
Abstract

Keywords
Neurons are metabolically active cells that generally are dependent on mitochondrial ATP production to meet their energetic needs. In contrast, other cell types with a high energy demand, including muscle or tumor cells, are better able to sustain ATP production through the glycolytic oxidation of stored glucose molecules when mitochondria are challenged or in oxygen debt. This places neurons at risk for overall cellular dysfunction when mitochondrial ATP production is compromised. Combining this metabolic susceptibility with the role that mitochondria play in the apoptotic pathway, it becomes clear that mitochondrial dysfunction is a particular liability for neurons. Mitochondrial electron transport, Ca2+ transport, free radical production, and participation in the apoptotic cascade were addressed in a companion review in the March 1999 issue of the Journal of Cerebral Blood Flow and Metabolism (Murphy et al., 1999). The current review addresses the evidence for mitochondrial failure in induction of neuronal death or dysfunction in ischemia/reperfusion injury as well as in chronic forms of neurodegeneration including Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), and Huntington's disease (HD).
MITOCHONDRIAL INVOLVEMENT IN ISCHEMIC NEURODEGENERATION
Substantial evidence indicates that mitochondrial dysfunction plays an important role in the pathophysiologic mechanism of acute neurodegeneration caused by trauma, stroke, and cardiac arrest (Hillered, 1986; Rosenthal and Fiskum, 1990; Sims and Zaidan, 1995 for review). Neuronal death in vivo typically is heterogeneous and exhibits morphologic characteristics that represent a continuum between necrosis and apoptosis (Portera-Cailliau et al., 1997; MacManus and Linnik, 1997). Rapid ischemic necrosis is primarily the result of anaerobic inactivation of mitochondrial oxidative phosphorylation, subsequent cellular deenergization, termination of active transport processes, and massive cellular influx of Ca2+ and other ions from activation of voltage-dependent ion channels and ligand-dependent channels (e.g., glutamate receptor operated channels). Calcium has the potential to overactivate degradative enzyme activities, including phospholipase, protease, and endonuclease activities, which ultimately cause widespread destruction of membranes and macromolecules (Siesjö, 1994; Werling and Fiskum, 1996) and, therefore, result in relatively rapid necrosis. Reperfusion of ischemic tissue can lead to swift reestablishment of Ca2+ homeostasis and potentially full recovery. However, ischemic alterations to mitochondrial oxidative phosphorylation, Ca2+ sequestration, and free radical production can result in sufficient chronic or secondary metabolic failure, oxidative stress, and altered cellular Ca2+ levels so that selectively vulnerable cells proceed toward some form of delayed death (Siesjo et al., 1995). Mitochondrial stress also can result in relatively subtle and reversible mitochondrial alterations that may specifically initiate the process of apoptosis without causing the level of metabolic failure that would induce necrosis (Siesjo et al., 1994).
Effects of cerebral ischemia and reperfusion on mitochondrial activities
Brain mitochondria are comparatively sensitive to ischemic injury and even exhibit substantial impairment in respiratory and electron transport chain complex activities after periods of moderately reduced cerebral blood flow, where there are no immediate changes in levels of ATP or phosphocreatine (Allen et al., 1993, 1995). It is therefore possible that the partial ischemia present within the penumbra surrounding a focal infarct as well as the variable degree of ischemia present within the focus of a stroke can result in mitochondrial alterations independent of the cellular energy state. The same argument also may apply to many areas of the forebrain that experience hypoperfusion for many hours after complete global cerebral ischemia, such as that which occurs after cardiac arrest. In either scenario, it is possible that incomplete ischemia can result in greater mitochondrial injury than complete ischemia as a consequence of the generation of reactive oxygen species (ROS) that attack mitochondrial lipids, proteins, and DNA and promote Ca2+-induced mitochondrial dysfunction (Rehncrona et al., 1979; Veitch et al., 1992; Allen et al., 1995).
In addition to brain mitochondria being sensitive to injury induced by incomplete ischemia, these organelles also exhibit rapid changes in electron transport and ion permeability characteristics within only a few minutes of complete cerebral ischemia. Mitochondrial injury in response to complete ischemia can be categorized and is dependent on the length of the ischemic period (Sims, 1991). In the first type of injury, which occurs after approximately 5 to 30 minutes of ischemia, depending on the animal model, maximal rates of respiration measured with isolated mitochondria or tissue homogenates are reduced by 25% to 75% (Ginsberg et al., 1977; Rehncrona et al., 1979; Hillered et al., 1985; White et al., 1985; Sims and Pulsinelli, 1987; Rosenthal et al., 1987; Sciamanna and Lee, 1992, 1993; Sun and Gilboe, 1994). Periods of complete ischemia in the range of 5 to 20 minutes represent those most relevant to human cardiac arrest when it is followed by successful cardiopulmonary resuscitation. Longer periods of ischemia generally are more applicable to the conditions present during a stroke. Most mitochondrial alterations that occur within the first 10 to 20 minutes of complete ischemia are reversible within an hour or two of reperfusion (Rehncrona et al., 1979; Sims and Pulsinelli, 1987; Rosenthal et al., 1987; Sims, 1991). However, the existence of these alterations during the critical, early period of reperfusion may contribute to the induction of a cascade of events that ultimately lead to one or more forms of delayed cell death or dysfunction and, consequently, neurologic impairment.
The second category of mitochondrial injury occurs during approximately 30 minutes to 2 hours of complete ischemia and probably is relevant to injury occurring in cells located within focal infarcts that undergo spontaneous reperfusion through vascular recanalization. This type of mitochondrial injury is represented by more extensive inhibition of electron transport throughout various regions of the electron transport chain (Linn et al., 1987). Unlike the alterations observed after short periods of complete ischemia, the respiratory impairment during this type of injury usually is only partially reversible without pharmacologic intervention and often becomes significantly worse within a few hours of reperfusion.
The third category of mitochondrial injury is characteristic of acute necrotic cell death in which prolonged periods of deenergization of approximately 2 hours or more cause generalized destruction to the structural and functional integrity of cellular membranes. All components responsible for oxidative phosphorylation, including the ATP synthase, are extensively damaged, and the inner membrane becomes so leaky to ions and molecules that an electrochemical gradient of protons sufficient to drive ATP synthesis and Ca2+ uptake cannot be generated. Reperfusion after these extensive periods of complete ischemia results in little improvement in mitochondrial activities and is not effective at reducing acute cell death. Nevertheless, some neuroprotective interventions may partially improve mitochondrial activities and reduce neuronal injury under these extreme conditions (Kuroda et al., 1996a, 1996b; Folbergrova et al., 1995). Such acute and mainly irreversible cell death may be applicable to cells within the core of focal ischemic infarcts that are deprived of O2 and glucose for several hours or more (Kuroda et al., 1996a). However, spontaneous reperfusion occurs even within the core of cerebral infarcts in humans after thromboembolytic stroke (Jorgensen et al., 1994) and, therefore, most acute ischemic mitochondrial injury that occurs during human stroke, cardiac arrest, and head trauma likely falls within the first two categories.
The fourth type of mitochondrial injury is characterized by reperfusion-dependent alterations. Depending on the duration and degree of ischemia, reperfusion may be accompanied by changes in the activity of specific electron transport chain components. As with acute ischemic injury, the flow of electrons through complex I is most sensitive to early, reperfusion-dependent inhibition after complete ischemia, although other complexes can be involved to a lesser extent (Sims, 1991; Sciamanna and Lee, 1993; Almeida et al., 1995). Such injury also is exacerbated when reperfusion follows incomplete rather than complete ischemia (Rehncrona et al., 1979). Complex IV (cytochrome oxidase [COX]) activity also declines significantly after complete ischemia or anoxia plus one to several hours of reperfusion in some models (Wagner et al., 1990; Nelson and Silverstein, 1994; Abe et al., 1995) but not in others (Sims, 1991, Canevari et al., 1997; Zaidan and Sims, 1997). Reperfusion periods of 24 hours or more result in additional generalized damage to the mitochondrial electron transport chain (Sims, 1991). Several laboratories describe an inhibition of the maximal activity of pyruvate dehydrogenase (PDH), a mitochondrial matrix enzyme, at periods of reperfusion of 30 minutes to 24 hours after 10 to 30 minutes of complete cerebral ischemia (Katayama and Welsh, 1989; Zaidan and Sims, 1993, 1997; Bogaert et al., 1994;). The anatomical distribution of injury to PDH also corresponds well with the spatial distribution of neuronal vulnerability to delayed death during ischemia and reperfusion (Zaidan and Sims, 1993).
