
Abstract
Release of cytochrome c (cyt c) into cytoplasm initiates caspase-mediated apoptosis, whereas activation of Akt kinase by phosphorylation at serine-473 prevents apoptosis in several cell systems. To investigate cell death and cell survival pathways, the authors studied release of cyt c, activation of caspase, and changes in Akt phosphorylation in rat brains subjected to 15 minutes of ischemia followed by varying periods of reperfusion. The authors found by electron microscopic study that a portion of mitochondria was swollen and structurally altered, whereas the cell membrane and nuclei were intact in hippocampal CA1 neurons after 36 hours of reperfusion. In some neurons, the pattern of immunostaining for cyt c changed from a punctuate pattern, likely representing mitochondria, to a more diffuse cytoplasmic localization at 36 and 48 hours of reperfusion as examined by laser-scanning confocal microscopic study. Western blot analysis showed that cyt c was increased in the cytosolic fraction in the hippocampus after 36 and 48 hours of reperfusion. Consistently, caspase-3–like activity was increased in these hippocampal samples. As demonstrated by Western blot using phosphospecific Akt antibody, phosphorylation of Akt at serine-473 in the hippocampal region was highly increased during the first 24 hours but not at 48 hours of reperfusion. The authors conclude that transient cerebral ischemia activates both cell death and cell survival pathways after ischemia. The activation of Akt during the first 24 hours conceivably may be one of the factors responsible for the delay in neuronal death after global ischemia.
Transient cerebral ischemia leads to delayed neuronal cell death in CA1 pyramidal neurons observed by light microscopic study after 2 to 3 days of reperfusion (Kirino, 1982; Pulsinelli et al., 1982; Smith et al., 1984). This delayed cell death provides an opportunity for studying the development of cell death after ischemia. The mechanisms underlying ischemic delayed neuronal death are not fully understood, Recent evidence suggests that brain ischemia activates cysteine proteases of the interleukin-1β family (caspases), which then trigger neuronal apoptosis (Yakovlev et al., 1997; Namura et al., 1998; Chen et al., 1998; Siesjo et al., 1998), However, the mechanism that causes the activation of caspase after brain ischemia still is unknown.
Mitochondria are believed to play a critical role in apoptosis in several cell systems, including neurons (Ankarcrona et al., 1995; Bernardi, 1996; Wadia et al., 1998; Kraemer et al., 1998; Yong and Cortopassi, 1998). When overloaded with calcium, subjected to oxidative stress, or exposed to dithiol cross-linking reagents, mitochondria are prone to open a membrane megachannel, referred to as a mitochondrial permeability transition (MPT) pore (Bernardi, 1996; Kraemer et al., 1998), This leads to swelling of the mitochondria, It has been hypothesized that long-term opening of the MPT may cause release of apoptogenic substances such as cytochrome c (cyt c) from mitochondria into the cytoplasm (Kroemer et al., 1998; Yong and Cortopassi, 1998; Green and Reed, 1998). Release of cyt c from mitochondria into cytoplasm, together with other apoptotic factors, activates caspase-3 and results in apoptosis (Liu et al., 1996; Li et al., 1997). Bcl-2 may prevent apoptosis, probably through the blocking of cyt c release (Yang et al., 1997; Kluck et al., 1997; Green and Reed, 1998). Cyclosporin A, a potent and virtually specific MPT blocker, is highly neuroprotective in conditions of transient cerebral ischemia (Uchino, et al. 1995; Li et al., 1996; Butcher et al., 1997; Siesjö et al., 1998).
Working parallel to factors constituting the cell death pathway, several molecules in the intracellular signaling pathways have been identified as survival factors that prevent cell death. These include some Bcl-2 family of proteins (e.g., Bcl-2 and Bcl-XL) and Akt kinase (Franke et al., 1997; Downward, 1998; Croder and Freeman, 1998). The Bcl-2 and Bcl-XL proteins have been shown to be expressed in surviving neurons but not in dying neurons after transient cerebral ischemia (Chen et al., 1997). Akt protein kinase is a serine/threonine kinase. When phosphorylated at Ser-473 by phosphatidylinositol 3-kinase, Akt kinase will transduce cell survival signals to prevent apoptosis in several cell systems (Dudek et al., 1997; Datta et al., 1997; Franke et al., 1997; Downward, 1998; Crowder and Freeman, 1998). For example, Akt phosphorylating Bad couples survival signals by releasing the Bad inhibition on Bcl-2 (Datta et al., 1997). Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor—dependent sympathetic neurons (Crowder and Freeman, 1998).
To understand cell death mechanisms after brain ischemia, we investigated the cell death and cell survival pathways in the hippocampus after transient cerebral ischemia. Our results suggest that transient cerebral ischemia activates the cell death pathway by release of cyt c and by activation of caspase as well as the cell survival pathway by activation of Akt. Activation of Akt may delay the cell death in hippocampal CA1 sector after transient cerebral ischemia.
EXPERIMENTAL PROCEDURES
Ischemic model
Male Wistar rats (250 to 300 g) were fasted overnight. All experimental procedures were approved by the subcommittee on animal studies of The Queen's Medical Center, Honolulu. Ischemia was induced as described previously (Smith et al., 1984). Anesthesia was induced with 3% halothane followed by maintenance of 1 % to 2% halothane in an oxygen/nitrous oxide (30%170%) gas mixture. Catheters were inserted into the external jugular vein, tail artery, and tail vein to allow blood sampling, arterial blood pressure recording, and drug infusion. Both common carotid arteries were encircled by loose ligatures. Fifteen minutes before ischemia induction and 15 minutes after ischemia, blood gases and pH were measured, and ventilation was adjusted to yield Pao2 above 90 mm Hg, Paco2 35 to 45 mm Hg, and pH 7.35 to 7.45 by altering the tidal volume of the ventilator. Bipolar EEG was recorded every 5 to 10 minutes before ischemia, continuously during the ischemic insult, and every 5 minutes after ischemia until the rat recovered from the anesthesia. At the beginning of a 30-minute steady-state period before induction of ischemia, the inspired halothane concentration was decreased to 0.5%, and 150 IU/kg heparin was administered intravenously. Blood was withdrawn from the jugular catheter to yield hypotension with a mean arterial blood pressure of 50 mm Hg, and both carotid arteries were clamped. Blood pressure was maintained at 50 mm Hg during the ischemic period by withdrawal or infusion of blood. At the end of the 15-minute period of ischemia, the clamps were removed, and the blood was reinfused through the jugular catheter, followed by 0.5 mL of 0.6 mol/L sodium bicarbonate. In all experiments, brain temperature, as measured subcutaneously on the skull bone, was maintained at 37°C before, during, and after ischemia with a heating lamp. Brain samples were collected during ischemia, as well as after 0.5, 4, 24, 36, and 48 hours of reperfusion. In the 30-minute recovery group, animals were maintained under anesthesia for the entire postishemic period before tissues were sampled. In animals with longer reperfusion times, halothane was discontinued at the end of ischemia, and all wounds were sutured. The animals were allowed 4, 24, 36, or 48 hours of reperfusion. These animals were reanesthetized, tracheotomized, and artificially ventilated at 30 minutes before the end of reperfusion, and the tissues were sampled at the end of reperfusion.
