
Abstract
Although degeneration of lower motor neurons is the most striking abnormality in amyotrophic lateral sclerosis (ALS), more subtle alterations may occur in the brain. Mutations in copper/zinc superoxide dismutase (Cu/Zn-SOD) are responsible for some cases of inherited ALS, and expression of mutant Cu/Zn-SOD in transgenic mice results in progressive motor neuron loss and a clinical phenotype similar to that of ALS patients. It is now reported that Cu/Zn-SOD mutant mice exhibit increased vulnerability to focal ischemic brain injury after transient occlusion of the middle cerebral artery. Levels of glucose and glutamate transport in cerebral cortex synaptic terminals were markedly decreased, and levels of membrane lipid peroxidation were increased in Cu/Zn-SOD mutant mice compared to nontransgenic mice. These findings demonstrate that mutant Cu/Zn-SOD may endanger brain neurons by a mechanism involving impairment of glucose and glutamate transporters. Moreover, our data demonstrate a direct adverse effect of the mutant enzyme on synaptic function.
Amyotrophic lateral sclerosis (ALS) is a progressive and always fatal neurodegenerative disorder that involves death of motor neurons in the spinal cord. Analyses of cerebrospinal fluid and postmortem spinal cord tissue from ALS patients have demonstrated increased levels of oxidative stress (lipid peroxidation and protein oxidation), (Beal et al., 1997; Ferrante et al., 1997a; Pedersen et al., 1998), increased levels of extracellular glutamate, and reduced levels of glutamate transporters (see Rothstein, 1995 for review), in ALS patients. Although the cause of most cases of ALS is unknown, some cases result from heritable mutations in Cu/Zn-SOD (Gurney et al., 1996). The Cu/Zn-SOD mutations do not compromise the normal dismutase activity of the enzyme but instead result in gain of an adverse property of the mutant protein which may involve increased peroxidase activity resulting in hydroxyl radical production (Wiedau-Pazos et al., 1996; Yim et al., 1997). Expression of ALS-linked Cu/Zn-SOD mutations in transgenic mice results in progressive degeneration of spinal cord motor neurons and associated paralysis (Gurney et al., 1994; Wong et al., 1995). Studies of spinal cord motor neurons from the ALS mice in cell culture and in vivo have provided evidence that the Cu/Zn-SOD mutations result in increased levels of oxidative stress and increased vulnerability to excitotoxicity (Ferrante et al., 1997b; Siklos et al., 1998; Roy et al., 1998; Kruman et al., 1999). The pathogenic action of Cu/Zn-SOD mutations in familial ALS does not result from a loss of dismutase function of the enzyme, but rather from gain of an adverse property of the mutant protein. Thus, there is no correlation between enzyme activity and disease severity in ALS patients and Cu/Zn-SOD mutant mice, and enzyme activity appears to be increased consistent with the level of enzyme overexpression (Gurney et al., 1994; Ripps et al., 1995; Wong et al., 1995). Moreover, Cu/Zn-SOD knockout mice are viable, live well beyond 1 year of age, and fail to develop motor neuron disease (Reaume et al., 1996). The specific alteration in the mutant enzyme has not been established but might result from enhanced hydroxyl radical production compared with the wild-type protein (Wiedau-Pazos et al., 1996; Yim et al., 1997).
Oxidative stress, impaired energy metabolism, and overactivation of glutamate receptors are increasingly implicated in the pathogenesis of neurodegenerative disorders ranging from Alzheimer's and Parkinson's diseases (Mattson, 1997; Jenner and Olanow, 1998) to stroke (Chan, 1996). Because glutamate receptors are concentrated in postsynaptic terminals, the excitotoxic process (be it apoptosis or necrosis) is initiated in synaptic compartments and propagates to the cell body (Mattson et al., 1998; Duan et al., 1999). Oxidative stress and lipid peroxidation can impair the function of glutamate and glucose transporters (Keller et al., 1997a; