
Abstract
The biochemical pathways to cell death in chronic and acute forms of neurodegeneration are poorly understood, limiting the ability to develop effective therapeutic approaches. As details of the apoptotic and necrotic pathways have been revealed, an appreciation for the decisive role that mitochondria play in life-death decisions for the cell has grown. As a result, the need has arisen to reevaluate the significance to cell viability of mitochondrial Ca2+ sequestration, reactive oxygen species generation, and the membrane permeability transition. This review provides basic information on these mitochondrial functions as they relate to control over cell death.
Keywords
The standard lecture on mitochondrial function in a basic biochemistry course used to be a relatively brief discourse on intermediary metabolism and aerobic ATP production, the brevity of which was appreciated by many students. Novel discoveries in the field have now forced the extension of the syllabus to include the control that mitochondria exert over life-and-death decisions in the cell, thereby making the experience less tedious for student and lecturer alike. As researchers, we recognize that our basic knowledge of mitochondrial function in both normal and pathologic conditions is expanding, yet many questions remain for expert and burgeoning mitochondriacs to answer. Further discoveries in this field will benefit our ability to develop appropriate and successful therapeutic strategies for neurodegenerative diseases, ischemia/reperfusion injury, cancer, and many other pathologic conditions in which mitochondrial function is altered.
This review addresses current concepts of mitochondrial function relevant to normal bioenergetic function, Ca2+ sequestration, free radical production, and regulation of the apoptotic pathway to provide a mitochondrial primer relevant to those interested in neurodegeneration. In the April 1999 issue of the Journal of Cerebral Blood Flow and Metabolism, a second review summarizes the current understanding of mitochondrial function during ischemic/reperfusion-induced neuronal death that accompanies stroke, cardiac arrest, and traumatic injury to the brain (Fiskum et al., 1999). This second review also addresses recent advances in understanding the role that mitochondrial dysfunction plays in chronic forms of neurodegeneration, including Alzheimer's disease, Parkinson's disease, amyotrophic lateral sclerosis, and Huntington's Disease. Our need to understand the underlying mechanisms of aging, including mitochondrial dysfunction and free radical-induced oxidative damage (Beal, 1995) is intensifying as the population is becoming more elderly. Experimental support continues to accumulate for a central role of defective energy metabolism, alteration in mitochondrial function leading to increased free radical production, and active mitochondrial participation in the apoptosis cascade in the pathogenesis of acute and chronic neurodegeneration.
NECROTIC VERSUS APOPTOTIC DEATH IN NEURODEGENERATION
It is reasonable that ischemic necrotic death, such as the necrosis that occurs at the core of an infarct, results from either acute or delayed loss of ionic homeostasis and osmotic cell lysis resulting from a lack of mitochondrial and glycolytic ATP production. Whether mitochondrial dysfunction contributes to necrotic neuronal death in chronic forms of degeneration is less clear. However, it is at least theoretically possible that defects in mitochondrial electron transport (Fiskum et al., 1999) could be severe enough to compromise total cellular ATP production and thereby result in necrosis. Such a proposal will remain primarily theoretical until we gain a better understanding of the mechanisms by which neurons are dying in chronic forms of degeneration.
For mitochondrial participation in apoptotic triggering (see Mitochondria as the Apoptotic Trigger) to be relevant to neurodegeneration, it must first be convincing that apoptosis is contributing to neuronal loss. Because of the current inability to easily discriminate between apoptotic and necrotic cells in tissue preparations, this topic is controversial but has been dealt with in a review by MacManus and Linnik (1997), which addresses the role that apoptotic death plays after cerebral ischemia. In addition, Martin et al. (1998) have proposed that death in neurodegeneration involving excitotoxicity exists along a continuum between apoptosis and necrosis. There also is evidence for apoptosis occurring in traumatic injury to CNS neurons (see Crowe et al., 1997, and references therein). Keeping the concerns over methodology in mind (MacManus and Linnik, 1997), the evidence for apoptotic death in chronic forms of neurodegeneration is accumulating. Apoptotic nuclei are evident in tissue from patients with Parkinson's disease (Mochizuki et al., 1996), amyotrophic lateral sclerosis (Yoshiyama et al. 1994), Huntington's disease (Thomas et al., 1995), and Alzheimer's disease (Smale et al., 1995; Anderson et al., 1996; Su et al., 1996; Su et al., 1997; Troncoso et al., 1996). It is possible that mitochondrial involvement in some forms of neurodegeneration, including Alzheimer's disease, arises primarily from chronic neuronal dysfunction associated with a state of near but not complete cell death, which induces functional impairment of the tissue.
The endogenous protein regulators of apoptosis are relevant to the current discussion. Providing a historical review of the Bcl-2 family of proteins and the caspase family of cysteine proteases, however, is beyond the scope of this work. The reader is urged to refer to several recent reviews on these subjects (Reed, 1997; Bredesen, 1996; Nicholson and Thornberry, 1997). The Bcl-2 family, which includes both inhibitors (such as Bcl-2 and Bcl-xL) and activators (such as Bax and Bad) are key regulatory molecules at the point in apoptosis where diverse pathways converge, potentially on the release of mitochondrial apoptogenic factors. Certain members of the caspase family of cysteine proteases (especially caspase-3 and caspase-9) lie downstream in the biochemical pathway from mitochondria and Bcl-2 family regulators (Chinnaiyan et al., 1996; Liu et al., 1996; Susin et al., 1996; Xiang et al., 1996; Kluck et al., 1997; Yang et al., 1997; Ellerby et al., 1997). Other caspases (especially caspase-8) lie upstream from mitochondrial events, as in the apoptotic pathway induced by tumor necrosis factor receptor family members (Boise and Thompson, 1997; Susin et al., 1997; Adachi et al., 1997).
MITOCHONDRIAL ELECTRON TRANSPORT AND THE PROTONMOTIVE FORCE
Critical to a discussion of mitochondria in cellular degeneration is an understanding of mitochondrial electron transport, and the resulting ability of mitochondria to sequester Ca2+ and to generate superoxide radical. Mitochondrial electron transport (Fig. 1) involves provision of reducing equivalents in the form of NADH or FADH2 to components of the chain as the result of the enzyme-catalyzed oxidation of fuel molecules. The resulting oxidation/reduction reactions successively shuttle these electrons from the component of more negative redox potential ultimately to O2. As a function of these redox reactions, protons are pumped by complex I, ubiquinone/complex III, and complex IV from the matrix to the cytosolic side of the inner mitochondrial membrane. As a result, the energy of fuel molecules is converted into a form of potential energy called the electrochemical gradient of protons (ΔμH+, expressed in kJ mol−1) or the protonmotive force (Δp, expressed in millivolts). Ultimately, the protonmotive force is used to perform work in the form of ATP synthesis by the ATP synthetase (termed oxidative phosphorylation), or ion, metabolite, and protein transport.