In addition to the forms of mitochondrial injury apparent with mitochondria isolated from brain samples after cerebral ischemia and reperfusion, other aspects of mitochondrial dysfunction are likely to exist in situ that are readily reversible and therefore not apparent once the tissue is processed and the mitochondria are isolated. For example, low tissue pH is known to inhibit respiration (Hillered et al., 1984, see later). It also is possible that other factors (e.g., the presence of free fatty acids and Ca2+ accumulated within the mitochondrial matrix) result in additional mitochondrial respiratory inhibition and membrane leakiness that is not apparent once the mitochondria are isolated and these factors have been lost (Hillered and Chan, 1988). The activity of COX in the brain after ischemia/reperfusion also may be grossly overestimated by O2 electrode and spectrophotometric measurements performed in vitro because of the fact that nitric oxide exerts a readily reversible but potent competitive inhibition with O2 for one or more active sites in this complex (Brown, 1995; Giuffre et al., 1996). Since at least one form of nitric oxide synthase appears to be mitochondrially localized (Bates et al., 1995a; Giulivi et al., 1998) and the generation of nitric oxide is greatly accelerated in the reperfused brain (Samdani et al., 1997), this agent may limit the rate of postischemic cerebral O2 consumption, particularly in regions and at times when tissue O2 concentrations are abnormally low from hypoperfusion (Bates et al., 1995b). Thus, the true degree of mitochondrial dysfunction within the brain is likely underestimated by measurements performed with mitochondria displaced from the extremely abnormal postischemic intracellular environment.
Consequences of mitochondrial injury
Consequences of mitochondrial injury after cerebral ischemia and reperfusion can be numerous and include metabolic failure, oxidative stress, exacerbation of excitotoxicity through impaired intracellular Ca2+ buffering, and promotion of apoptosis through release of apoptogenic factors such as cytochrome c (Fig. 1). There is considerable evidence that mitochondrial dysfunction can limit the return and continued maintenance of normal levels of ATP and other high-energy metabolites after both global and focal cerebral ischemia (Chang et al., 1992; Katayama and Welsh, 1989; Kuroda et al., 1996a; Siesjö et al., 1995). A decrease in the rate at which mitochondria can generate ATP also may be responsible for the chronic postischemic tissue lactic acidosis observed in some models of ischemic brain injury (Rosenthal et al., 1992). Support for the possibility that inhibition of PDH is a least partially responsible for postischemic lactate accumulation comes from observations demonstrating reduction of tissue lactate levels and improvement of neurologic outcome by administration of agents such as acetylcarnitine and 1,3-butanediol, which have the potential to generate acetyl coenzyme A, a product of the PDH reaction (Marie et al., 1987; Rosenthal et al., 1992; Gueldry and Bralet, 1994; Aureli et al., 1994; Shuaib et al., 1995). The finding that brain pyridine nucleotides and electron transport chain components are hyperoxidized for a substantial period during reperfusion also indicates that a primary metabolic defect resides at one or more reactions that generate NADH for use by the electron transport chain (Rosenthal et al., 1995; Pérez-Pinzòn et al., 1997). It remains to be established whether the degree of inhibition of the activity of PDH or any or the components of the electron transport chain is sufficient to be responsible for postischemic metabolic derangements and whether these relations specifically contribute to the selective vulnerability of different neuronal subclasses to delayed postischemic death.
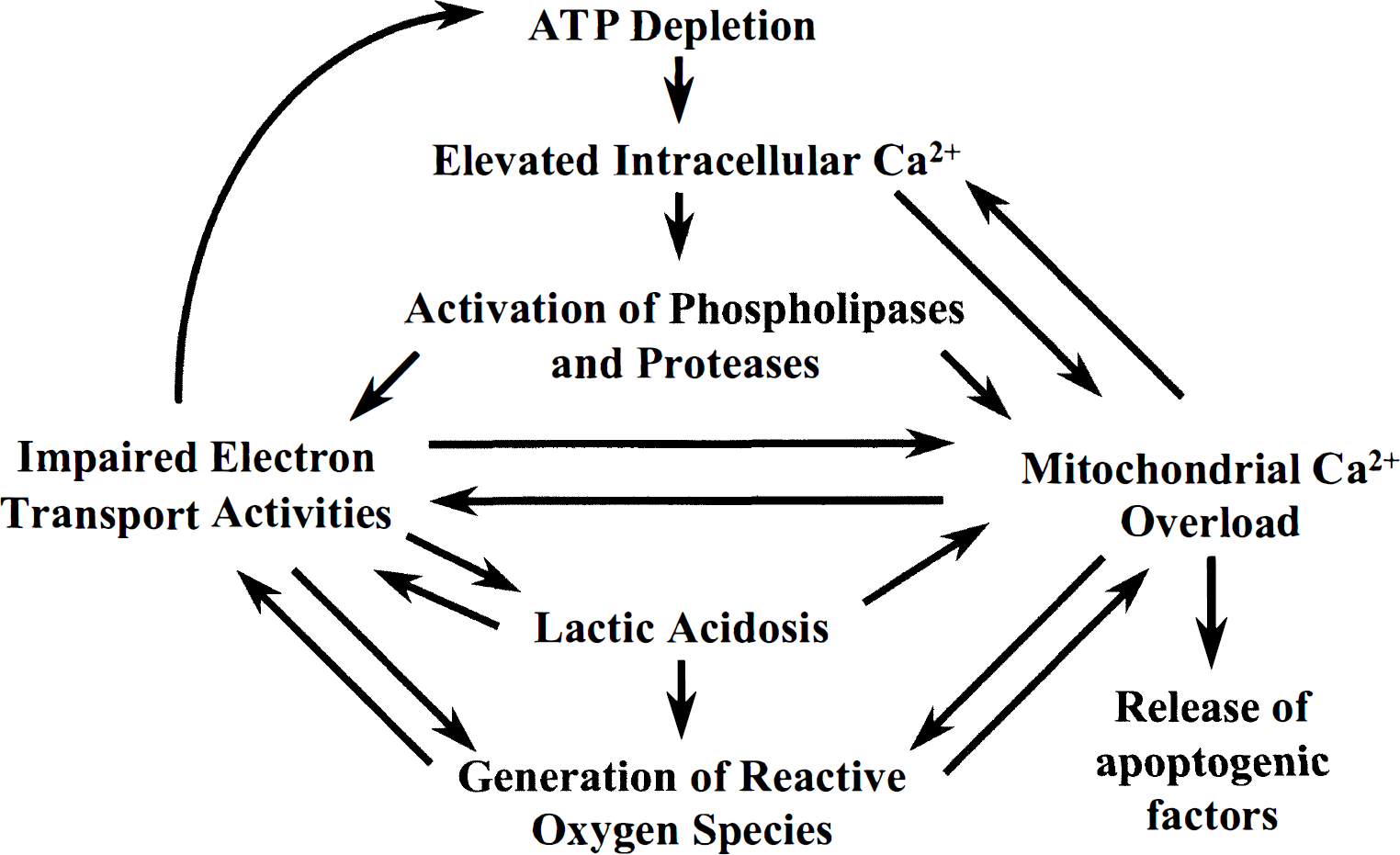
Relations between altered mitochondrial activities and abnormal neuronal energy metabolism, Ca2+ homeostasis, reactive oxygen species generation, and release of apoptogenic factors during cerebral ischemia and reperfusion.