Tissue samples for Western blot analysis of cyt c were obtained by decapitation under anesthesia. The dorsal hippocampus was immediately isolated to prepare subcellular fractions. All procedures were conducted in a cold room. Nonfrozen brain tissue was used for analysis of cyt c release with Western blot because freezing tissue causes release of cyt c from mitochondria (data not shown). We did not divide the dorsal hippocampus into CA1 and dentate gyrus (DG) regions for analysis because time-consuming dissection may have caused cyt c release. Tissue samples for analysis of Akt phosphorylation were obtained by freezing the brain in situ with liquid nitrogen to prevent protein dephosphorylation from occurring after decapitation and tissue isolation (Hu et al., 1994, 1998). The brains were perfused with ice-cold 4% phosphate-buffered paraformaldehyde in phosphate-bufered saline (PBS) for immunocytochemical study, and perfused with 2% paraformaldhyde/2.5% glutaraldhyde in 0.1 mol/L cacodylate buffer for electron microscopic examination. The brains were sectioned with a Leica Vibratome (Heidelberg, HRB, Germany) at a thickness of 50 μm for immunocytochemical study or of 200 μm for electron microscopic study. Sham-operated rats were subjected to the same surgical procedures but without clamping of the arteries.
Electron microscopic studies
Tissue sections from experimental and control animals were stained by the conventional osmium—uranium—lead method. Briefly, coronal brain sections were cut at a thickness of 200 μm with a Vibratome through the level of the dorsal hippocampus and postfixed for 1 hours with 4% glutaraldehyde in 0.1 mol/L cacodylate buffer (pH 7.4). Sections were postfixed for 2 hours in 1 % osmium tetroxide in 0.1 mo1/L cacodylate buffer, rinsed in distilled water, and stained with 1 % aqueous uranyl acetate overnight. The tissue sections then were dehydrated in an ascending series of ethanol to 100%, followed by dry acetone, and embedded in Durcopan ACM. Thin sections were counterstained with lead citrate before examination in the electron microscope.
Immunocytochemistry
Double-label fluorescence immunocytochemical study was performed on coronal brain sections (50 μm) from a sham-operated control group, and animals subjected to 15 minutes of ischemia followed by 24, 36, and 48 hours of reperfusion. A monoclonal antibody against cyt c was obtained from PharMingen (San Diego, CA, U.S.A.). The brain sections were washed twice in PBS for 5 minutes at room temperature (RT) and then in PBS containing 0.2% TX100 for 30 minutes. Nonspecific binding sites were blocked in 3% bovine serum albumin (BSA) in PBS/0.2% TX100 for 30 minutes. The brain sections were incubated with a primary antibody for cyt c diluted 1:500 in PBS/0.1 % TX100 and 1 % BSA overnight at 4°C, then washed in PBS containing 0.1 % TX100, three times 10 minutes each at RT. The sections were labeled for 2 hours at RT with fluorescein-labeled anti-mouse secondary antibody and propidium iodide to stain nuclear. For Akt immunolabeling, brain sections were incubated with poly clonal Akt antibody (1:500) overnight. After several washes, the sections were incubated for 2 hours at 4°C with a monoclonal antibody against microtubule-associated protein 2 (MAP-2, Sigma) diluted 1:300 in PBS/0.1% TX100 and 1% BSA. Fluorescein-labeled anti-rabbit and lissamine rhodamine—labeled anti-mouse antibodies were diluted 1:200 in PBS containing 0.1 % TX100 and 1% BSA and applied for 1 hour at RT. The sections for both cyt c and Akt were washed several times in PBS/0.1 % TX100 and mounted on glass slides; coverslips then were applied using Gelvatol. The slides were analyzed on a BioRad MRC 1024 laser-scanning confocal microscope.
Preparation of subcellular fractions
Subcellular fractions were prepared from sham-operated animals and from ischemic brains after 30 minutes and 4, 24, 36, and 48 hours of reperfusion. Samples were prepared from four animals in each group. Preparation of subcellular fractions was carried out at 4°C. Brain tissues were homogenized using a Dounce homogenizer (15 strokes) in 10 volumes of homogenization buffer containing 15 mmol/L Tris base/HCl, pH 7.6, 1 mmol/L DTT, 0.25 mol/L sucrose, 1 mmol/L MgCl2, 1.25 μg/ml pepstatin A, 10 μg/ml leupeptin, 2.5 μg/ml aproptonin, 0.5 mmol/L PMSF, 2 mmol/L ethylenediamine tetraacetic acid, 1 mmol/L EGT A, 0.1 mol/L Na3 VO4, 50 mmol/L NaF, and 2 mmol/L sodium pyrophosphate. The homogenates then were centrifuged at 1000 × g at 4°C for 10 minutes. The pellets were discarded, and the supernatants were centrifuged at 17,000 × g at 4°C for 20 minutes to get the cytosolic fraction in the supernatant and the crude mitochondrial fraction in pellets. The protein concentration was determined by the method of Lowry.
Western blot analysis
Western blot analysis was carried out on 15% SDS—polyacrylamide gel electrophoresis (SDS-PAGE) for cyt c and cyt c oxidase (subunit IV) and 8% SDS-PAGE for Akt and phospho-Akt according to the method of Laemmli (1970). One sample containing 50 μg of protein from cytosolic fraction or 10 μg of mitochondrial fraction was applied to each lane in a slab gel of SDS-PAGE. After electrophoresis, proteins were transferred to an immobilon-P membrane (Millipore). The membrane was incubated overnight at 4°C with primary antibodies against cyt c, cyt c oxidase (subunit IV), Akt, or phospho-Akt, each at a dilution of 1:2000. Monoclonal cyt c and cyt c oxidase antibodies were obtained from PharMingen (San Diego, CA, U.S.A.) and Molecular Probes (Eugene, OR, U.S.A.) respectively. Polyclonal antibodies to Akt and phospho-Akt were obtained from the New England Biolabs (Beverly, MA, U.S.A.). The antibody against Akt does not distinguish between phosphorylated and unphosphorylated forms, whereas the antibody to the phosphorylated forms recognizes only the phosphorylated forms. The membranes then were incubated for 45 minutes at RT with horseradish peroxidase—conjugated secondary antibodies. The blots were developed using the ECL detection method (Amersham). Four animals in each experimental group were used to analyze the levels of each protein by Western blot; each protein band on the Western blots was derived from one animal. Each immunoblot was performed using the samples from two animals in each experimental group. The density of protein bands in some experiments was evaluated according to the method we described previously (Hu et al., 1994).