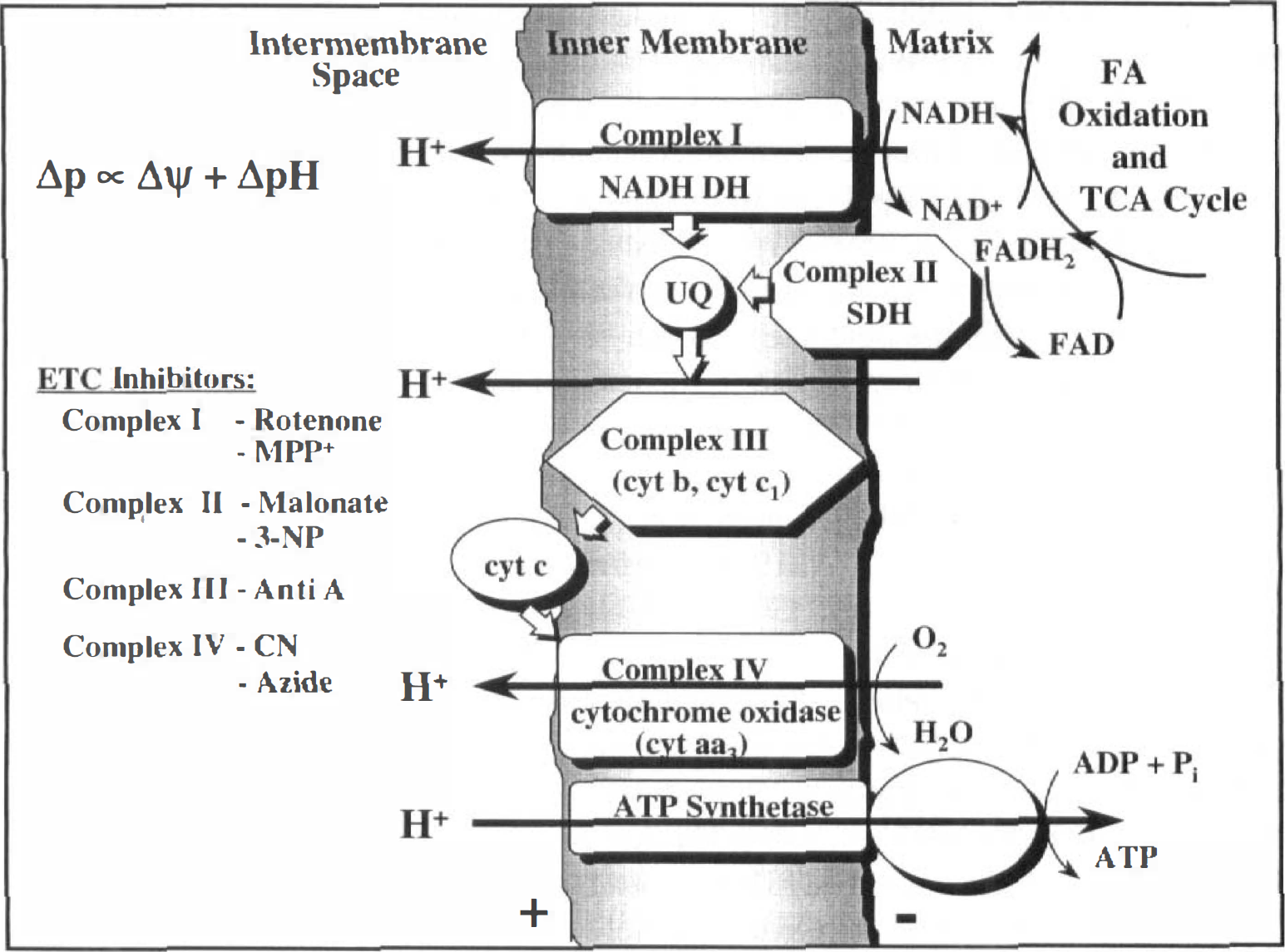
Mitochondrial electron transport, the electrochemical gradient of protons, and oxidative phosphorylation (see Nicholls and Ferguson [1992] for details). The flavin moiety of succinate dehydrogenase is a covalently bound cofactor of the enzyme, which is embedded in the inner membrane. Depending on the tissue type, additional complexes exist (not shown), which can donate electrons to ubiquinone. DH, dehydrogenase (oxidoreductase); UQ, ubiquinone; Δp, protonmotive force; Δψ, membrane potential; MPP+, 1-methyl-4-phenylpyridinium; 3-NP, 3-nitropropionic acid; anti A, antimycin A; CN, cyanide.
The gradient generated by mitochondrial electron transport has both an electrical (Δψ) and a chemical component (ΔpH). Under normal conditions, the contribution of the pH gradient to the total protonmotive force is relatively small. For instance, it has been estimated that when the total protonmotive force under state 4 (resting) conditions is 200 mV, Δψ is approximately 170 mV and the pH gradient is approximately 0.5 pH units (alkaline inside) or equivalent to 30 mV (Nicholls and Ferguson, 1992). This issue is relevant because certain transport processes are driven primarily by Δψ (for instance, Ca2+ uptake), and some are driven primarily by ΔpH. For instance, ΔpH drives phosphate transport, which functions in isolated mitochondria to maintain ΔpH at a relatively constant value during changes in respiration-induced proton pumping), and drives K+/H+ antiport, which is involved in mitochondrial volume regulation (Brierley et al., 1994).
The activity of the electron transport chain can be measured using an O2 electrode with isolated mitochondria or cells in which the plasma membrane has selectively been made permeable with digitonin, a technique most amenable to suspensions of cells. Such assays yield information about the maximal rates of electron transfer, as well as the degree of coupling of electron transport to ATP production. Rates of state 3 (phosphorylating) respiration are measured in the presence of oxidizable substrates and ADP and reflect the maximal rate of coupled respiration, that is, when electron transport is coupled to ATP synthesis. This rate typically is limited in healthy mitochondria by the kinetics of either the ATP synthetase or the rate of adenine nucleotide translocase (ANT)-mediated provision of ADP for synthetase activity through antiport of extramitochondrial ADP and intramitochondrial ATP. However, under pathologic conditions (including Ca2+ or free radical-mediated injury), the activity of certain components of the electron transport chain may become rate limiting, placing a restriction on the protonmotive force that can be generated or potentiating mitochondrial free radical production (see later). State 4, or “resting” respiration, can be measured in the absence of ADP or the presence of the ATP synthetase inhibitor oligomycin, and reflects the rate of leakage of protons back across the inner mitochondrial membrane into the matrix. State 4 rates increase when the integrity of the inner mitochondrial membrane has been compromised, such as during opening of the mitochondrial permeability transition pore (see later) or as a result of Ca2+ or free radical-induced damage. The maximal rate at which substrate oxidation and electron flow through the chain to O2 can take place in the absence of the rate restrictions imposed by the synthetase, or the ANT can be measured by the addition of an uncoupler such as carbonyl cyanide p-trifluoromethoxyphenyl hydrazone (FCCP). Such agents act as ionophores to allow H+ to move down their concentration gradient into the matrix, thus dissipating Δp and removing the driving force for mitochondrial work. Such measurements identify specific sites of alterations in mitochondrial function (Adachi et al., 1997; Murphy et al., 1990) and greatly enhance understanding of measurements of Δψ. Fluorometric measurements of Δψ can be ambiguous under certain circumstances, because a drop in membrane potential can arise for numerous reasons, including Ca2+ transport, respiratory uncoupling, or respiratory inhibition.
What is the consequence of altered electron transport to neuronal viability? If severe enough, electron transport inhibition compromises the ability to generate an electrochemical gradient and, therefore, decreases the rate of ATP synthesis (Davey and Clark, 1996). This, in turn, may establish conditions for secondary excitoxicity (Henneberry, 1997). In addition, mitochondrial free radical generation may be enhanced (see Mitochondrial Reactive Oxygen Species Generation). Ultimately, either apoptotic or necrotic death, or chronic cellular dysfunction may result. The pathway taken by the cell may be partly dependent on the cellular ATP level, a function of the rates of mitochondrial and glycolytic ATP production and the rate of energy utilization. Studies support that apoptosis occurs only if there is sufficient ATP to execute the process (Eguchi et al., 1997; Leist et al., 1997). Electron transport inhibition with cyanide when combined with glucose deprivation can result in either acute cell death (within 1 hour) if ATP levels are depleted for a sufficient amount of time, or delayed cell death (within 24 hours) if electron transport inhibition and ATP depletion is brief (Myers et al., 1995). When electron transport is inhibited and glycolytic ATP production continues by provision of glucose, the outcome also can be either necrotic or apoptotic death, presumably dependent on the ability of glycolysis to meet cellular energy demands. Chronic inhibition of succinate dehydrogenase with 3-nitropropionic acid (a model of Huntington's disease) can induce apoptotic (Behrens et al., 1995; Pang and Geddes, 1997) and necrotic (Pang and Geddes, 1997) death in primary cultures of neurons. Apoptosis may be induced in vitro or in vivo by administration of the complex I inhibitor 1-methyl-4-phenylpyridinium (a model of Parkinson's disease) or the complex II inhibitor 3-nitropropionic acid (Dipasquale et al., 1991; Hartley et al. 1994; Sheehan et al., 1997; Sato et al., 1997; Tatton and Kish, 1997). In addition, chronic hypoxic treatment of several cell types induces delayed cell death (Jacobson and Raff, 1995; Shimizu et al., 1995). Relevant to later discussion, in many of these studies, substrates for cytoplasmic ATP production were provided. Therefore, a mitochondrial membrane potential can be maintained as a result of reversal of the proton-pumping activity of the ATP synthetase. The inclusion of oligomycin, an inhibitor of the synthetase, blocks this process (Budd and Nicholls, 1996a).