In addition to the influence that ischemic and post-ischemic mitochondrial injury has on cerebral energy metabolism, mitochondrial alterations may significantly influence postischemic intracellular Ca2+ homeostasis. Because of the technical difficulties involved in measuring intracellular Ca2+ levels within the brain, the primary evidence for importance of mitochondrial Ca2+ transport in determination of neuronal viability after reperfusion comes indirectly from studies reviewed in the March 1999 issue of Journal of Cerebral Blood Flow and Metabolism (Murphy et al., 1999) demonstrating the importance of mitochondrial Ca2+ accumulation in excitotoxic death (see also Reynolds and Hastings, 1995, Dugan et al., 1995, Ankarcrona et al., 1995, Budd and Nicholls, 1996; Stout et al., 1998). Mitochondrial sequestration clearly is taking place during reperfusion, since mitochondria isolated from reperfused brain contain higher loads of Ca2+ (see later, Zaidan and Sims, 1994). However, damage to mitochondria lowers the capacity for energy-dependent Ca2+ sequestration by isolated brain mitochondria after as little as 10 minutes of global cerebral ischemia (Sciamanna et al., 1992; Fiskum et al., 1997). A 5-hour period of reperfusion after 30 minutes of forebrain ischemia in the rat also has been reported to greatly inhibit the ability of isolated mitochondria to actively accumulate and retain Ca2+ (Sciamanna and Lee, 1993). Whether this reduction in active Ca2+ uptake and retention limits the rate and extent of normalization of intracellular Ca2+ during cerebral reperfusion is unknown. Regardless, this reduction in the ability to accumulate supranormal levels of extramitochondrial Ca2+ is associated with an increased sensitivity to Ca2+-induced respiratory inhibition and to Ca2+-stimulated release of the apoptotic factor cytochrome c (Fiskum et al., 1997).
Mitochondria are a primary source of ROS in normal, healthy tissue. Although mitochondria often are also proclaimed to be the primary generators of ROS during reperfusion, there are only a few key studies performed in vivo that support this hypothesis. Using microdialysis procedures, Piantadosi and Zhang (1996) demonstrated that cerebral cortex free radical production increases during reperfusion and that addition of a complex I electron transport chain inhibitor substantially attenuates this elevation. Other evidence obtained with electron spin resonance spectroscopy indicates that radicals are generated at the level of the electron transport chain component coenzyme Q during cerebral hypoxia and reoxygenation (Hasegawa et al., 1993). As with the potential for mitochondrial involvement in secondary metabolic failure and altered intracellular Ca2+ levels, additional studies are needed to establish direct relations between mitochondrial free radical production and oxidative reperfusion injury and to determine how such interactions relate to selective neuronal vulnerability to delayed death.
Mediators of mitochondrial injury
Several interrelated factors have been proposed to contribute to mitochondrial impairment after cerebral ischemia and reperfusion. These factors include low intracellular pH, high levels of intracellular Ca2+, and oxidative stress caused by elevated production of ROS. Since these are the same pathologic conditions that are exacerbated by mitochondrial dysfunction, a vicious cycle of subcellular injury and abnormal intracellular conditions can be generated (Fig. 1).
Lactic acidosis has long been considered as a mechanism responsible for mitochondrial injury during cerebral ischemia. Support for this hypothesis has come from experiments in which increased mitochondrial respiratory impairment has been associated with increased systemic acidosis during the ischemic period (Rehncrona et al., 1979). These conditions were invoked by inducing incomplete ischemia rather than by completely eliminating cerebral blood flow. Since incomplete ischemia likely also promotes the generation of ROS, the exacerbation of mitochondrial injury may have been caused by increased oxidative stress rather than aggravated acidosis. Other studies demonstrate that hyperglycemia before complete cerebral ischemia exacerbates accumulation of brain lactate and prolongs the reperfusion time necessary for full recovery of mitochondrial respiratory characteristics (Kim et al., 1996; Hillered et al., 1985). Experiments performed with isolated brain mitochondria have clearly demonstrated that respiration is sensitive to inhibition by acidosis, with state 3 O2 consumption being inhibited by 70% at pH 6.1 compared with that observed at pH 7.2 (Hillered et al., 1984). Early, postischemic brain acidosis would therefore be expected to impair oxidative phosphorylation and limit the rate at which the concentrations of high-energy phosphate-containing compounds return to normal; however, measurements of postischemic ATP and phosphocreatine levels have not supported this hypothesis (Hillered et al., 1985). Acidic pH also may affect other mitochondrial activities such as ROS production and Ca2+ sequestration. Although under some circumstances acid pH reduces the maximal capacity for mitochondrial Ca2+ sequestration (Åkerman, 1978), low pH also may inhibit the mitochondrial membrane permeability transition (Bernardi et al., 1992). Because the disadvantages or advantages of mild acidosis during and after cerebral ischemia are controversial (Levine, 1993; LaManna et al., 1995; Schurr et al., 1997), more information on the effects of pH on the normal and abnormal activities of brain mitochondria should be obtained.
Mitochondrial energy transduction is highly dependent on membrane-linked oxidation/reduction reactions and ion transport activities. It is therefore not surprising that ischemic mitochondrial derangements have been associated with alterations to membrane lipids. Free fatty acids are released from phospholipids and accumulate rapidly in the brain after just a few minutes of cerebral ischemia (Bazan, 1970; Katsura et al., 1993). Several studies demonstrate that ischemic brain mitochondria specifically undergo phospholipid degradation and accumulate free fatty acids and that such accumulation can be at least partially responsible for the respiratory impairment evident after cerebral ischemia (Hillered and Chan, 1988; Nakahara, et al., 1991; Sun and Gilboe, 1994a). Moreover, postischemic normalization of brain mitochondrial lipids has been associated with normalization of mitochondrial respiration (Sun and Gilboe, 1994b).
Mitochondrial lipids also are subject to oxidative modifications during cerebral ischemia/reperfusion. Brain mitochondrial malondialdehyde, an oxidized lipid degradation product, accumulates both during reperfusion and during complete global ischemia (Sun and Gilboe, 1994a, 1994b). Substantial evidence points to membrane lipid oxidation as an important component of reperfusion brain injury (Traystman et al. 1991; Chan, 1996; Liu et al., 1998), although the degree to which mitochondrial lipid oxidation contributes to injury is unknown. Mitochondrial lipid oxidation can occur during ischemia alone and continues during reperfusion (Sun and Gilboe, 1994a). Mitochondrial malondialdehyde levels are lowered by postischemic treatment of rats with platelet activating factor antagonists (Sun and Gilboe, 1994b). Also, the protection against reperfusion-dependent respiratory impairment afforded by either preischemic or postischemic treatment of rats with the free radical scavenger ascorbate may be due to inhibition of mitochondrial lipid oxidation (Sciamanna and Lee, 1993).