Caspase-3 activity
Ac-DEVD-AMC[N-acetyl-Asp-Glu-Val-Asp-AMP(7-amino-4-methylcoumarin] (PharMingen, component no. 6633KC) was used to determine caspase activity. Ac-DEVD-AMC has been reported to have linear Michaelis-Menton kinetics with a Michaelis constant of 10 μmol/L for caspase-3. Caspase-3 cleaves the tetrapeptide between D and AMC, releasing the fluorescent AMC, which can be quantified by spectrofluorometry, using an excitation wavelength of 380 nm and an emission wavelength of 440 nm. For each reaction, a 25-μL sample (200 μg of protein), 200 μL of reaction buffer (20 mmol/L HEPES [pH 7.5], 10% glycerol, 4 mmol/L DTI), and 5 μL (l μg/μL) of reconstituted Ac-DEVD-AMC was added to each well of a 96-well plate. The reaction mixture was incubated for 1 hour at 37°C. The AMC liberated from the Ac-DEVD-AMC was measured using a well plate reader of a spectrofluorometer (LS-50B, PERKIN ELMER). In some experiments, extracts were preincubated with 5 μL (0.1 μg/μL) of the caspase-3 inhibitor Ac-DEVD-CHO [N-acetyl-Asp-Glu-Val-Asp-CHO (aldehyde)].
RESULTS
Fifteen minutes of transient cerebral ischemia caused more than 95% neuronal death in CA1 pyramidal neurons after 48 or 72 hours of reperfusion in the two-vessel occlusion model (Smith et al., 1984). This pattern of neuronal death was confirmed by examination of cresyl violet—stained sections from postischemic animals (data not shown).
At the electron microscopic level, we found most neurons to possess relatively normal cell membranes and nuclei before 24 hours of reperfusion, but dark material, monoribosomes, and membranous vesicles were found in the CA1 cell body after 4 hours of reperfusion, as described previously (Petito and Pulsinelli, 1984; Kirino et al., 1984; Deshpande et al., 1992; Rafols et al., 1995). At 36 hours of reperfusion, further increases in dark material and membranous vesicles were observed in the neurons. In addition, the damaged mitochondria were found to coexist with morphologically intact mitochondria in most CA1 neurons after ischemia (Figs. 1 and 2, arrows). The cisterns in the mitochondria appeared to be damaged. The morphologic features of the cell nuclei appeared normal in these neurons (Figs. 1 and 2). At 48 hours of reperfusion, some CA1 neurons became dark in the cytoplasm, indicating that these neurons were degenerated (data not shown). These changes were rarely seen in DG granule cells after ischemia. Also, these findings are in line with several previous studies (Petito and Pulsinelli, 1984; Kirino et al., 1984; Deshpande et al., 1992; Rafols et al., 1995).
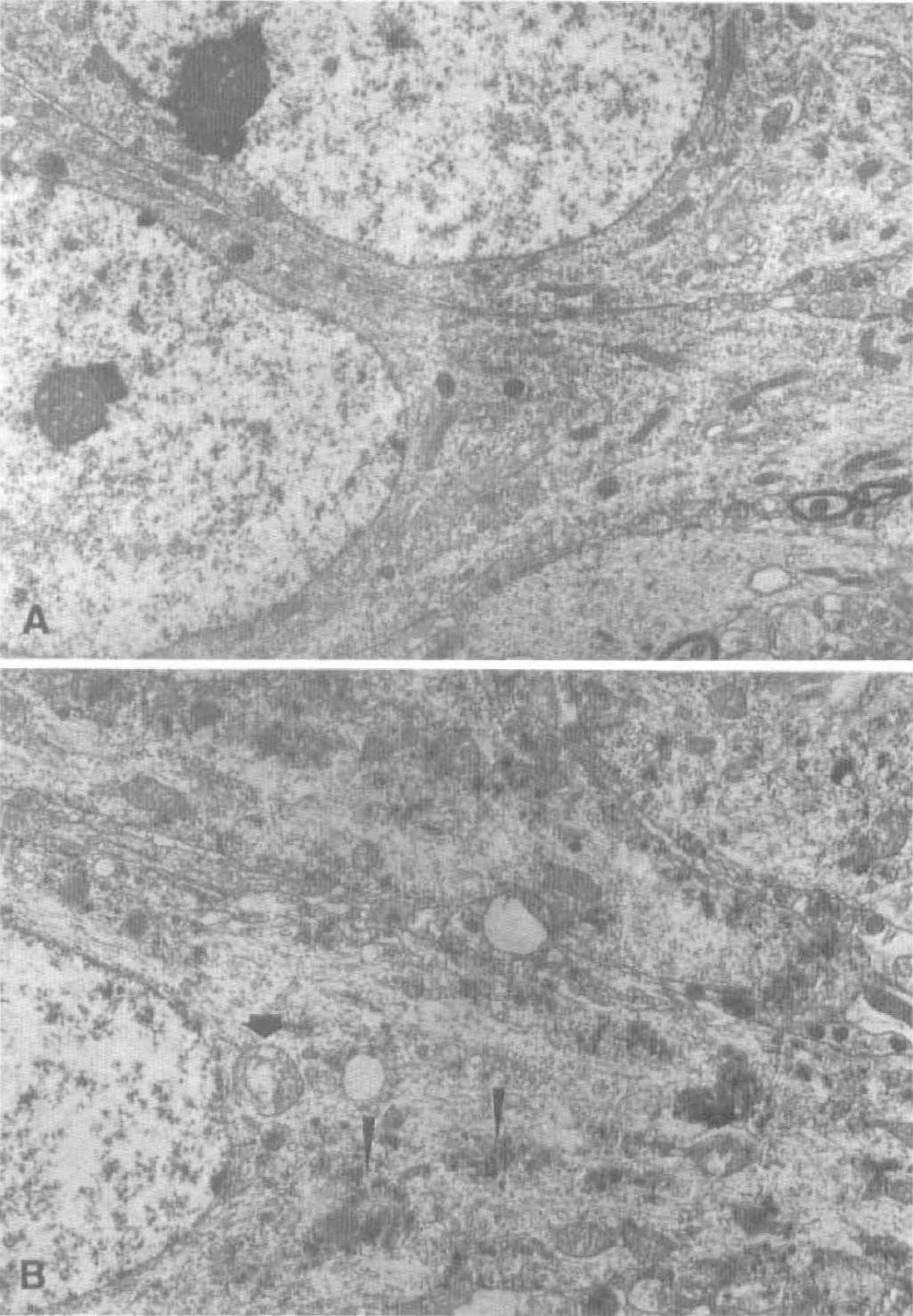
Electron micrographs of osmium-uranyl-Iead—stained pyramidal neurons in the dorsal hippocampal CA1 area from sham-operated rats
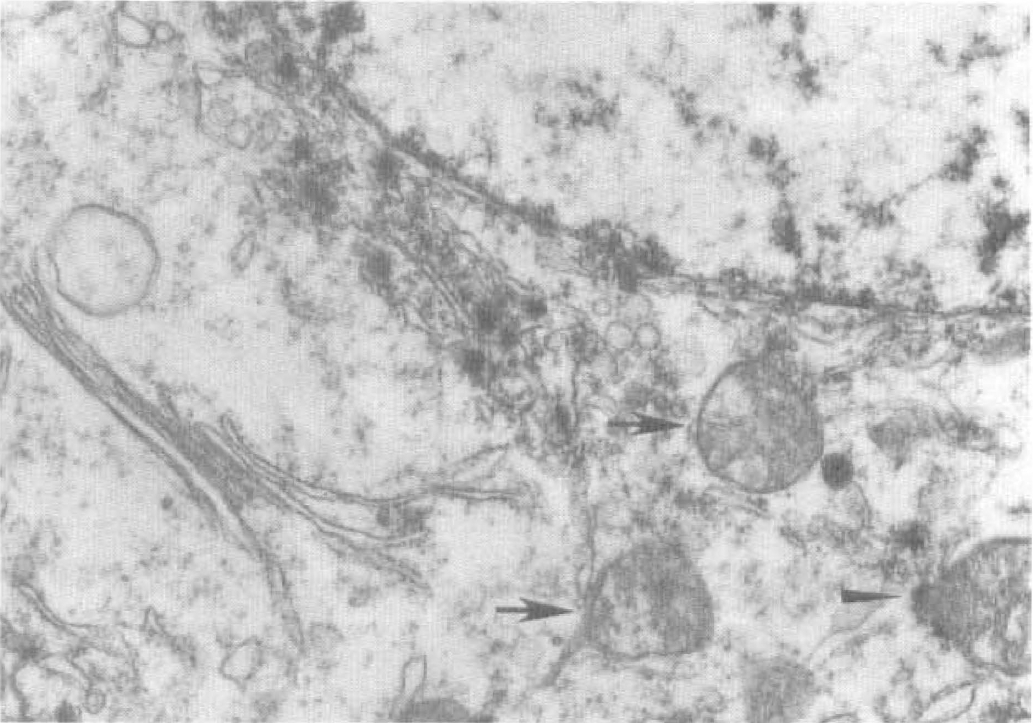