MITOCHONDRIAL CA2+ TRANSPORT
The electrical component of the proton gradient, Δψ, can be used to drive the uptake of Ca2+ into the matrix (see Gunter et al., 1994; and Gunter and Pfeiffer, 1990, for review). Fig. 2 depicts the transport processes involved in mitochondrial Ca2+ homeostasis in electrically excitable cells. Mitochondrial Ca2+ transport is relevant because of the role of excitotoxic and ischemic events in acute forms of neurodegeneration. It also is potentially applicable if mitochondrial defects in chronic forms of degeneration are severe enough to compromise normal fluctuations in ion gradients in response to neurotransmitter stimulation. Mitochondrial Ca2+ uptake occurs when the extramitochondrial concentration rises above what is termed the mitochondrial “buffer” or “set point,” the concentration at which the Ca2+ uniporter becomes more active than the Ca2+ efflux mechanism. Nicholls and Scott (1980) estimate the set point of brain mitochondria to be approximately 0.5 μmol/L. However, enhanced sensitivity to the concentration of adenine nucleotides (Rottenberg and Marbach, 1990a,b) and spatiotemporal variables imply that mitochondrial Ca2+ sequestration may occur in subpopulations of mitochondria in a manner that is not predicted by the overall cytoplasmic Ca2+ value (Rizzuto et al., 1998). Although Na+, polyanions such as spermine, and adenine nucleotides can alter the buffer point in vitro (Gunter and Pfeiffer, 1990; Rottenberg and Marbach, 1990a,b; Murphy and Fiskum, 1988), it is not known to what degree this value varies physiologically, nor is it known how it varies from one neural cell type to another. Because of the rapid kinetic capability of the uniporter (which may have two kinetic modes of operation [Sparagna et al., 1995]) and the slow Vmax of the Ca2+ efflux mechanism (a Na+/Ca2+ antiporter in mitochondria of electrically excitable cells), net Ca2+ sequestration can occur. Also, because of the velocity of the uniporter, Ca2+ uptake will deplete Δψ and stimulate electron transport until extramitochondrial Ca2+ is lowered below the set point. Sequestered Ca2+ will then exist in equilibrium with protein and lipid Ca2+ binding sites and osmotically inactive Ca2+ (the latter of which represents most of matrix calcium). Provided that extramitochondrial Ca2+ remains below the set point and the load of mitochondria Ca2+ is physiologic, matrix free [Ca2+] then will slowly decrease, the rate of which is dependent on extramitochondrial [Na+], ΔpH, and, possibly, other physiologic effectors that await characterization.
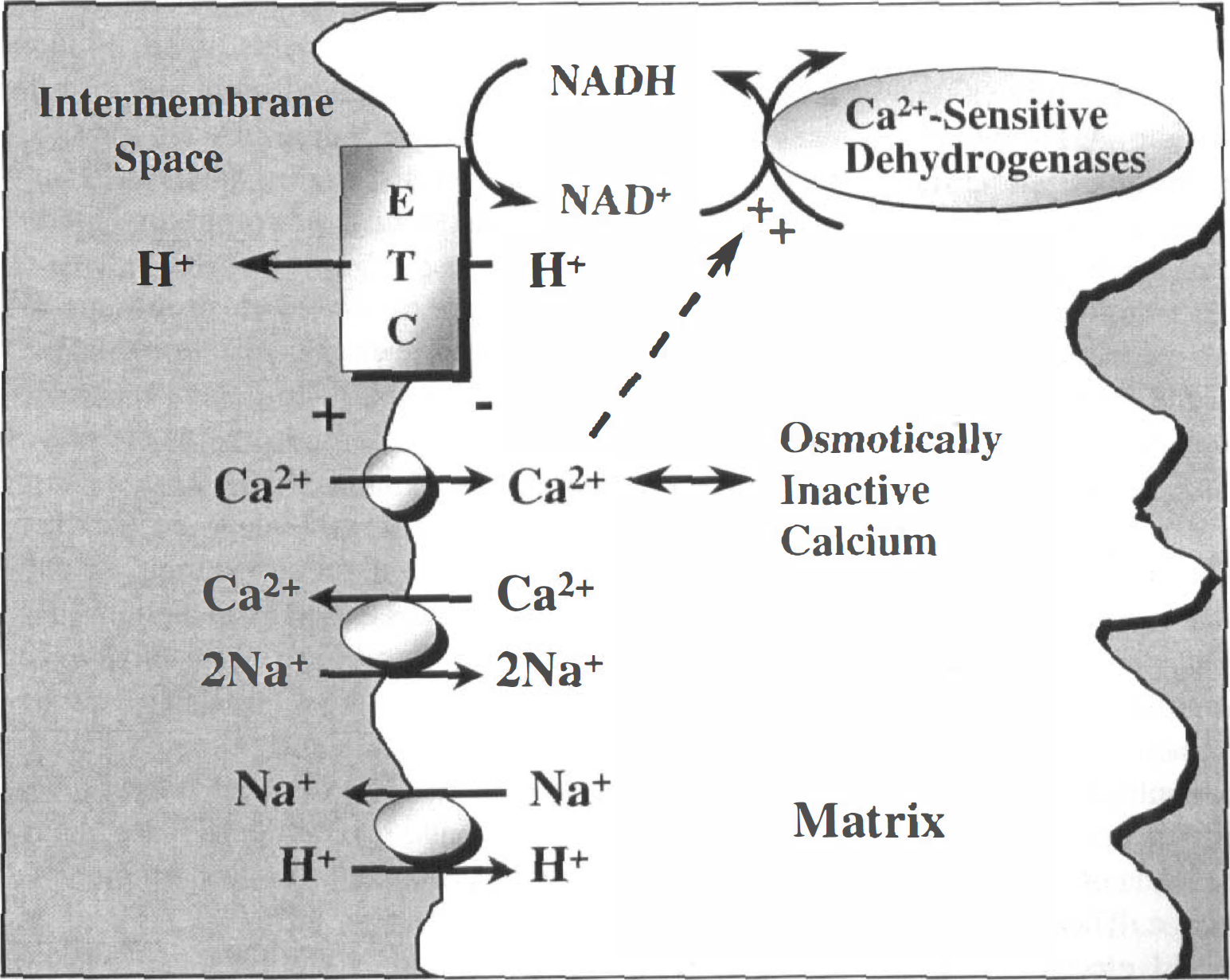
Mitochondrial Ca2+ transport mechanisms. The Ca2+ efflux mechanism depicted is believed to be that which operates in the mitochondria of electrically excitable tissues. Alternatively, for instance, in liver mitochondria, efflux may be mediated by Ca2+/2H+ antiport. Efflux through the Na+/Ca2+ exchanger is influenced by the Na+/H+ antiporter that is driven by ΔpH. See Gunter and Pfeiffer (1990) and Gunter et al. (1994) for further discussion.
Relatively low levels of mitochondrial Ca2+ sequestration, such as that which follows normal depolarization, acts in a bioenergetic signaling capacity by stimulating matrix Ca2+-sensitive dehydrogenase activity. This increases provision of reducing equivalents to the electron transport chain, enhancing ATP synthesis to meet the increased cellular energy demand (Gunter and Pfeiffer, 1990). Higher levels of sequestration have been reported to induce Ca2+-induced Ca2+ release, which may play an important role in the propagation of Ca2+ signals (Ichas and Mazat, 1998). Above a certain level, however, Ca2+ sequestration can begin to compromise mitochondrial function, resulting in respiratory inhibition or uncoupling of oxidative phosphorylation, formation of nonspecific pores (see later), or osmotic lysis. Respiratory inhibition (if severe enough) and uncoupling lower the measured mitochondrial membrane potential. There-fore, a Ca2+-induced drop in Δψ may be followed by Ca2+-induced injury to electron transport and the inner mitochondrial membrane, or Ca2+-induced cytochrome c release, which could sustain the loss of membrane potential.