In addition to oxidative modification to lipids, indirect evidence indicates that certain mitochondrial proteins may be subject to oxidative injury. Bogaert and colleagues (1994) demonstrated that the activity of PDH is reduced during reperfusion in a model in which sitespecific brain protein oxidation occurs (Liu et al., 1993). Bogaert and coworkers (1994) and Tabatabaie and associates (1996) also have demonstrated that this protein is particularly sensitive to oxidative inactivation. However, it remains to be established whether the mechanism responsible for the loss of this important enzyme activity in vivo is through oxidative injury, proteolysis, or other means. It is also possible that oxidative molecular alterations are additionally responsible for injury to the electron transport chain. Exposure of isolated brain mitochondria to experimental conditions that generate superoxide and hydroxyl radicals results in patterns of respiratory injury similar to those observed in vivo (Hillered and Ernster, 1983). Specifically, state 3 respiration is dramatically inhibited with little or no increase in state 4 respiration, and the inhibition is more profound when NADH-linked substrates (e.g., malate) rather than FADH-linked substrates (e.g., succinate) are oxidized. Direct oxidation of electron transport proteins may occur during reperfusion but is less likely to happen after ischemia alone, since oxidation of brain proteins has only been detected after at least a few hours of reperfusion (Oliver et al., 1990; Liu et al., 1993). Mitochondrial proteins may possibly undergo oxidative injury, including that resulting from sulfhydryl oxidation as a consequence of postischemic exposure to peroxynitrite, which is formed from a reaction between superoxide and nitric oxide (Radi et al., 1991a). Although isolated mitochondria and mitochondria present within cultured neurons are sensitive to respiratory inhibition by peroxynitrite (Radi et al., 1994), the primary site of inhibition appears to be one or more of the components that catalyze succinate/cytochrome c oxidoreductase activity, whereas complex I electron transport activity is resistant to inactivation by peroxynitrite (Bolaños et al., 1994, 1995). Since complex I activity is substantially more sensitive than complex II and III to injury caused by global ischemia and reperfusion, peroxynitrite-mediated mitochondrial damage may be more relevant to prolonged focal cerebral ischemia in which substantial loss of succinate dehydrogenase activity has been observed (Kuroiwa et al., 1994). As previously indicated, inhibition of electron transport activities evident after both ischemia alone and after ischemia/reperfusion also could be an indirect consequence of mitochondrial membrane lipid oxidation (Soussi et al., 1990), which can be catalyzed by peroxynitrite (Radi et al., 1991b).
Several lines of evidence suggest that Ca2+ plays an important role in mitochondrial injury during cerebral ischemia and reperfusion. Preischemic treatment of animals with agents that either directly or indirectly inhibit cellular Ca2+ influx results in substantial protection against mitochondrial respiratory impairment after 10 minutes of cardiac arrest (Rosenthal et al., 1987). Ca2+-induced injury during ischemia could occur by several different mechanisms, including stimulation of phospholipid degradation through activation of phospholipases (Rordorf et al., 1991) or induction of proteolysis through activation of proteolytic enzymes such as calpains (Aguilar et al., 1996). Such mechanisms may respond directly to an increase in the cytosolic free Ca2+ concentration or be initiated by mitochondrially accumulated Ca2+.
Since the driving force for mitochondrial Ca2+ uptake is the respiration-generated membrane potential that exists across the inner membrane, the absence of O2 during complete cerebral ischemia should severely limit mitochondrial Ca2+ accumulation. Nevertheless, a small but significant time-dependent net accumulation of mitochondrial Ca2+ occurs in complete cerebral ischemia (Sciamanna et al., 1992; Zaidan and Sims, 1994). Substantially more Ca2+ is found in brain mitochondria isolated after only a few minutes of reperfusion (Zaidan and Sims, 1994). These observations with isolated mitochondria are supported by cytochemical staining procedures demonstrating early reperfusion-dependent mitochondrial calcium deposits that disappear within a few hours of reperfusion and reappear after 1 to 2 days of recirculation (Hossmann et al., 1985; Dux et al., 1987). Substantial accumulation of calcium by brain mitochondria within the first few minutes of reperfusion would be expected, since electrophysiologic measurements of intraneuronal Ca2+ indicate an increase in concentration from a baseline of less than 0.2 μM to greater than 30 μM in hippocampal neurons after 5 to 10 minutes of complete ischemia (Silver and Erecinska, 1990).
During excessive reperfusion-dependent mitochondrial Ca2+ sequestration, mitochondrial may undergo reversible or irreversible damage by several different mechanisms. Reperfusion-dependent, Ca2+-mediated mitochondrial injury can be described as “mitochondrial Ca2+ overload” (Fig. 1) and may include the membrane permeability transition as well as molecular alterations that can be independent of mitochondrial swelling and insensitive to inhibitors of the permeability transition (e.g., cyclosporin A) (Murphy et al., 1999). Siesjö and colleagues suggest that mitochondrial Ca2+ overload together with oxidative stress induce the permeability transition and bioenergetic failure after several hours of reperfusion (Siesjö et al., 1995; Kuroda et al., 1996a; Folbergrová et al., 1997). Although a delayed permeability transition is consistent with the temporal course of delayed tissue and mitochondrial Ca2+ accumulation and with some measures of mitochondrial respiratory impairment, many of the intracellular conditions present during the first 10 to 30 minutes of reperfusion suggest that a transition could occur within this period. These conditions include exposure of respiring mitochondria to high levels of Ca2+ and abnormal levels of ROS in the presence of a relatively oxidized redox state of pyridine nucleotides (Silver and Ericinska, 1990; Eleff et al., 1991; Piantidosi and Zhang, 1996; Rosenthal et al., 1995). Mitochondrial Ca2+ overload may also contribute to the release of cytochrome c and other apoptogenic factors from the miochondria into the cytosol (Fig. 1), as observed after both focal and global cerebral ischemia (Fujimura et al., 1998; Perez-Pinzon et al., 1999). As discussed in Murphy et al. (1999), this Ca2+-induced phenomenon may be the consequence of the permeability transition or due to the activation of a specific channel located in the mitochondrial outer membrane.
MITOCHONDRIA IN CHRONIC FORMS OF NEURODEGENERATION
Correlation of mitochondrial dysfunction with aging
Substantial evidence indicates that mitochondrial function declines with age. The fact that normal aging is the most reliable and robust risk factor for neurodegenerative diseases is likely beyond coincidence. It has been suggested that mitochondrial DNA (mtDNA) is both more susceptible to oxidative damage and is repaired less efficiently. Studies of hydrogen peroxide-treated human fibroblasts show threefold more damage to a fragment of mtDNA compared with a fragment of nDNA, and the damage was much more persistent (Yakes and Van Houten, 1997). Numerous studies shown an age-dependent accumulation of mtDNA deletions in both animals and man (reviewed in Beal, 1995).
A critical issue is whether mtDNA deletions or point mutations have functional consequences. This was addressed in an elegant study of Laderman and colleagues (1996). A novel technique pioneered by King and Attardi is to transfer mitochondria from patients to mtDNA-deficient cells (ρ° cells) (King and Attardi, 1989). Cell lines can be depleted of mtDNA by exposing them to low concentrations of ethidium bromide. Ethidium bromide is concentrated within mitochondria and intercalates into mtDNA at concentrations that inhibit replication of mitochondrial but not nuclear DNA. Exposed cells assume an anaerobic phenotype and become auxotrophic for uridine and pyruvate. Cybrids are formed by fusing mtDNA-depleted cells with the platelets of enucleated cells (containing mitochondria) of patients. Removal of uridine and pyruvate from the medium causes death of any untransformed cells and thus serves as a selection tool.
The resulting cybrids enable determination as to whether any defects in oxidative phosphorylation are attributable to alterations in a patient's mtDNA, since the patient's mitochondria now function in the presence of a different nuclear genome. Laderman and colleagues took enucleated fibroblasts (containing mitochondria) of normal human individuals aged 20 weeks to 103 years and fused them with ρ° cells. The ensuing cybrid cell lines showed an age-dependent decrease in O2 consumption. Although there was an age-dependent decrease in mtDNA content, this did not correlate with O2 consumption. These findings therefore support the notion that age-dependent accumulation of structural damage to the mitochondrial genome has functional consequences.