High-magnification electron micrographs of pyramidal neurons in the hippocampal CA1 region from a rat subjected to 15 minutes of ischemia followed by 36 hours of reperfusion. Examples of two damaged mitochondria surrounding a nucleus are shown. Membranes and internal cisterns of these mitochondria are damaged (arrows). Dark material clustered with some unidentified membranous structures. Some dark material was observed close to the mitochondria (arrowhead). Scale bar = 0.1 mm.
To test whether cyt c was released from mitochondria after ischemia, we double-stained brain sections with an antibody against cyt c and with propidium iodide (Fig. 3). We examined these sections by laser-scanning confocal microscopic study. Brain sections were from sham-operated control animals (C) and from animals with 15 minutes of ischemia followed by 24, 36, and 48 hours of reperfusion (Fig. 3). Figure 3A is an image of CA1 neurons and Fig. 3B is of DO granule cells. No obvious changes in the intensity of propidium iodide staining, which stains nucleic acids, were seen in the hippocampus between sham-operated control and postischemic brain sections after 15 minutes of ischemia (Figs. 3A and 3B, red). A punctuate immunolabeling with a monoclonal anti—cyt c antibody, likely representing mitochondria, was clearly visible in neurons from sham-operated control sections (Fig. 3A and 3B, green). The punctuate immunolabeling pattern changed to a more evenly diffuse cytoplasm staining in several CA1 neurons at 36 hours of reperfusion, implying that cyt c might be released from damaged mitochondria into the cytoplasm (Fig. 3A, 36 hours and 48 hours, green). There were 7.3 ± 3.1 (mean ± SD, n = 3) of the diffusing stained neurons in the CA1 region at less than 1 mm of optical slices, as examined by confocal microscopic study at 36 hours of reperfusion after 15 minutes of ischemia. The average number of neurons in the optical slices was 325 ± 55 (mean ± SD, n = 3) in the CA1 region. Therefore, the diffusing stained neurons were about 2% of the CA1 neurons at 36 hours of reperfusion. Such diffusing stained neurons are likely to represent a massive mitochondrial destruction and release of large amounts of cyt c into the cytoplasm. The mitochondrial damage at the electron microscopic level shown in Fig. 2 may not be reflected as such diffusing neurons in confocal microscopic examination. This is because such mitochondrial damage shown in Fig. 2 was seen in most CA1 neurons at 36 hours of reperfusion. Some neurons in the CA1 region were degenerated and lost cyt c immunostaining after 48 hours of reperfusion after 15 minutes of ischemia, judged by their condensed nuclei stained with propidium iodide (Fig. 3A, 48 hours, red). There were no obvious changes in the pattern of cyt c immunostaining in DG after ischemia (Fig. 3B).