On the cellular level, the consequences of extensive mitochondrial Ca2+ sequestration (which has a significantly greater capacity than accumulation by the endoplasmic reticulum) to neuronal cytosolic Ca2+ levels, mitochondrial membrane potential, and mitochondrial free radical production indicate that mitochondrial Ca2+ loading likely is a critical event in the determination of neuronal viability after excitotoxic stress (Stout et al., 1998; Budd and Nicholls, 1996b; Harrington et al., 1996; Schinder et al., 1996; White and Reynolds, 1996; Wang and Thayer, 1996; Ankarcrona et al., 1995; Reynolds and Hastings, 1995; Dugan et al., 1995, Bindokas et al., 1995). Hartley and coworkers (1993) suggest that the total amount of Ca2+ that enters the cell correlates better with toxicity than the cytosolic Ca2+ load, as though an unidentified noncytosolic pool was responsible for toxicity. Although conventional dyes such as fura-2 may have underestimated the level of Ca2+ reached in the cytoplasm after NMDA exposure (Hyrc et al., 1997), it is tempting to modify the excitotoxic theory of neuronal death to more specifically state that the degree of mitochondrial Ca2+ loading after a stimulus may be the determining factor in cell death. White and Reynolds (1996) indicate that in forebrain neurons responding to excitotoxic concentrations of glutamate, inhibition of the mitochondrial Na+/Ca2+ exchanger (the Ca2+ efflux mechanism) with CGP-37157 enhances the rate and proportion of mitochondria that lose membrane potential. Ankarcrona et al. (1995) suggest that those cerebellar granule cells treated with a high concentration of glutamate in which mitochondria depolarize and never recover membrane potential go on to die by necrosis. In contrast, those neurons that lose mitochondrial membrane potential during glutamate exposure, but recover Δψ within 30 minutes after removal of excitotoxin, die by apoptosis. Stout et al. (1998) and Budd and Nicholls (1996b) have made the significant finding that dissipation of Δψ in neurons (which prevents mitochondrial Ca2+ sequestration) during glutamate exposure prevents cell death. These data argue that glutamate toxicity is relatively unrelated to the peak and plateau values of cytoplasmic Ca2+ but is directly related to mitochondrial Ca2+ sequestration.
MITOCHONDRIAL REACTIVE OXYGEN SPECIES GENERATION
Excessive Ca2+ accumulation can potentiate neuronal mitochondrial superoxide production (Dykens, 1994; Reynolds and Hastings, 1995; Dugan et al., 1995, Bindokas et al., 1996). Studies initiated approximately 25 years ago (Boveris and Chance, 1973; Cadenas and Boveris, 1980) characterized the ability of isolated mitochondria to generate superoxide, typically measured as H2O2 (because of spontaneous dismutation of superoxide and endogenous activity of Mn-superoxide dismutase). Experiments with isolated heart and liver mitochondrial suggest that these organelles produce superoxide radical through either autooxidation of the flavin component of complex I of the electron transport chain (NADH dehydrogenase) or through autooxidation of ubisemiquinone at complex III (Fig. 3; Boveris and Chance, 1973; Boveris et al., 1976; Turrens and Boveris, 1980). In general, early studies suggest that H2O2 production is maximal under conditions of severe mitochondrial respiratory inhibition when the electron carriers responsible for free radical production are maximally reduced (such as in the presence of the complex III inhibitor antimycin A). Such data predict that if damage or mutation occurs to an electron transport chain component (or if a component such as cytochrome c is lost from the intermembrane space) that forces a proximal free radical producing site to shift to a more reduced state, then mitochondrial reactive oxygen species (ROS) generation will be enhanced. For instance, if electron transport by complex III or IV or cytochrome c becomes severely rate limiting, then free radical production at ubiquinone (and possibly complex I) may be enhanced (see Cai and Jones, 1998). Therefore, since the redox status of electron transport chain components when mitochondria are in state 3 or are uncoupled is relatively oxidized, then free radical production under these conditions is minimal (Boveris and Chance, 1973). Somewhat counterintuitively, these early studies indicate that the rate of H2O2 production could be enhanced when antimycin A was combined with an uncoupling agent (a protonophore) (Boveris and Chance, 1973). Consistent with the conclusions of the early work, recent studies by Kowaltowski and others (1995, 1996a,b) describe that Ca2+ stimulation of H2O2 production by uncoupled liver mitochondria is associated with a shift in the redox state of ubiquinone to a more reduced level. Studying conditions relevant to ischemia/reperfusion injury and excitotoxicity, Dykens (1994) has demonstrated that isolated brain mitochondria increase ROS generation when exposed to high extramitochondrial Ca2+, Na+, and ADP. Furthermore, evidence was provided that mitochondrial free radical production is likely self-amplifying because of the susceptibility of electron transport to free radical-induced inhibition (Dykens, 1994). Cai and Jones (1998) argue that during staurosporine-induced apoptosis in HL-60 cells, mitochondrial superoxide generation is stimulated by the loss of cytochrome c from the intermembrane space. Whether cytochrome c release is involved in the enhanced ROS production described in some of the aforementioned studies requires further investigation.
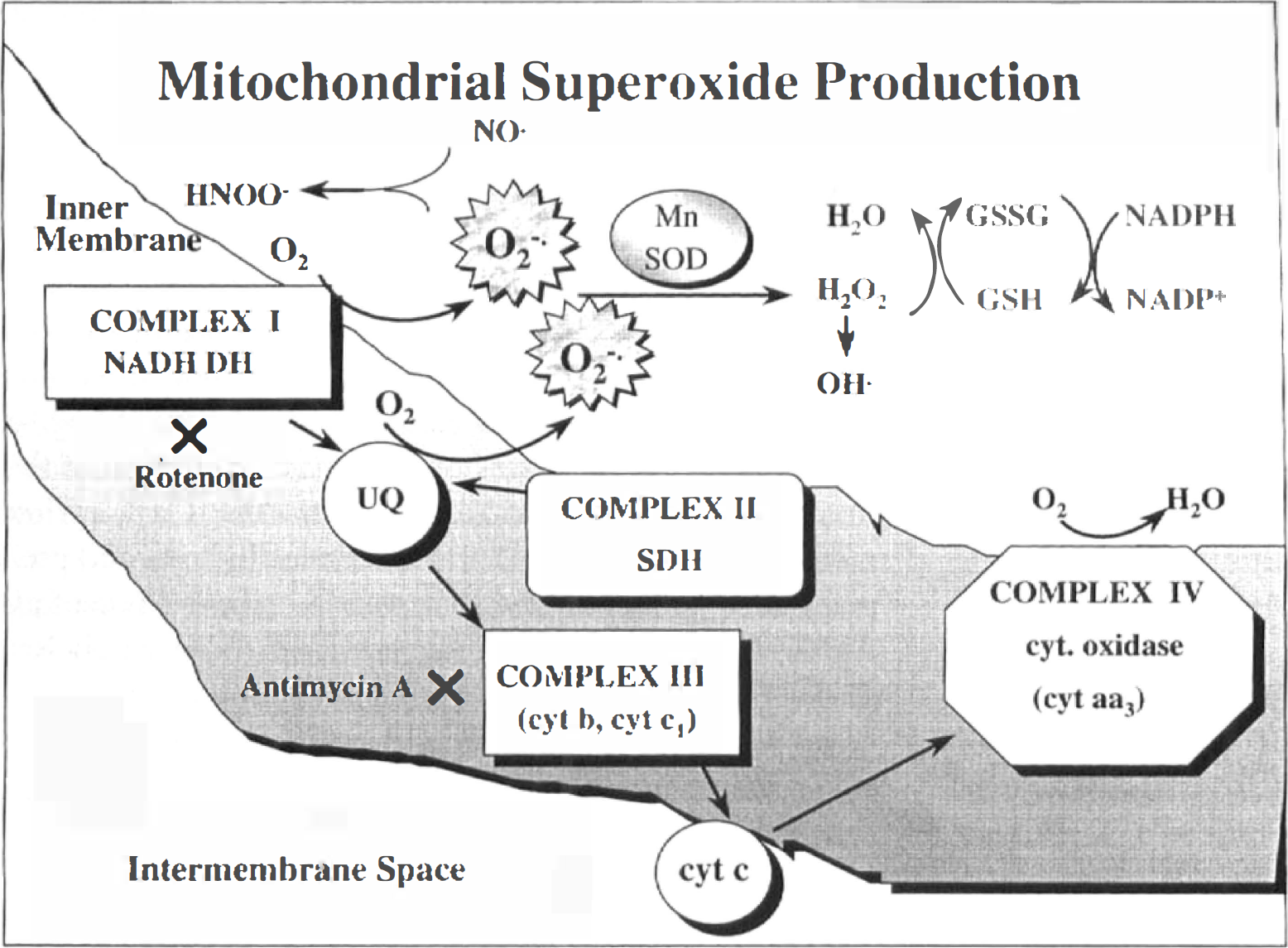
Mitochondrial superoxide production. HNOO−, peroxynitrite; NO·, nitric oxide; O2−·, superoxide; OH·, hydroxyl radical; Mn SOD, manganese superoxide dismutase; GSSG, glutathione disulfide; GSH, glutathione.