An interesting animal model of aging is that which occurs in prosimian primates (Schmechel et al., 1996). These animals show accelerated brain aging starting in early adulthood. They initially develop dystrophy of cholinergic axons and generalized gliosis at 10% of maximal life span, followed by extensive cerebral amyloidosis, which commences at 50% maximal life span. Consequently, these animals represent a partial model for both accelerated aging and for AD. Studies show that these primates have mitochondrial abnormalities in both the heart and renal proximal tubules as early as 1 year of age. In cholinergic neurons, which show axonal dystrophy, there are both profiles of degenerating mitochondria as well as spheroids, which are immunoreactive for manganese superoxide dismutase (SOD). Mitochondrial abnormalities occurred early and were closely associated with axonal dystrophy. These findings link mitochondrial dysfunction to both age-related axonal dystrophy and potentially to β-amyloid deposition.
Mitochondria in Alzheimer's disease
Alzheimer's disease is the most common of the neurodegenerative diseases. Advancing age is the most important risk factor, with a 10-fold increase in prevalence between ages 60 and 80 years, and a prevalence of 47% for patients older than age 85 years (Evans et al., 1989). The essential clinical features are an early impairment of recent memory followed by progressive intellectual dysfunction. The pathologic hallmarks consist of deposition of amyloid in the form of senile plaques and the development of neurofibrillary tangles. Familial AD is autosomal dominant and accounts for about 5% of all cases. These patients were found to have mutations in either the amyloid precursor protein or in novel proteins termed presenilins. Both of these genetic defects may be associated with increased production of β-amyloid1–42, which shows an increased tendency to aggregate.
Most cases of AD, however, appear to be sporadic, although there is an increased incidence in families. If mitochondrial defects play a role in this form of AD, it would be expected that there would be evidence for maternal inheritance, since the mitochondrial genome is inherited solely from the mother. Several studies suggest that this is the case. These studies show an increased female-to-male ratio of 3.6:1 or 3.8:1 in the parental generation of probands (Duara et al., 1993; Edland et al., 1996). In 10 families with one affected parent and at least two affected siblings, the ratio of mothers to fathers was 9:1 (Edland et al., 1996).
Evidence for metabolic deficits in AD comes from positron emission tomography (PET) studies, which show reduced glucose metabolism in the temporoparietal cortex. Changes occur early in the disease when there is minimal cognitive impairment. The PET studies also show increased O2 utilization in comparison with glucose utilization (Fukuyama et al., 1994). This finding was confirmed in studies of arteriovenous extraction of O2 and glucose (Hoyer, 1993; Ogawa et al., 1996). The molar ratio of O2 to glucose utilization in AD patients was 9.86 compared with 5.63 for seven age-matched controls. Abnormal glucose metabolism in brain biopsy specimens has been interpreted as partial uncoupling of mitochondria (Sims et al., 1987). Phosphorus magnetic resonance spectroscopy shows reduced phosphocreatine in cerebral cortex of mildly demented AD subjects (Pettegrew et al., 1995) and increased inorganic phosphate (Pi) in frontal and temporoparietal cortex (Brown et al., 1989). In frontal cortex, the phosphocreatine-inorganic phosphate ratio (PCr/Pi) was significantly reduced in AD compared with elderly controls (Smith et al., 1995). Other evidence for mitochondrial abnormalities comes from ultrastructural studies, which show paracrystalline inclusions in AD mitochondria (Saraiva et al., 1985).
Studies of electron transport enzymes in AD show reduced complex IV activity. Parker and colleagues report that complex IV activity was reduced in platelet mitochondria (Parker et al., 1989), which was disputed (Van Zuylen et al., 1992) but confirmed in a follow-up study (Parker et al., 1994). In postmortem brain tissue, there is reduced complex IV gene expression and enzyme activity in frontal and temporal cortex (Kish et al., 1992; Chandrasekaran et al., 1996). We found consistent reductions in complex IV activity in four cortical regions and no consistent alterations in the activities of complexes I, II-III or V (Mutisya et al., 1994). A study of highly purified mitochondria from AD brain tissue shows reduced catalytic activity and perturbed enzyme kinetics, yet normal amounts of cytochrome aa3, the heme-containing component of complex IV (Parker et al., 1994). This suggests that reduced complex IV enzyme activity is a consequence of abnormal catalytic activity rather than decreased enzyme levels.
What is the source of the reduced complex IV activity in AD brain and platelets? It was demonstrated that COX defects can be transferred to cybrids from AD platelets and that the ensuing cybrid cell lines show increased free radical production (Davis et al., 1997; Swerdlow et al., 1997). This suggests that a mtDNA defect results in decreased COX activity. Initial work suggests that point mutations in the COX1 and COX2 genes segregate with most AD patients (Davis et al., 1997), but more recent evidence shows that the presumed mutations are polymorphisms in a nuclear pseudogene (Hirano et al., 1997; Wallace et al., 1997). These polymorphisms therefore cannot be responsible for decreases in complex IV activity in AD. The etiology of the defect in COX activity and mitochondrial function in AD cybrids therefore requires further investigation.
Several other mtDNA mutations were reported in association with AD. A ND2 mtDNA mutation in AD was found in one study (Lin et al., 1992); however, this was not confirmed in two other studies (Kosel et al., 1994; Petruzzella et al., 1992). Shoffner and colleagues found a mutation in nucleotide 4336 of the tRNAgln sequence in patients with AD and PD. The mutation was found in 5.2% of patients with AD-PD and in 0.7% of controls (Shoffner et al., 1993). This finding was confirmed in a follow-up study (Hutchin and Cortopassi, 1995) but disputed in another (Wragg et al., 1995).
The consequences of COX defects in the cybrid cell lines on intracellular Ca2+ buffering have been examined. The AD cybrids show elevated basal cytosolic Ca2+ concentrations and enhanced sensitivity to inositol-1,4,5-triphosphate-mediated Ca2+ release, and they recover more slowly from the increased Ca2+ levels (Sheehan et al., 1997). These findings are consistent with findings in AD fibroblasts (Gibson et al., 1996; Huang et al., 1991; Ito et al., 1994) and with the finding of decreased Ca2+ uptake in mitochondria from AD fibroblasts (Kumar et al., 1994). Impaired Ca2+ buffering also is observed in fibroblasts from patients with mitochondrial encephalopathy lactic acidosis and strokes (MELAS) syndrome (Moudy et al., 1995).
If mitochondrial dysfunction truly plays a central role in the pathogenesis of AD, then an increase in oxidative damage might be expected as a consequence. Mitochondrial DNA would be expected to be particularly susceptible, since it is located close to the inner mitochondrial membrane. Consistent with this possibility, there is a threefold increase in oxidative damage to mtDNA in postmortem tissue from AD patients compared with age-matched controls (Mecocci et al., 1994). Other studies found increased concentrations of malondialdehyde and protein carbonyl groups and oxidative damage to nuclear DNA (reviewed in Beal, 1995; Lyras et al., 1997). Novel spin trapping techniques showed oxidative damage to both lipids and proteins (Hensley et al., 1995). There also is evidence for oxidative damage at a cellular level. Neurofibrillary tangle-bearing neurons show immunostaining with antibodies to advanced glycation end products, hemeoxygenase-1, malondialdehyde, 4-hydroxynonenal, protein carbonyl groups, carbonylated neurofilaments, and 3-nitrotyrosine (Smith et al., 1995; Smith et al., 1994; Smith et al., 1996; Yan et al., 1994; Schipper et al., 1995; Premkumar et al., 1995; Good et al., 1996; Sayre et al., 1997; Smith et al., 1997). Interestingly, these techniques all show increased staining in cell bodies and neurites rather than senile plaques, suggesting an intracellular source of free radicals.