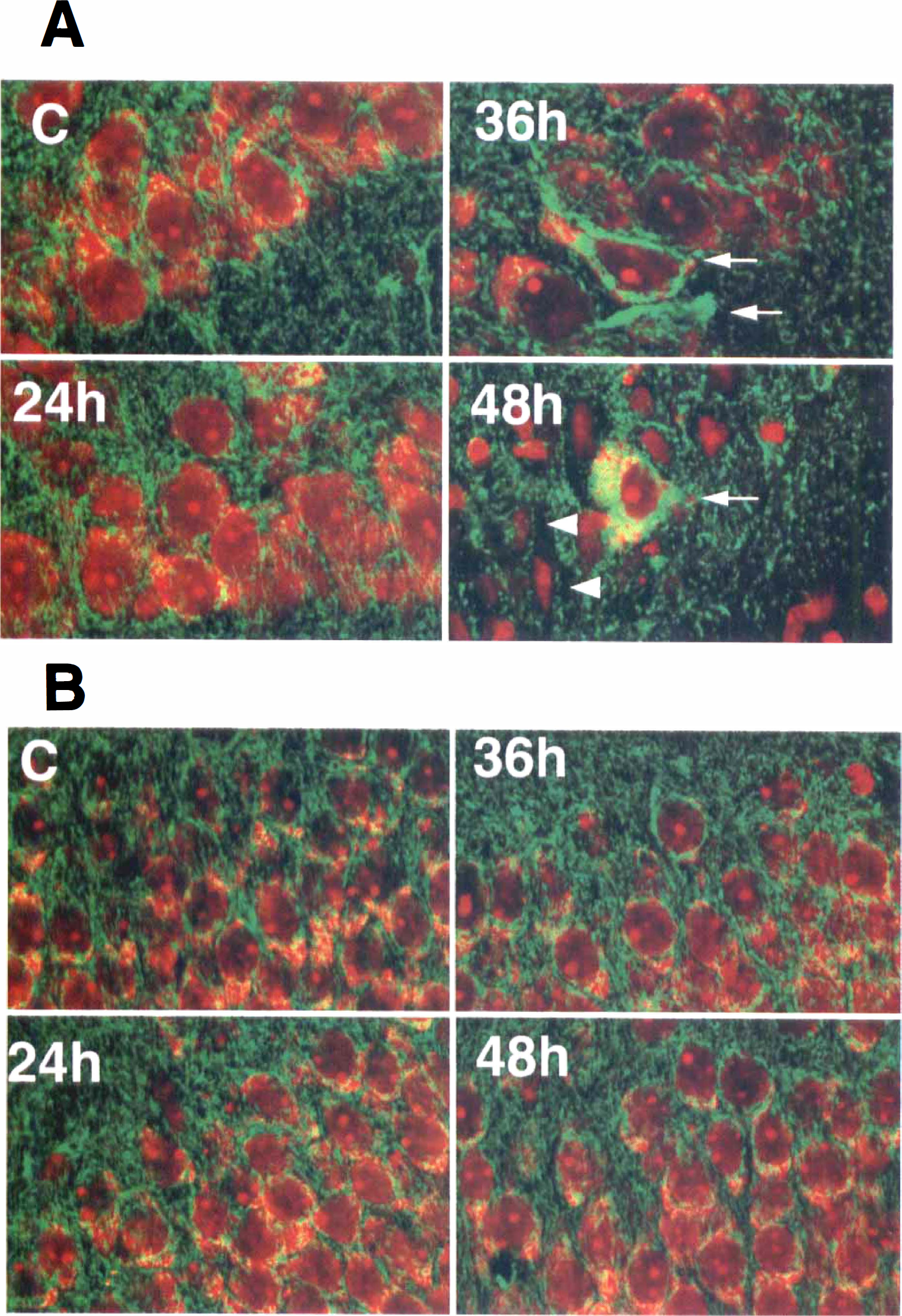
Oouble-immunostaining images for cytochrome c (cyt c) (green) and propidium iodide (red) in CA1
The cytosolic fractions prepared from rat hippocampi isolated immediately after decapitation were used for Western blot analysis of cyt c. Figure 4 demonstrates that the level of cyt c was only slightly increased in the cytosolic fraction after 36 hours of reperfusion in the hippocampal samples, where most of neurons were still intact. A marked increase of cytosolic cyt c in the CA1 region was seen at 48 hours of reperfusion when some neurons were degenerated (Fig. 3A, 48 hours, and Fig. 4B). The levels of cyt c were not measurably decreased in the mitochondrial fraction at 36 or 48 hours of reperfusion (Fig. 4A). This suggests that cyt c may be released from a small portion of damaged mitochondria at 36 and 48 hours of reperfusion. To elucidate whether other mitochondrial protein was released from mitochondria, we determined the cyt c oxidase level in the cytosolic and mitochondrial fraction in Western blots using anti—cyt c oxidase subunit IV antibody. The cyt c oxidase subunit IV was detected only in the mitochondrial fraction but not in the cytosolic fraction (Figs. 4C and 4D). This suggests that cyt c oxidase was not co-released with cyt c from mitochondria.
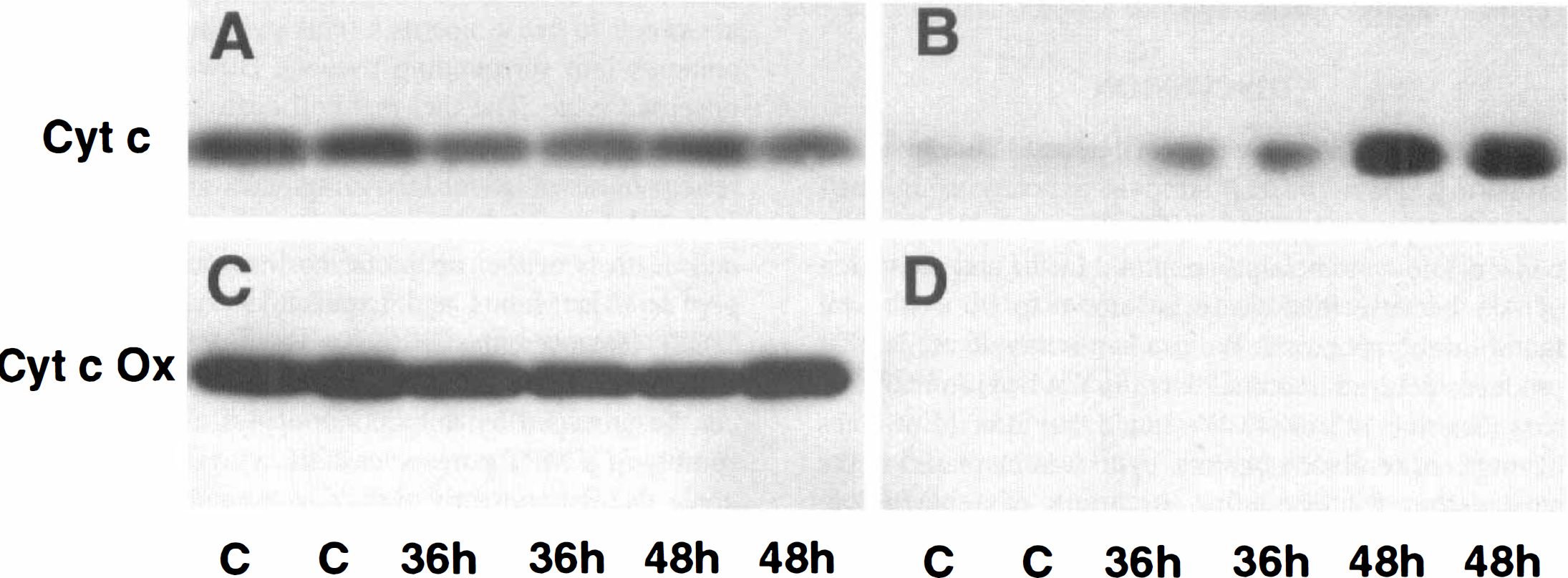
Western blots of cyt c
To follow-up the results of cyt c release, we determined caspase-3–like activity using a synthetic substrate (PharMingen). Figure 5 illustrates that caspase-3–like activity was significantly increased at 36 and 48 hours, respectively, but not at 24 hours of reperfusion after ischemia, suggesting that the caspases may be activated by cyt c released into the cytoplasm.