On the intact cell level, fluorescence imaging studies document Ca2+ sequestration-induced stimulation of mitochondrial ROS generation relevant to neurodegeneration. Using primary cultures of neurons, Reynolds and Hastings (1995), Dugan et al. (1995), and Bindokas et al. (1996) have provided evidence for NMDA receptor activation-induced increases in ROS generation by a source that appears to be mitochondrial. This response was dependent on extracellular Ca2+ and was insensitive to inhibitors of nitric oxide synthase or arachidonate metabolism (Reynolds and Hastings, 1995; Dugan et al., 1995). In contrast, K+ depolarization did not induce mitochondrial ROS generation (Dugan et al. 1995; Bindokas et al., 1996). As a cautionary note, work by Budd et al. (1997) indicates that certain ROS-sensitive fluorescent dyes can yield misleading data under conditions in which mitochondria may depolarize. In vivo data supporting the premise that mitochondria are a primary source of ROS in ischemia/reperfusion injury has been provided only recently and is reviewed in Fiskum et al. (1999).
MITOCHONDRIAL MEMBRANE PERMEABILITY TRANSITION
If the degree of mitochondrial Ca2+ loading during excitoxic events is critical to determining whether a neuron will remain viable or will undergo either necrosis or apoptosis, then what is the critical Ca2+-stimulated mitochondrial event? The answer to this question is currently unresolved; however, it is known that excessive Ca2+ sequestration, as well as other stimuli, can induce the mitochondrial membrane permeability transition in mitochondria of certain tissues. This event represents the opening of a nonspecific, voltage-sensitive proteinacious pore (PTP), which allows solutes of less than 1500 daltons (d) (including Ca2+, glutathione, and pyridine nucleotides) to equilibrate across the membrane. A lower conductance state of the pore may be induced by more physiologic levels of mitochondrial Ca2+ sequestration, which may operate to release sequestered Ca2+ (Ichas and Mazat, 1998). Opening of the high-conductance pore induces a maximal rate of substrate oxidation and O2 consumption in a futile attempt by the mitochondria to establish an electrochemical gradient. Provided that the transition occurs in a sufficient population of mitochondria, this event should induce a decrease in the measured Δψ. Pore formation also sustains a drop in Δψ induced by Ca2+ or other stress that compromises membrane potential. In other words, a measured drop in Δψ in response to high extramitochondrial Ca2+ may have two phases, the first involving a Ca2+ uniporter-induced drop, followed by a second phase resulting from Ca2+ stimulation of pore formation. However, a measured drop in mitochondrial membrane potential is not automatically indicative of pore formation. Based on the behavior of isolated mitochondria suspended in isotonic buffers, sustained PTP opening or, for that matter, any significant loss in mitochondrial inner membrane integrity in an intact cell, may result in mitochondrial swelling (which is the result of a high concentration of relatively fixed charges associated with the proteins and phospholipids within and surrounding the matrix). Whether swelling results from pore formation in an intact cell is still debated. Δψ can be recovered after pore closure, provided that pore opening does not persist to a point at which swelling induces irreparable damage to membranes or the pyridine content is insufficient.
Comprehensive reviews of PTP regulation have been provided (Bernardi et al., 1994; Zoratti and Szabó, 1995). Pore formation has been studied primarily by measuring large-amplitude swelling of isolated mitochondria in sucrose-containing buffers. The pore can be induced by a significant increase in matrix Ca2+ (especially in the presence of inorganic phosphate) and is potentiated by a decrease in Δψ or by treatment with prooxidants. Characterized prooxidants include tert-butylhydroperoxide (which results in glutathione or pyridine nucleotide oxidation), metabolic oxidants (such as acetoacetate or oxaloacetate), quinone cycling compounds, and sulfhydryl derivatizing or oxidizing agents such as phenylarsine oxide or diamide (Fig. 4). Evidence also suggests that a step in the process of pore formation involves mitochondrial free radical production. Kowaltowski and others (1996a,b) have found that pore formation in isolated Ca2+-loaded rat liver mitochondria in response to either inorganic phosphate or a loss in Δψ induced by uncoupler is dependent on mitochondrial free radical production.
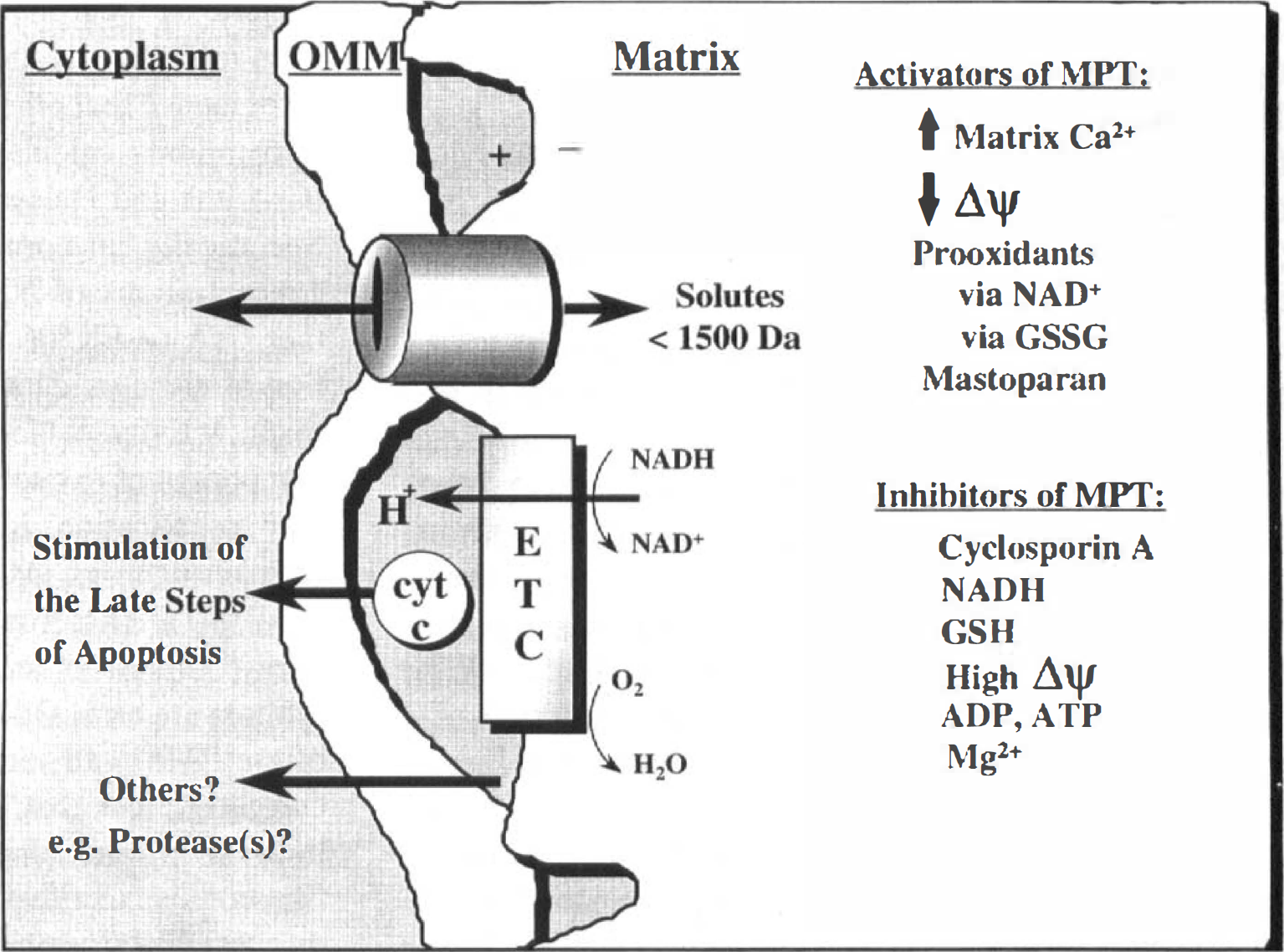
The mitochondrial membrane permeability transition and apoptogen release. See Zoratti and Szabo (1995) for further discussion. ETC, electron transport chain.