Since the most consistent pathologic abnormality in AD is accumulation of amyloid in senile plaques, an important issue is whether impaired oxidative metabolism can contribute to both β-amyloid deposition and the generation of neurofibrillary tangles. Gabuzda and colleagues first showed that impairment of COX in vitro leads to an increase in c-terminal fragments of the amyloid precursor protein that contain the β-amyloid peptide (Gabuzda et al., 1994). An increase in intracellular β-amyloid1–42 was found after exposure of cultured guinea pig neurons to hydrogen peroxide (Ohyagi and Younkin, 1996), and oxidative stress increased β-amyloid in mammalian lenses (Frederikse et al., 1996). Increased accumulation of intracellular β-amyloid also was observed in cultured astrocytes from patients with Down's syndrome, which have increased free radical production (Busciglio and Yankner, 1995). Oxidative damage leads to cross-linking and impaired solubility of β-amyloid, which may contribute to its deposition (Dyrks et al., 1992; Dyrks et al., 1993). In vivo studies show that an impairment of oxidative metabolism produced by thiamine deficiency leads to an accumulation of amyloid precursor protein in abnormal neurites (Calingasan et al., 1996; Calingasan et al., 1995). Oxidative injury may contribute to the production of paired helical filaments, since oxidation of cysteine 322 on tau forms intermolecular cross-bridges, and this leads to generation of paired helical filaments in vitro (Schweers et al., 1995). Impaired ATP production also can activate the PK40 kinase, which can convert tau into a paired helical filament-like form (Roder and Ingram, 1991; Roder et al., 1993). These studies therefore suggest that energy impairment associated with COX deficiency could be responsible for amyloid deposition in AD.
Cell loss in AD, which may include apoptotic death (see Murphy et al., 1999), greatly exceeds the number of neurofibrillary tangles that accumulate, and more than 50% of the neurons are lost in the superior temporal gyrus (Gomez-Isla et al., 1997). Mitochondrial dysfunction may contribute to this cell loss.
Mitochondria in Parkinson's disease
Much of the interest in the association of neurodegen-eration with mitochondrial dysfunction and oxidative damage developed from studies of parkinsonism induced by 1-methyl-4-phenyl-1,2,3,5-tetrahydropyridine (MPTP) (Bloem et al., 1990). This MPTP was inadvertently synthesized as a byproduct of synthesis of “designer” opiates. It led to an outbreak of parkinsonism in southern California after injection by intravenous drug abusers. It was subsequently shown to produce parkinsonism in both mice and primates. The MPTP molecule is converted to 1-methyl-4-phenylpyridinium (MPP+) by monoamine oxidase B. Then, MPP+ is taken up by the dopamine transporter and accumulates in mitochondria, where it inhibits complex I activity by binding reversibly to a site at or near the rotenone binding site. The MPP+ toxicity is associated with oxidative damage, since MPP+ induces superoxide formation and increases lipid peroxidation.
The most consistent defect in oxidative phosphorylation in PD is reduced complex I activity (Beal, 1995). In studies of PD brain tissue, complex I activity is reduced in the substantia nigra and is not altered in other brain regions. Three well-controlled studies show reduced complex I activity in PD platelets, including unmedicated patients (Parker et al., 1989; Benecke et al., 1992; Haas et al., 1995). We found reduced coenzyme Q10 levels in PD platelet mitochondria, which correlate with reduced complex I activity (Schults et al., 1997). A decrease in complex I activity also was reported in some studies of skeletal muscle and lymphocytes. Immunostaining for complex I subunits and for α-ketoglutarate dehydrogenase, a regulatory enzyme of the tricarboxylic acid cycle, is reduced in PD substantia nigra (Hattori et al., 1991; Mizuno et al., 1994).
If a mtDNA defect plays a role in the pathogenesis of PD, it should be possible to identify evidence for maternal inheritance. In five families in which a parent and multiple siblings had PD, the affected parent was the mother (Wooten et al., 1997). There also appeared to be an early age of onset in children of affected mothers. Several mtDNA point mutations were reported to be associated with PD, but this has not been confirmed (Lucking et al., 1995; Ozawa et al., 1991).
Mitochondrial complex I deficits from platelets of PD patients were expressed in cybrid cell lines, and the complex I deficit was associated with increased free radical production, leading to apoptotic death (Swerdlow et al., 1996; Gu et al., 1998). Furthermore, these cybrid cell lines showed increased vulnerability to MPP+, and the cell lines also have impaired Ca2+ buffering (Sheehan et al., 1997).
Other evidence of impaired oxidative phosphorylation in PD is decreased cortical and subcortical glucose metabolism as assessed by PET, and increases in occipital lobe lactate using chemical shift magnetic resonance spectroscopy (Bowen et al., 1995; Eberling et al., 1994). An increase in basal ganglia lactate concentrations was found in 12 of 24 patients studied (Chen et al., 1994). One study using phosphorus magnetic resonance spectroscopy showed a decreased PCr/Pi ratio in resting forearm muscle of PD patients; however, this was not seen in other studies (Penn et al., 1995; Taylor et al., 1994).
Direct evidence for oxidative damage in PD comes from studies showing elevations in both malondialdehyde and in cholesterol hydroperoxides (Dexter et al., 1994). Furthermore, concentrations of 8-OHdG are increased threefold to fourfold in the caudate and substantia nigra of PD patients (Sanchez-Ramos et al., 1994). Reduced concentrations of glutathione are found both in the substantia nigra of PD patients and in incidental Lewy body disease, which may be a presymptomatic phase of PD (Sofic et al., 1992; Sian et al., 1994; Dexter et al., 1994). Increased nitrosyl complexes were recently found in PD substantia nigra (Shergill et al., 1996). An immunohistochemical study shows increased 4-hydroxynonenal protein adducts, a marker of lipid peroxidation, in PD substantia nigra neurons (Yoritaka et al., 1996) and Lewy bodies stain with antibodies to hemeoxygenase-1 and glycation end products (Castellani et al., 1996).
Mitochondria and oxidative damage in amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis is a prototypical neurodegenerative disease of late life characterized by progressive muscle weakness, atrophy, and spasticity, which ultimately leads to paralysis and death within 3 to 5 years (Brown, 1995). The essential neuropathologic features consist of a loss of motor neurons in the anterior horns of the spinal cord, brain stem, and motor cortex. In the motor neurons, there are abnormal accumulations of neurofilaments as well as proximal axonal swellings (Rouleau et al., 1996). Ninety percent of ALS cases are sporadic (SALS) with no identifiable genetic or environmental risk factors. The remaining 10% of cases show familial inheritance, and one fourth of these are associated with missense mutations in the antioxidant enzyme copper/zinc superoxide dismutase (SOD1) (Rosen et al., 1993).
The observation that mutations in SOD cause familial ALS (FALS) suggests that a loss of function of this enzyme might lead to oxidative damage of the motor neuron. However, several factors argue that the disease arises from an adverse property of the mutant SOD1 protein. These include the dominant inheritance pattern in FALS; the lack of correlation between enzyme activity and disease severity (Bowling et al., 1995), and the observation that overexpression of the mutant enzyme in transgenic mice (which have increased SOD activity) leads to motor neuron degeneration (Gurney et al., 1994; Ripps et al., 1995; Wong et al., 1995). With rare exceptions, the mutations occur outside of the active site of the enzyme; all mutations that have been studied appear to alter the stability of the protein backbone and to reduce the half-life of the molecule (Borchelt et al., 1994). These changes may relax the conformation of the active channel, thereby allowing increased access of hydrogen peroxide or peroxynitrite to the active site copper. This is predicted to increase generation of hydroxyl radicals or nitronium ions, which can nitrate proteins (Beckman et al., 1993). In vitro studies show that SOD with two different FALS mutations generates hydroxyl radicals more readily than the wild-type molecule (Wiedau-Pazos et al., 1996; Yim et al., 1996). We confirmed this in vivo using microdialysis in transgenic mice carrying the G93A mutation (Bogdanov et al., 1998).