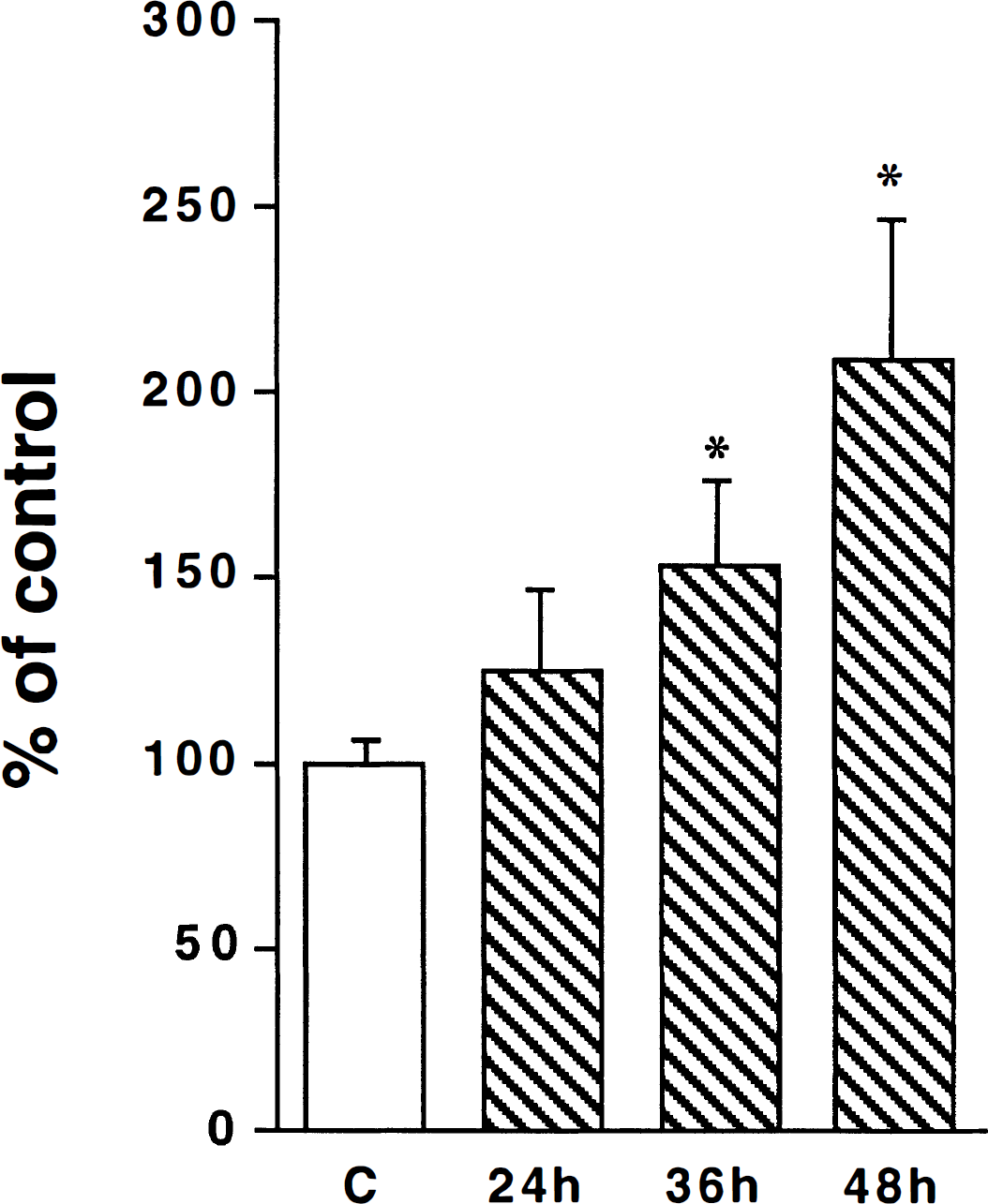
Changes in caspase-3–like activity after ischemia. The brain samples are from sham-operated rats (C) and rats subjected to 15 minutes of transient ischemia followed by 24, 36, and 48 hours of reperfusion, respectively. Ten micrograms of protein in the samples were used for the assay. Data are expressed as mean ± SD (n = 4). *P < 0.05 denotes a significant difference between control and postischemic groups analyzed by analysis of variance followed by Fisher's protected least significant difference test.
To study the activation of Akt protein kinase, we analyzed both total Akt and phospho-Akt in the postischemic hippocampal tissues by Western blot analyses. Total Akt was unchanged in hippocampal CA1 (Fig. 6A) and DG (Fig. 6B) regions after ischemia, but phospho-Akt was dephosphorylated to an undectable level at the end of 15 minutes of ischemia (0 minutes of reperfusion). This may be because of the lack of ATP results in relative activation of phosphatases at the end of 15 minutes of ischemia (Hu et al., 1998). The Phospho-Akt was rapidly rephosphorylated over the control levels at 30 minutes and 4 hours of reperfusion (Figs. 6C and 6D). Phosphorylation of Akt returned to the control level at 24 hours (Figs. 6C and 6D), with no secondary increase at 36 and 48 hours of reperfusion (data not shown). The density of phospho-Akt bands were evaluated and demonstrated in Fig. 6E. No significant differences were observed in the levels of the Akt phosphorylation between the CA1 region and the DG area after ischemia (Fig. 6E).