Physiologically, pore formation can be inhibited by high Δψ, elevated NAD(P)H/NAD(P)+, sufficient glutathione to maintain critical sulfhydryls in a reduced state, extramitochondrial Mg2+, and ATP or ADP. In addition, data have been provided that a decrease in matrix pH inhibits pore formation (Bernardi et al., 1992). Pharmacologically, the pore typically can be inhibited by the immunosuppressant agent cyclosporin A (CsA), phospholipase A2 inhibitors, and sulfhydryl reducing agents (at certain concentrations). CsA has been proposed to inhibit the PTP by binding to cyclophilin D in the mitochondrial matrix (Nicolli et al., 1996; Connern and Halestrap, 1996). Other biological activities of CsA include binding to cytosolic cyclophilins. This CsA-cyclophilin complex can bind and inhibit calcineurin, a cytoplasmic Ca2+/calmodulin-dependent serine/threonine phosphatase (type 2B), which is enhanced in concentration in neural tissue (Steiner et al., 1992). The inhibition of calcinerin activity appears to be responsible for the immunosuppressant properties of CsA (see Kunz and Hall, 1993).
The protein components of the pore are currently unknown; however, there is compelling evidence that the ANT is involved in pore formation (Zoratti and Szabó, 1995), and directly binds cyclophilin-D (Woodfield et al., 1998). Bongkrekic acid, which stabilizes the translocator in the “M” conformation (for matrix-facing), is a good PTP blocker, whereas atractyloside and carboxy-atractyloside, which stabilize the ANT in the “C” (cytosol-facing) conformation, are good pore inducers. Another proposed component is the voltage-dependent anion channel (see Zoratti and Szabó, 1995). The complex of the ANT, voltage-dependent anion channel, and an 18-kd protein has been suggested to form the mitochondrial benzodiazepine receptor of rat brain (McEnery et al., 1993). Potential regulatory components include mitochondrially associated hexokinase or creatine kinase. The possibility that other large conductance pores may form in the mitochondrial membrane under certain conditions also should be considered. Sokolove and Kinally (1996) suggest that amphiphilic peptides, which structurally mimic the signal sequence for mitochondrial protein import, can induce the formation of a pore that appears to be distinct from the PTP, most notably in requirement for rather than inhibition by Δψ.
Although the characteristics of the PTP have been studied for years, direct evidence that the PTP opens in intact cells has been provided only recently. Researchers have used either a fluorescent dye (Zahrebelski et al., 1995; Nieminen et al., 1995; Nieminen et al., 1997; Bradham et al., 1998) or a radiolabel (Pastorino et al., 1994) to detect an increase in matrix-accessible space that was inhibited by agents known to block the pore in isolated mitochondria. These studies provide evidence that the PTP plays a critical role in hepatocyte necrosis (in response to tert-butylhydroperoxide, chemical hypoxia/aglycemia, or rotenone) and apoptosis (in response to tumor necrosis factor). Numerous, less-direct studies document an ability of pore inhibitors, including CsA to, protect cells (especially hepatocytes) or perfused tissues from prooxidants, anoxia, Ca2+ mobilizing agents, or ischemia/reperfusion.
A definitive role of the PTP in induction of neuronal death is less clear. Studies of a neural version of the PTP using isolated brain mitochondria suggest that physiologic concentrations of Mg2+ and adenine nucleotides are potent inhibitors of the transition (Rottenberg and Marbach, 1990b; Kristal and Dubinsky, 1997; Andreyev et al., 1998). However, no direct measurements of increased matrix-accessible space in the neuronal death pathway have been reported. Kristal and Dubinsky (1997) report Ca2+- or phosphate-induced changes in morphologic features of mitochondria in cultured astrocytes and isolated brain mitochondria that were sensitive to agents known to block large-amplitude swelling in liver mitochondria. Consistent with pore formation in neurons are observations that CsA or inhibitors of phospholipase A2 can attenuate or delay glutamate-induced losses in Δψ (Hoyt et al., 1997; White and Reynolds, 1996; Schinder et al., 1996; Nieminen et al., 1996), and can block excitoxic neuronal death (Dawson et al. 1993; White and Reynolds, 1996; Schinder et al. 1996; Ankarcrona et al., 1996). CsA also protects against death and seizure activity after transient forebrain ischemia or hypoglycemia (Uchino et al., 1995; Li et al., 1997b; Friberg et al., 1998). However, recent evidence for involvement of CsA-inhibitable calcineurin activity in Ca2+-mediated cell death (Shibasaki and McKeon, 1995; Ankarcrona et al., 1996) suggests that this ability to inhibit excitotoxic neuronal death does not result exclusively from inhibition of the PTP. Such skepticism is supported by evidence that other compounds such as FK506, which also inhibits calcineurin yet has no effect on the PTP (Griffiths and Halestrap, 1991), can be neuroprotective. Specifically, in cortical neurons exposed to excitotoxic concentrations of NMDA, calcineurin inhibition by either CsA or FK506 is known to inhibit dephosphorylation of nitric oxide synthase, thereby inhibiting its enzymatic activity, which can contribute to the free radical component of excitoxic death (Dawson et al., 1993). Further complications include reports of cytotoxic effects of CsA on neurons (McDonald et al., 1996; Mosieniak et al., 1997). Therefore, conclusive identification of the pore components and development of pore-specific inhibitors are critical to the ability to clearly define the role of the PTP in all forms of cell death. Although it is not currently clear that the PTP is completely responsible, there is substantial evidence that altered mitochondrial function is a critical event in triggering apoptosis, as discussed later.
MITOCHONDRIA AS THE APOPTOTIC TRIGGER
Correlative evidence that altered mitochondrial function is an early, potentially causal event in the apoptotic pathway in a variety of cell types is plentiful. These data include measurements of early decreases in tetrazolium bromide reduction (Vukmanovic and Zamoyska, 1991; Deckwerth and Johnson, 1993; Jacobson et al., 1994; Ankarcrona et al., 1995), which may be indicative of decreased mitochondrial respiratory function (Slater et al., 1963). Others have measured a decrease in respiratory rates (Krippner et al., 1996) or the degree to which oxidative phosphorylation (ATP production) is coupled to electron transport (Myers et al., 1995; Vayssiere et al., 1994; Cai and Jones, 1998). In addition, numerous studies of diverse cell types responding to widely divergent apoptotic stimuli indicate an early drop in the mitochondrial membrane potential (reviewed by Kroemer et al., 1997). As discussed earlier, such a drop might be expected in Ca2+-mediated forms of apoptosis. However, many of these apoptotic paradigms involved signals devoid of a pathologic increase in cytosolic Ca2+ (Baffy et al., 1993; Koh et al., 1995).