Several studies suggest the involvement of energy metabolism defects in the pathogenesis of SALS, including observations of reduced glucose metabolism in the cerebral cortex of SALS patients in PET studies (Hatazawa et al., 1988; Dalakas et al., 1987). We found that complex I activity was significantly elevated by 55.3% in frontal cortex (Brodmann area 6) of familial ALS patients with the A4V SOD1 mutation (Bowling et al., 1993). In motor cortex (Brodmann area 4), we found a significant increase in complex I and II-III activities, whereas in parietal cortex there was an increase in complex I activity, and in cerebellum, complex II-III activity was increased (Browne et al., 1998). Interestingly, complex I activity also was significantly increased in cerebral cortex of transgenic mice with the G93A human SOD1 mutation. This mouse, as well as other mice with SOD1 mutations, shows mitochondrial swelling and vacuolization as a prominent early pathologic feature (Gurney et al., 1994; Wong et al., 1995, Dal Canto and Gurney, 1994). Furthermore, an increase in mitochondrial vacuolization immediately precedes a rapid phase of development of motor weakness (Kong and Xu, 1998). There is no precedent for increased electron transport activity in neurologic illnesses, and we can only speculate that this may be a compensatory phenomenon, perhaps for a damaged inner mitochondrial membrane. Expression of the G93A SOD1 mutation in vitro leads to a decline in mitochondrial membrane potential and sensitizes mitochondria to depolarization produced by valinomycin (Carri et al., 1997).
We did not find alterations in electron transport enzymes in SALS; however, it is still possible that subtle abnormalities may occur. If electron transport defects are heterogeneous among patients, it may not be possible to detect them in postmortem tissue because of an averaging effect. Peripheral blood lymphocytes from SALS patients show increased cytosolic Ca2+ and altered responses to uncouplers of oxidative phosphorylation (Curti et al., 1996). Ultrastructural studies of mitochondria of anterior horn cells from both FALS patients and SALS patients show mitochondrial abnormalities (Hirano 1991; Sasaki et al., 1990; Sasaki and Iwata, 1996). Liver biopsy specimens show large mitochondria and intramitochondrial inclusions in SALS patients (Masui et al., 1985). Mitochondria with abnormal protrusions were observed in a FALS patient who was later determined to have an SOD1 mutation (Hirano, 1991). Studies of muscle biopsy specimens of SALS patients showed increased mitochondrial volume and Ca2+ levels (Siklos et al., 1996).
Mitochondrial dysfunction could contribute to the pathogenesis of both FALS and SALS by leading to increased free radical production and oxidative damage. Furthermore, it could contribute to slowed axonal transport, leading to the accumulation of neurofilaments, which have been observed in both SALS and FALS (Rouleau et al., 1996; Sasaki and Iwata, 1996; Shibata et al., 1996). Studies of motor neurons in SALS patients show an accumulation of mitochondria in proximal axons, consistent with impaired axonal transport (Sasaki and Iwata, 1996).
Studies of oxidative damage show increased protein carbonyl groups in the motor cortex and spinal cord of sporadic ALS patients (Bowling et al., 1993; Shaw et al., 1995). We measured concentrations of 3-nitrotyrosine and its major metabolite, 3-nitro-4-hydroxy-phenylacetic acid, in the thoracic and lumbar spinal cord of both sporadic ALS patients and three FALS patients with SOD1 mutations (Beal et al., 1997). Increased concentrations of both 3-nitrotyrosine and 3-nitro-4-hydroxyphenylacetic acid were found in both the sporadic and the FALS patients. We also examined whether there was evidence for increased oxidative damage at the cellular level using antibodies to 3-nitrotyrosine, hemeoxygenase-1, and malondialdehyde. Both the sporadic and the familial ALS patients showed increased staining in anterior horn motor neurons (Ferrante et al., 1997).
Mitochondria in Huntington's disease
Huntington's disease is an autosomal dominant inherited neurodegenerative illness characterized by both a movement disorder and dementia. The pathologic hallmark is a degeneration of the basal ganglia and, to a lesser extent, cerebral cortex. Interestingly, the pathologic process within the basal ganglia is highly selective, with interneurons, such as those containing the histochemical marker NADPH-diaphorase, being spared. The genetic defect in the illness has been elucidated as being an expansion of a CAG trinucleotide repeat sequence in a gene whose protein product was designated huntingtin. How the gene defect leads to neuronal degeneration remains obscure. The CAG repeats in the huntingtin protein are expressed, leading to an expanded stretch of glutamines within the protein. The mRNA for huntingtin is widely expressed in the CNS, without pronounced regional differences; however, the protein is preferentially expressed in the medium-sized spiny projection neurons of the basal ganglia and cortical pyramidal projection neurons that degenerate in HD (Ferrante et al., 1997).
Several studies using PET showed reduced glucose metabolism in the basal ganglia of HD patients (reviewed in Beal, 1995). Using proton magnetic resonance spectroscopy, we found elevated lactate concentrations in both occipital cortex and in the basal ganglia of symptomatic HD patients (Jenkins et al., 1993). Some asymptomatic gene carriers also show elevated lactate in the basal ganglia (Jenkins et al., 1998). The increased lactate concentrations correlate well with the duration of disease, implying that normal energy metabolism is progressively impaired by the disease process. An increase in lactate in frontal and occipital cortex and cerebellum was reported by others (Harms et al., 1997; Martin et al., 1996). We found a decrease in the PCr/Pi ratio in resting gastrocnemius muscle of HD patients, providing evidence for a systemic defect in energy metabolism (Koroshetz et al., 1997). This observation is consistent with reports of progressive weight loss in HD patients despite increased caloric intake (O'Brien et al., 1990). The CSF measurements show an increased lactate-pyruvate ratio in HD patients (Koroshetz et al., 1997).
Several studies have examined oxidative phosphorylation enzymes in postmortem brain tissue. Reduced succinate dehydrogenase activity was found in two studies (Butterworth et al., 1985; Stahl and Swanson, 1974). A recent study showed a 55% to 60% decrease in complexes II and III activities in HD caudate, and a 33% decrease in complex IV activity (Gu et al., 1996). We found that complex II-III activity was reduced by 29% in the caudate and 67% in the putamen, but unaffected elsewhere (Browne et al., 1997). Complex IV also was reduced 62% in the putamen. Complex I activity and citrate synthase activities showed no changes. The most consistent biochemical enzymatic defects in HD therefore appear to involve succinate dehydrogenase and complex II-III activity (of which succinate dehydrogenase is a component) of the electron transport chain. This is of interest, since we found that systemic administration of complex II-III inhibitor 3-nitropropionic acid produces striatal lesions in rats and primates that closely mimic HD (Beal et al., 1993; Brouillet et al., 1995). Furthermore, an inherited defect in succinate dehydrogenase is associated with basal ganglia degeneration (Bourgeron et al., 1995).
Studies of cultured fibroblasts from HD and control subjects are consistent with a mitochondrial defect. Greenamyre and colleagues examined the consequences of Ca2+ influx induced by ionomycin on mitochondrial membrane potential (Gutekunst et al., 1996). Normal fibroblasts show depolarization and recovery, but HD fibroblasts show a failure to recover fully after the second application of ionomycin.
One proposed mechanism by which the HD gene defect could lead to impaired energy metabolism is by an interaction with glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Huntingtin binds with high affinity to GAPDH, a critical component of the glycolytic pathway that catalyzes the oxidation and phosphorylation of glyceraldehyde 3-phosphate to 1,3-bisphosphoglycerate (Burke et al., 1996). Furthermore, both GAPDH and α-ketoglutarate dehydrogenase can be inactivated by transglutaminase when pathologic increases in polyglutamine repeats are present in vitro (Cooper et al., 1997). Intrastriatal administration of the GAPDH inhibitor iodoacetate produces dose-dependent striatal excitotoxic lesions, which correlate with inhibition of enzyme activity (Matthews et al., 1997a). However, spectrophotometric assays of GAPDH activity failed to detect differences between HD and control postmortem brain samples (Browne et al., 1997).