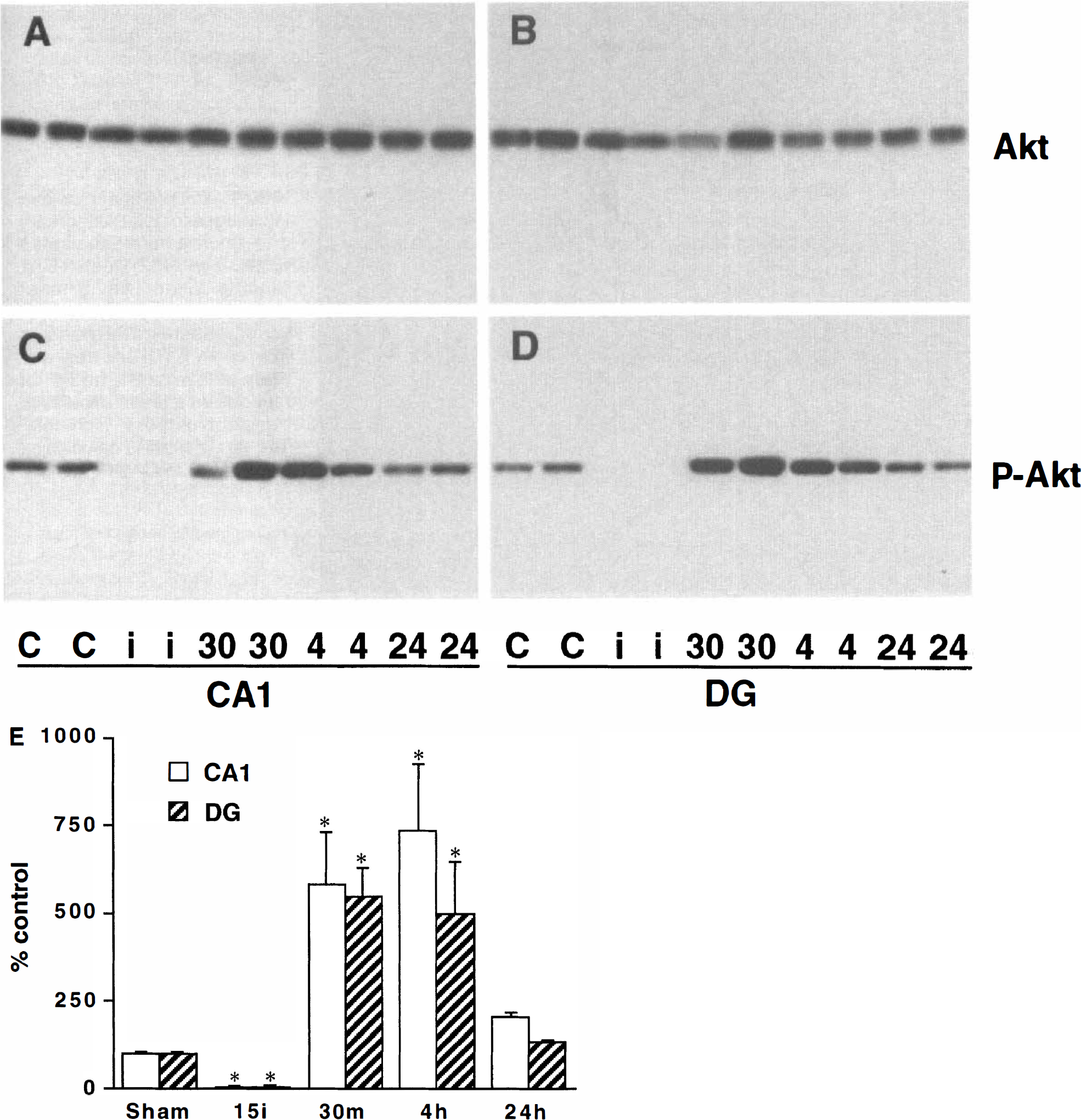
Western blots of total Akt
Total Akt and MAP-2 double-immunostaining on the brain sections showed that Akt was colocated with MAP-2 in CA1 neurons, as examined by laser scanning confocal microscopic study (Fig. 7).
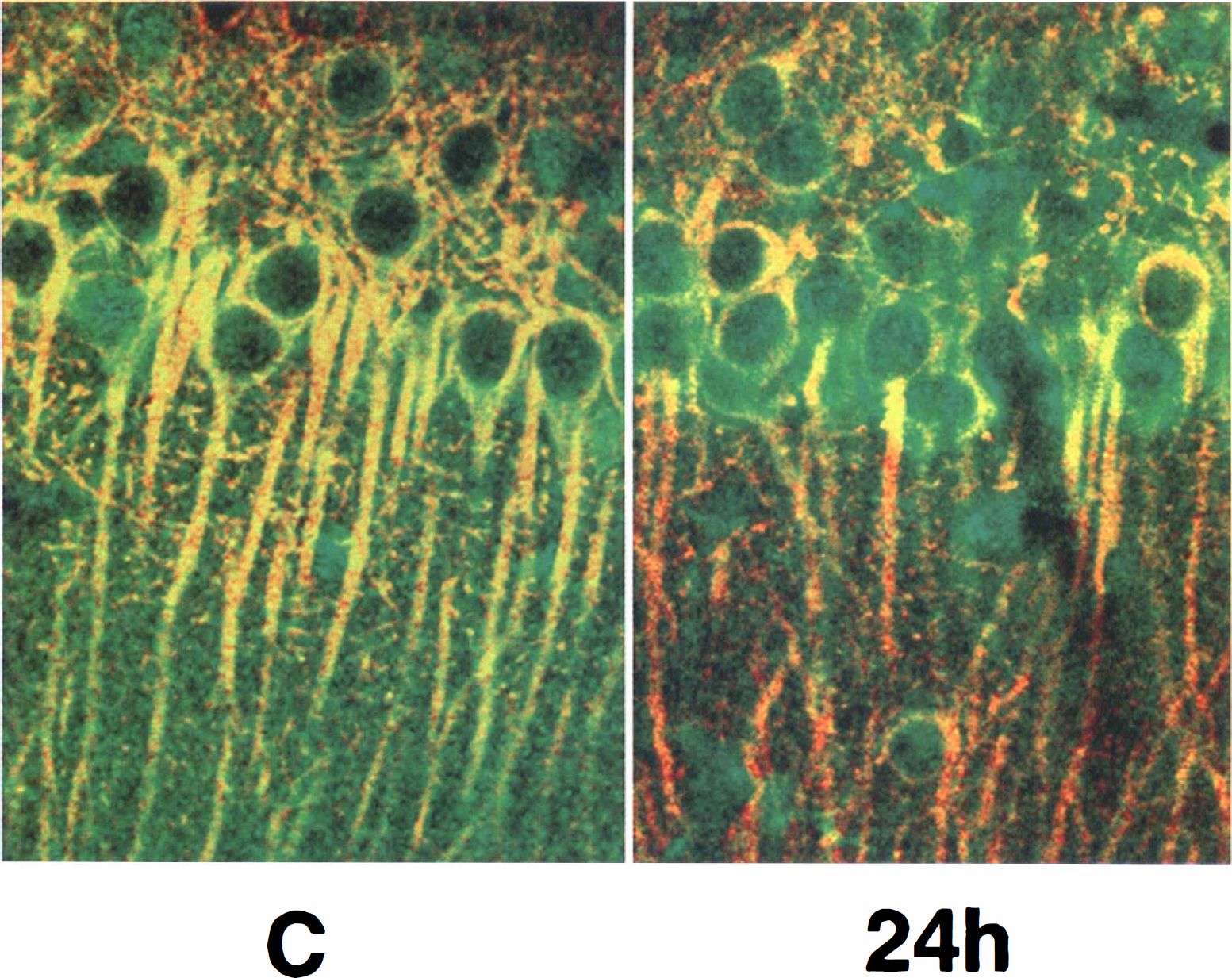
Double-immunostaining images for total Akt (green) and microtubule-associated protein 2 (MAP-2) (red) in CA1 regions taken with a laser scanning confocal microscope. Sections are shown from controls (C) and from animals allowed 24 hours of reperfusion after 15 minutes of transient cerebral ischemia. The Akt protein is colocalized with MAP-2 in CA1 neurons.