These data are simply correlative. Mitochondrial function has been observed to change early in the apoptotic process, well before evidence of nuclear condensation or loss of plasma membrane integrity. The most compelling evidence for a causal role for altered mitochondrial function in apoptosis has been provided by cell-free reconstitution assays (Newmeyer et al., 1994; Liu et al., 1996; Zamzami et al., 1996b; Yang et al., 1997; Kluck et al., 1997; Ellerby et al., 1997). Initially, Newmeyer and coworkers (1994) subfractionated egg extracts and found that when cytosol and nuclei were recombined, a dense organelle fraction enriched in mitochondria was required for the induction of nuclear condensation or fragmentation. Others have confirmed this result, adding the caveat that the mitochondria capable of inducing nuclear changes must be either damaged or functionally altered (Liu et al., 1996; Zamzami et al., 1996). Liu et al. (1996) suggest that damaged mitochondria release cytochrome c, which ultimately stimulates the caspase family protease CPP-32 (caspase-3) and nuclear condensation when in the presence of cell cytosol and deoxyadenosine triphosphate (dATP). Cytochrome c is an approximate 13-kd electron transport protein normally localized to the mitochondrial intermembrane space, where it electrostatically associates with the outer leaflet of the inner mitochondrial membrane (Fig. 4). Kluck et al. (1997) and Yang and coworkers (1997) have confirmed this potential activity of cytochrome c by assessing the translocation of this protein into the cytosolic fraction of cells treated with the protein kinase inhibitor staurosporine, etoposide, H2O2, or ultraviolet radiation. Ellerby et al. (1997) found similar results in a neural cell version of this assay using staurosporine, tamoxifen, mastoparan, or atractyloside as the inducing agents. Microinjection of cytochrome c can induce apoptosis in some cells with the exception of those lacking expression of caspase-3 (Li et al., 1997a; Zhivotovsky et al., 1998; Duckett et al., 1998). The studies using reconstitution assays also provide evidence that the mechanism by which the antiapoptotic protein Bcl-2 protects from apoptosis is through inhibition of cytochrome c release from mitochondria (however, see Adachi et al., 1997). These studies suggest, as do others (Newmeyer et al., 1994; Susin et al., 1996), that Bcl-2 must be localized to mitochondria to have a significant antiapoptotic effect. Using a cell-free system in which cell cytosol is not required, Susin and associates (1996, 1997) suggest that functionally altered mitochondria release apoptosis inducing factor (AIF), which is a protease (approximately 50 kd) that can stimulate the late stages of apoptosis. Reports also document mitochondrial localization of caspase-3 and caspase-7 (Mancini et al., 1998; Chandler et al., 1998). Therefore, mitochondria can release apoptogenic factors that activate the late steps of apoptosis.
How can the data from cell-free systems supporting a role for mitochondria in apoptosis be extrapolated to physiologic systems? To be convincing, cytochrome c activation of these late stages of apoptosis must be specific, and translocation to the cytosol must occur in intact cells. Lending an indication of specificity, Yang et al. (1997) have indicated that apocytochrome c, (which lacks the heme-containing moiety normally inserted by heme lyase in the intermembrane space) cannot activate caspase-3. Ellerby et al. (1997) found that lysines on the surface of the cytochrome c molecule are critical in the process of caspase-3 activation, since acetylated cytochrome c was unable to stimulate the activation of caspase-3. Numerous studies have now documented the release of cytochrome c to the cytoplasm in various apoptotic models, including NGF withdrawal-induced neuronal apoptosis (Neame et al., 1998; Deshmukh and Johnson, 1998). The latter study supports that cytosolic cytochrome c is not sufficient for execution of the apoptotic scheme in sympathetic neurons. Cytochrome c also redistributes to the cytoplasm following cerebral ischemia in rats (Fujimura et al., 1998; Pérez-Pinzón, 1999), although it cannot be eliminated that this process occurs as a consequence of osmotic lysis of mitochondria during necrotic cell death. Additional translocation data in intact tissues is warranted, and it should be confirmed that the increase in cytosolic cytochrome c is derived from mitochondrial release of heme-containing protein, as opposed to an increase in cytosolic apocytochrome c.
The biochemical role that cytochrome c plays in activating the late steps of apoptosis is unclear, but details are being revealed. At least three cytosolic proteins (termed Apaf-1, Apaf-2, and Apaf-3 for apoptotic protease activating factors) have been identified by Wang's group to be required for caspase activation in their apoptosis reconstitution assay (Liu et al., 1996; Zou et al., 1997). The group identified Apaf-2 as cytochrome c (Liu et al., 1996) and found that cytochrome c can bind to Apaf-1 (Li et al., 1997c), which shares homologic features with the apoptotic death protein in the nematode Caenorhabditis elegans, termed CED-4 (Zou et al., 1997). Evidence has been presented that Apaf-3 is caspase-9, which binds to Apaf-1 in the presence of cytochrome c and dATP, and ultimately activates caspase 3 (Li et al., 1997c; Zou et al., 1997). Redox activity of cytochrome c is not critical to caspase-3 activation (Hampton et al., 1998). A link in the pathway beyond cytochrome c activation of caspase-3 to DNA fragmentation has been provided in a study by Liu et al. (1997) in which a cytosolic heterodimeric protein, called DNA fragmentation factor, was identified and was found to be cleaved by caspase-3, allowing the DNA fragmentation factor to initiate DNA degradation in intact nuclei.
One of the most intriguing issues to be resolved is the mechanism by which cytochrome c or AIF leaves the intermembrane space. Kroemer et al. (1997) propose that opening of the PTP is responsible for both the drop in Δψ measured in many cell types undergoing apoptosis and for the release of apoptogens that stimulate the late steps of apoptosis for which others have provided evidence (Kantrow and Piantadosi, 1997; Scarlett and Murphy, 1997). Because of a lack of definitive measurement of pore formation in intact cells (a drop in the membrane potential and mitochondria swelling are not exclusively indicative of PTP formation), these data are primarily correlative. How does the opening of a pore linking the matrix to the cytoplasmic space control the loss of proteins from the intermembrane space (Fig. 4)? The pore, as characterized, is not large enough to allow cytochrome c or AIF to directly traverse. However, Ca2+- or PTP-induced mitochondrial swelling could rupture the outer membrane, thereby releasing cytochrome c and other intermembrane space proteins, given the small amount of surface area of the outer versus the inner membrane (Chappel and Crofts, 1965; Pfeiffer et al., 1995). Such swelling could be accompanied by a more persistent drop in Δψ (even if PTP opening is transient), since the loss of cytochrome c from the intermembrane space could become rate-limiting for respiration (for instance, see Kim et al. 1997; Cai and Jones, 1998; Krippner et al., 1996; Adachi et al., 1997).
There are two current conflicts associated with the theory that the PTP, or other nonspecific pore formation, is responsible for apoptogen release in all forms of apoptotic death. First, mitochondrial swelling should theoretically accompany nonspecific pore formation. Yet, swelling is not a hallmark morphologic change associated with apoptotic death. In fact, the lack of organellar swelling is noted as a general characteristic of apoptosis (Kerr et al., 1972). Morphologic descriptions of apoptosis may have not have detected mitochondrial swelling because this may be an early, transient event in vivo. Mitochondria have been documented to swell early in the pathway of Fas-mediated apoptosis (Krippner et al., 1996; Vander Heiden et al., 1997). Others have found mitochondrial swelling in HIV-induced apoptosis (Carbonari et al., 1997). The possibility also exists that swelling of only a small fraction of the mitochondrial population may be required to release sufficient apoptogen, although this seems to be a risky strategy for survival in an evolutionary sense. It may be relevant that conditions have been described for the release of matrix components from isolated mitochondria in the absence of detectable large-amplitude swelling, yet electron micrographs indicate that a fraction of the mitochondria under these conditions are slightly swollen (Reed and Savage, 1995). So, it is possible that mitochondrial swelling does take place in apoptosis but largely goes undetected.