Few studies explore oxidative damage in HD. In HD caudate, there was a significant 21% increase of 8-OHdG in nuclear DNA (Browne et al., 1997). A sufficient amount of mtDNA for measurements was available only in cerebral cortex and cerebellum. In parietal cortex, there was a twofold significant increase in 8-OHdG in mtDNA (Polidori and Beal, unpublished findings). There were increased mtDNA deletions in HD cerebral cortex (Horton et al., 1995), although this was not found in another study (Chen et al., 1995). In preliminary studies, there was increased staining of HD striatal neurons with antibodies to hemeoxygenase-1, 8-OHdG, and malondialdehyde-modified protein (Ferrante et al., 1996).
There is evidence for a metabolic defect in a transgenic model of HD. Transgenic mice were made by expressing numerous CAG repeats (130 to 140) in exon 1 of the HD gene (Mangiarini et al., 1996). The mice develop normally for about 8 weeks followed by onset of a movement disorder with tremors and subsequent seizures. The mice die at about 12 weeks of age. Neuropathologic examination shows small brains, although there does not appear to be basal ganglia degeneration. Notice that the animals show a progressive weight loss starting at 8 weeks of age despite an apparently increased caloric intake. This is consistent with increased caloric expenditure, which occurs in HD patients (O'Brien et al., 1990).
Other neurologic diseases
Several neurodegenerative diseases other than HD are associated with an expanded CAG trinucleotide repeat, suggesting that there may be common pathologic mechanisms in all of these diseases. In several of these, there is evidence for metabolic defects. In spinocerebellar ataxia 1, there are reduced levels of α-ketoglutarate dehydrogenase in postmortem tissue (Mastrogiacomo et al., 1996). In spinocerebellar ataxia 2, muscle biopsy specimens show areas devoid of mitochondria or with focal accumulations and autophagic vacuoles, which may contain mitochondria (Forsgren et al., 1996). In spinocerebellar ataxia 3 (Machado-Joseph Disease), CSF lactate/pyruvate is reported to be elevated (Matsuishi et al., 1996). Overexpression of a portion of this gene containing the CAG repeat leads to apoptotic cell death in cultured neurons and to Purkinje cell degeneration in transgenic mice (Ikeda et al., 1996).
Friedrich's ataxia is autosomal recessive and evidence suggests a loss of function. Evidence shows that the Friedrich's ataxia protein, frataxin, has a mitochondrial localization (Babcock et al., 1997). The homologous gene in yeast regulates mitochondrial iron transport, and its dysfunction leads to accumulation of mitochondrial iron and oxidative damage (Babcock et al., 1997; Priller et al., 1997). In postmortem tissue, α-ketoglutarate dehydrogenase levels are reduced (Mastrogiacomo et al., 1996). Endomyocardial biopsy specimens show reduced complex I, complex II-III, and aconitase activities, consistent with oxidative damage to iron-sulphur clusters (Rotig et al., 1997). Wilson's disease may involve a similar mechanism, since the disease-associated gene encodes a copper-transporting P-type ATPase that was recently localized to mitochondria (Lutsenko and Cooper, 1998). A defect in platelet complex I activity was found in idiopathic dystonia (Benecke et al., 1992; Schapira et al., 1997). Patients with Leber's optic atrophy with dystonia, which is caused by point mutations in the ND 6 subunit of complex I (Jun et al., 1994), develop progressive basal ganglia degeneration (Bruyn et al., 1992; Novotny et al., 1986). Another neurodegenerative disease that recently has been shown to caused by mitochondrial impairment is hereditary spastic paraplegia (Casari et al., 1998). This disease was shown to be caused by either a deletion or point mutations in a protein termed paraplegia, which is a mitochondrial metalloprotease. Patients show ragged red fibers and bizarre mitochondrial morphologic features in muscle biopsy specimens.
THERAPEUTIC CONSEQUENCES
A novel therapeutic strategy for either acute or chronic neurodegeneration is to use compounds to improve mitochondrial metabolism to compensate for injury or disease-related defects. As described earlier, acetyl-L-carnitine provides neuroprotection after cerebral ischemia by a mechanism that appears to compensate for reperfusion injury to the mitochondrial PDH enzyme (Rosenthal et al., 1992; Bogaert et al., 1994). Other putative neuroprotective agents may have direct or indirect effects on mitochondrial oxidative phosphorylation. The electron transport chain component coenzyme Q10, or ubiquinone, is a potent antioxidant that protects against glutamate toxicity in vitro (Favit et al., 1992). It produces dose-dependent protection against striatal lesions produced by the succinate dehydrogenase inhibitor malonate, and it protects against malonate-induced ATP depletions (Beal et al., 1994). It also protects against both MPTP and 3-nitropropionic acid neurotoxicity, and it extends survival in the transgenic mouse model of FALS (Matthews et al., 1998a). In HD patients, oral administration results in significant decreases in occipital cortex lactate concentrations, which reverse after withdrawal of therapy (Koroshetz et al., 1997). Coenzyme Q10 exerts additive effects with N-methyl-D-aspartate antagonists against malonate lesions (Schulz et al., 1996). This has led to a proposal to test coenzyme Q10 and an N-methyl-D-aspartate antagonist both alone and in combination in the treatment of HD. Another approach that we recently examined is to use creatine administration to increase phosphocreatine levels and thereby buffer against energy depletion. Oral administration protects against striatal lesions produced by both malonate and 3-nitropropionic acid (Matthews et al., 1998a). Furthermore, we showed that it protects against phosphocreatine and ATP depletions as well as increases in 3-nitrotyrosine concentrations, which we believe, occur downstream of energy depletion.
As discussed, there is substantial evidence that oxidative damage occurs in acute and chronic neurodegenerative diseases, which may be a consequence of mitochondrial dysfunction. This suggests that free radical scavengers may be effective. Free radical spin traps attenuate malonate, 3-nitropropionic acid, and MPTP neurotoxicity (Schulz et al., 1996). A critical mediator of oxidative damage may be peroxynitrite, which is generated by the reaction of nitric oxide with superoxide radical (O2−). Mitochondrial dysfunction leads to increased peroxynitrite by increasing mitochondrial O2− generation and by raising cytosolic Ca2+ levels, which then activates neuronal nitric oxide synthase. 7-Nitroindazole, a relatively selective neuronal nitric oxide synthase inhibitor, dose-dependently blocks MPTP neurotoxicity, and it protects against malonate and 3-nitropropionic acid neurotoxicity (Schulz et al., 1995a, 1995b). Mice that are deficient in neuronal nitric oxide synthase are resistant to MPTP toxicity (Przedborski et al., 1996), and S-methyl-thiocitrulline, another relatively selective neuronal nitric oxide synthase inhibitor, also protects against MPTP neurotoxicity in mice (Matthews et al., 1997b). 7-Nitroindazole produced protection against dopamine depletion, loss of substantia nigra neurons, and motor and cognitive deficits induced by MPTP in baboons (Hantraye et al., 1996). Other antioxidants, such as those present in extracts from the Ginkgo Biloba tree, also have been purported to protect against indices of oxidative injury in mitochondria from the brains of old animals (Sastre et al., 1998).
Other potential agents for use in amelioration of acute and chronic neurodegeneration include neurotrophic factors, which can be effective in animal models involving mitochondrial dysfunction (Kirschner et al., 1996), and nonimmunosuppressive agents, which target the mitochondrial PTP to attempt to block its potential contribution to necrotic and apoptotic death. Current data suggest that strategies designed to improve mitochondrial respiratory function, decrease ROS production, and inhibit mitochondrial apoptogen release might all prove to be useful in the treatment of neurodegenerative diseases associated with mitochondrial dysfunction.