses the BH 1 to 3 domains that are essential for it to heterodimerize with proapoptotic Bcl-2 family members. Thus, it can be inferred that, like its homologues such as Bcl-2 and Bcl-xL, Bcl-w may regulate apoptosis through the mitochondrial pathway by opposing the effect of proapoptotic proteins. However, the prosurvival effect by Bcl-w or its homologues may or may not be related to its direct binding to proapoptotic proteins. It has been shown that Bcl-w protected against apoptosis induced by overexpression of Bax or Bad but not that induced by Bak or Bik, although Bcl-w was able to heterodimerize with all four proteins (Holmgreen et al., 1999). On the other hand, A1 protein, a Bcl-w homologue, although unable to bind to Bax or Bad, blocked Bax- and Badinduced cytotoxicity (Holmgreen et al., 1999). Furthermore, certain BH mutants of Bcl-xL that cannot bind to Bax or certain other proapoptotic proteins retain significant antiapoptotic activity (Cheng et al., 1996; Kelekar et al., 1997). Therefore, alternative mechanisms by which Bcl-2 members regulate apoptosis that are independent of protein-protein heterodimerization must exist. One such mechanism that has been proposed is the channel-forming activity of these proteins. Both pro- and anti-apoptotic Bcl-2 members are capable of acting as ion channels in mitochondrial membranes (Antonsson et al., 1997; Minn et al., 1997; Schendel et al., 1997; Schlesinger et al., 1997), and the functional states of these channels may largely determine the changes in mitochondrial membrane permeability during apoptosis (Green and Reed, 1998). Therefore, Bcl-w may possess the neuroprotective effect through the mitochondrial pathway via multiple mechanisms.
Our in vitro data strongly support a role for Bcl-w in regulating cell death through the mitochondria. As shown in Fig. 6, the recombinant rat Bcl-w protein decreased Bax- or Ca2+-induced release of cytochrome c from isolated brain mitochondria. Furthermore, Bcl-w inhibited Ca2+-induced mitochondrial permeability transition in a dose-dependent manner (Fig. 6), an effect that resembles the mitochondrial permeability transition pore inhibitor cyclosporin A. These findings bear significance in the context of ischemic brain injury. Mitochondrial release of cytochrome c and other apoptosis inducers such as caspase-9 has recently been detected in ischemic neurons (Fujimura et al., 1998, 1999; Krajewski et al., 1999), suggesting a role for mitochondrial damage in activating effector caspases. In addition, calcium accumulation in mitochondria and cyclosporin A-inhibitable mitochondrial permeability transition has been implied in the development of cerebral infarction and delayed death of hippocampal neurons after transient ischemia (Uchino et al., 1998; Matsumoto et al., 1999). Accordingly, Bcl-w, upon overexpression and translocation to mitochondria, could protect the mitochondria from damage induced either by proapoptotic proteins or by calcium accumulation and consequently reduce cell death.
In summary, transient focal ischemia induced the overexpression of the antiapoptotic protein Bcl-w in rat brain. Increased expression of Bcl-w was detected mainly in neurons and other cell types that survived ischemia but not in cells that developed DNA fragmentation. As Bcl-w is a mitochondrial membrane-bound protein and Bcl-w can inhibit mitochondrial damage induced by Bax or calcium in vitro, these results thus support a role for Bcl-w in protecting neurons against ischemia and reperfusion injury via mitochondrial mechanisms. Further investigation is warranted to confirm these effects by Bcl-w in vivo.
Footnotes
Acknowledgments
The authors thank Cristine O'Horo and Jingyun Luan for their excellent technical support.