The second problem with nonspecific pore formation as the sole mechanism for apoptogen release is that several researchers have presented data arguing that there is no measureable drop in Δψ before cytochrome c release (Yang et al., 1997; Kluck et al., 1997; Kim et al. 1997; Vander Heiden et al., 1997; Richards et al., 1997; Bossy-Wetzel et al., 1998; Cai and Jones, 1998). These studies raise several issues, both technical and theoretical. Regardless of the caveats that follow, these studies present the possibility that the specific apoptotic stimulus may determine whether there is a measureable loss in Δψ before cytochrome c translocation. It may be reasonably theorized that stimuli that involve significant mitochondrial Ca2+ loading or prooxidant stress induce the release of cytochrome c through PTP stimulation. Alternatively, the signaling process to mitochondria from stimuli such as staurosporine or trophic factor withdrawal is less clear and could involve novel pathways impinging on mitochondrial function and cytochrome c release. Technically, several issues that should be considered when evaluating these data. For measurement of mitochondrial membrane potential to be informative, mitochondria must be provided with a respiratory substrate and with a dye that is membrane potential-sensitive without secondary effects on the sulfhydryl redox status of the mitochondria. Otherwise, the data are uninterpretable. The methods also should be quantitative and sensitive at levels of high membrane potential, since it is possible that a drop in Δψ resulting from a permeability transition may only have to occur in a fraction of the mitochondrial population to propagate the apoptotic signal. This is complicated by the fact that as a subpopulation of mitochondria depolarize, others with normal Δψ may accumulate the excess cationic dye, ultimately resulting in little or no detectable change in fluorescence. Evaluation of the behavior of the fluorescent dye over a range of uncoupler concentrations is important to understanding the effect of dye quenching on the fluorescent signal as mitochondria depolarize. That is, the fluorescence of rhodamine 123 can increase as mitochondria depolarize from dequenching of the dye (Emaus et al., 1986). This behavior of the dyes makes increases in fluorescence difficult to interpret with respect to changes in Δψ.
As reports now indicate that cytochrome c can be released in the absence of the permeability transition as conventionally defined (Andreyev et al., 1998), identification of the mechanism responsible for such release is imperative. In this circumstance, either swelling takes place that does not compromise Δψ, or a specific mechanism for translocation of apoptogens across the outer membrane must exist. With regard to swelling, mechanisms of the control of mitochondrial volume have been described, and resistance of isolated mitochondria to hypotonic stress is known to involve activity of the K+/H+ antiporter that is driven by ΔpH (Brierley et al., 1994; Halestrap, 1989). If an apoptotic signal involves a swelling stress to mitochondria, then observations of hyperpolarization before apoptogen release may be real. That is, providing that the total protonmotive force remains constant, ΔpH could collapse in an attempt at volume regulation, whereas Δψ would commensurately increase. In considering some type of specific channel, the largest known channel in the outer membrane is formed by mitochondrial porin, also known as the voltage-dependent anion channel, which may accommodate molecules as large as 8 kd under certain conditions (De Pinto and Palmieri, 1992). This may not be large enough to translocate the approximately 15-kd cytochrome c and is not large enough for the approximately 50-kd AIF. Other theoretical possibilities include reversal of the protein import process (Mayer et al. 1995) or specific facilitation by membrane components. It also has been proposed that the mitochondrial outer membrane is a target for specific proteolysis early in Fas-induced apoptosis (Susin et al., 1997; Krippner et al., 1996).
Development of a theoretical mechanism for apoptogen release also must consider current x-ray and NMR data suggesting that Bcl-xL shares some structural homologic features to the domains of bacterial toxins that form pores in membranes (Muchmore et al., 1996). This predicted function has been confirmed in electrophysiologic studies demonstrating ion conductance (most predominant at acidic pH) by recombinant Bcl-xL and Bcl-2 in phospholipid bilayers (Minn et al., 1997; Schendel et al., 1997; Schlesinger et al., 1997). Antonsson and others (1997) have shown that recombinant Bax forms channels in liposomes, allowing preloaded carboxyfluorescein (molecular weight ∼376d) to be released at physiologic pH, although acidic pH significantly enhanced the rate and extent of release. This Bax-induced release of fluorescent dye was inhibitable by recombinant Bcl-2. Recent evidence suggests that Bax may induce cytochrome c release and caspase activation in a CsA-sensitive manner, suggestive of induction of the PTP (Pastorino et al., 1998; Jürgensmeier et al., 1998; Narita et al., 1998). In contrast, Eskes et al. (1998) have suggested that the effects of Bax are not mediated by the PTP as conventionally defined. Since Bax may play an important role in trophic factor withdrawal-induced apoptosis in neurons (White et al., 1998), a clearer understanding of the mechanism of action of Bax is warranted.
How could channel formation in mitochondria be incorporated into a hypothesis for regulated control over apoptosis? It is difficult to devise a theoretical scenario in which an antiapoptotic protein would form a K+ or Cl− channel that could operate to the advantage of the mitochondria, given that the ultimate function of these organelles is highly dependent on the impermeability of the inner mitochondrial membrane to such ions. For instance, a K+-specific channel would allow this ion to move down the electrical gradient, dissipating Δψ and potentially inducing mitochondrial swelling, which might be more appropriate as a theoretical mechanism for a proapoptotic factor such as Bax. Theoretically, it is possible to ascribe K+ or Cl− channel function to anti-apoptotic proteins if they are linked to energy-dependent translocation of osmotically active ions out of the matrix (possibly to enhance mitochondrial volume homeostasis). It is known that the mitochondria of control and bcl-2-overexpressing cells have virtually identical respiratory properties under control conditions (Myers et al., 1995; Murphy et al., 1996; Murphy and Fiskum, 1996); therefore, there is no current evidence that antiapoptotic proteins such as Bcl-2 act to enhance respiration or decrease the level of uncoupling under control conditions. However, it is clear that Bcl-2 preserves electron transport function in response to stress (Murphy et al., 1996a,b; Myers et al., 1995; Shimizu et al., 1998). Whether this ability to maintain electron transport in response to prooxidant or Ca2+-induced stress is solely the result of the ability of Bcl-2 to block cytochrome c release requires further investigation.
There also is current evidence that kinase-mediated signal transduction cascades regulate the function of Bcl-2 and its proapoptotic homologue, Bad, at the mitochondria (Gajewski and Thompson, 1996). Survival factor signaling through the interleukin-3 receptor on hematopoietic cells results in the phosphorylation of Bad, preventing apoptosis potentially by inhibiting Bad's sequestration of Bcl-2 or Bcl-xL through heterodimerization (Zha et al., 1996). Wang and others (1996) have provided evidence that Bcl-2 is involved in targeting Raf-1 (a kinase known to be involved in ras-mediated growth factor signaling) to mitochondria, which may ultimately result in phosphorylation and inhibition of Bad. Furthermore, serine phosphorylation of Bcl-2 has been reported to alter its antiapoptotic activity (Haldar et al., 1997), and deletion of a regulatory region of Bcl-2, which contains multiple serine/threonine phosphorylation sites, extends the ability of Bcl-2 to prevent apoptosis (Chang et al., 1997).
Translocation of regulatory molecules to allow for specific protein-protein interactions at the mitochondrial membrane may be important in apoptogen release. Specifically, Bax and Bcl-xL shift from cytosolic- to membrane-bound forms during thymocyte apoptosis, whereas Bcl-2 is found consistently to be membrane bound (Hsu et al., 1997).
Unanswered questions
Critical issues with regard to the mechanism of mitochondrial apoptogen release remain to be resolved. If there are exceptions to permeability transition-mediated apoptogen release, are the exceptions relevant pathophysiologic conditions? That is, does staurosporine-induced apoptosis mimic a disease process that makes this alternative mechanism a target for therapeutics? Is this alternative mechanism mediated by mitochondrial swelling or by some more specific event allowing for apoptogen release? Alternatively, is the permeability transition mediating all forms of apoptogen release, and in a subset of apoptosis paradigms this occurs to an undetectable portion of the total mitochondrial population? On a more basic level, we need to understand the degree to which the apoptotic pathway contributes to neuronal dysfunction in neurodegenerative diseases. Is early dementia in Alzheimer's disease caused by an abortive apoptotic pathway that creates chronic cell dysfunction? These are only a few of the critical questions for understanding the role that mitochondria play in neurodegeneration, and the answers will facilitate rationale therapeutic design.