DISCUSSION
In the current study, we investigated factors involved in the cell death and cell survival pathway using post-ischemic hippocampal tissues. We selected release of cyt c that is known to be a proapoptotic factor and activation of Akt because this kinase is known to be a survival factor during apoptosis. We used an ischemic model that produces delayed neuronal death in CA1 pyramidal neurons (Smith et al., 1984). We found that after 15 minutes of transient cerebral ischemia, cyt c was increased in the supernatant fraction after 36 hours of reperfusion, whereas Akt was activated before 24 hours of reperfusion. Consistently, mitochondria were swollen, and some of them were visibly damaged in most CA1 neurons after 36 hours of reperfusion, as demonstrated by electron microscopic study. Caspase-3-like activity was increased after 36 hours of reperfusion. These data suggest that cyt c/caspase-mediated cell death is triggered as late as 36 hours of reperfusion, whereas Akt kinase, a cell survival factor, becomes active in the early phase before 24 hours of reperfusion after ischemia, as determined by its phosphorylation at serine-473.
The cascade of events encompassing damage of mitochondrion, release of cyt c, activation of caspase-3, and fragmentation of DNA has been considered an important signaling pathway for cell death (Ankarcrona et al., 1995; Bernardi, 1996; Wadia et al., 1998; Kroemer et al., 1998; Yong and Cortopassi, 1998; Green and Reed, 1998). Long-term opening of the MPT pore or long-term mitochondrial swelling may result in damage of mitochondria and release of cyt c (Kreomer et al., 1998; Green and Reed, 1998). This seems to be in line with the results of the current study that swollen, visibly damaged and normal mitochondria all are present in the same CA1 neurons after 36 hours of reperfusion, as observed by electron microscopic study. Our data support that cyt c was released from damaged mitochondria because (1) electron microscopy showed that mitochondria were damaged, (2) cyt c was increased only after 36 hours of reperfusion when the neurons were dying or died, and (3) the cyt c level in the mitochondrial fraction was not obviously decreased after ischemia. This means that most of the mitochondria still may be intact, whereas others are starting to release apoptotic factors such as cyt c in the neuron to initiate cell death. If this is a correct interpretation, cell death may result from partial mitochondrial damage.
It is currently debated whether cell death from transient cerebral ischemia is of apoptotic or necrotic type (MacManus and Linnik, 1997; Siesjo, et al., 1998). It is widely held that some ischemic insults, particularly those which are of brief or moderate density, activate mechanisms of apoptosis, whereas dense, long-lasting insults are prone to cause necrosis (cell swelling to release its contents into surrounding tissues). However, this is an unsettled issue. The fact that cell death caused by transient ischemia with reperfusion may reveal electrophoretic patterns of ladder DNA fragments in the absence of morphologic criteria of apoptosis suggests that ischemic cell death is neither apoptotic nor necrotic but an entity per se (MacManus and Linnik, 1997; Siesjo, et al., 1998). However, the distinction between apoptosis and necrosis is not critical because both types of cell death can be preceded by mitochondrial dysfunction with assembly of a MPT pore, release of cyt c or other apoptogenic factors, and activation of caspase-3–like proteases (Hirsch et al., 1997; Green and Reed, 1998; Kreomer et al., 1998). The current results should be discussed in this context. Thus, it is not contradictory that our data failed to show coalescence and margination of chromatin in the electron microscopy or punctuate DNA fragmentation (indicative of apoptosis), whereas at the same time, the results showed mitochondrial damage, a sign of necrotic cell damage. It is also not contradictory that signs of cyt c release and caspase activation were observed. It is thus possible that cell death after transient ischemia is a combination of necrosis and apoptosis and is initiated by a slow type of mitochondrial failure. On the other hand, the activated caspases may cause further damage of mitochondria and subsequent cell death (Ankarcrona et al., 1995; Namura et al., 1998; Chen et al., 1998; Wadia et al., 1998; Kreomer et al., 1998; Green and Reed, 1998; Siesjö, et al., 1998). Conceivably, the mitochondrial damage observed after 36 hours of reperfusion reflects such a slow type of mitochondrial failure, which then leads to secondary and terminal bioenergetic compromise (Pulsinelli and Duffy, 1983).
When phosphorylated at serine-473, Akt protein kinase is a cell survival factor in several cell systems. As mentioned earlier, phosphorylation of Akt can release the inhibition of Bcl-2. Thus, Bcl-2 can protect mitochondria (Datta et al., 1997; Crowder and Freeman, 1998). Our data show that Akt was highly phosphorylated at serine-473 in the hippocampal regions during the first 24-hour period of reperfusion. If such activation of Akt is a survival factor for hippocampal neurons after ischemia as in other cell systems, it may protect mitochondria through the Bcl-2 family of proteins and prevent the release of cyt c in the early period of reperfusion after transient cerebral ischemia.
Besides Akt activation, changes of several other factors may contribute to the delay of selective neuronal death after ischemia. For instance, brain-derived neutrophic factor, trkB, and phosphorylation of cAMP responsive element binding protein all are highly increased predominantly in DG neurons, but they are barely expressed in CA1 neurons during the postischemic phase (Lindvall et al., 1992; Hu et al., 1994; Hu et al., 1998). In contrast, phosphorylation of ATF-2, a stress factor, is highly increased in CA1 neurons (Hu et al., 1998). Release of cyt c, as well as expression of proapoptotic members of the Bc1-2 family (Chen et al., 1998; Hara et al., 1996; Gillardon et al., 1996) may be what causes the balance between antiapoptotic and proapoptotic factors to be disturbed in favor of ischemic cell death.
In summary, the current study demonstrates that transient cerebral ischemia not only initiates a cell death process, but also activates a cell survival factor. The release of cyt c and activation of caspase-like proteases in the later phase may overcome the early activation of survival factors such as Akt kinase after ischemia and result in delayed cell death.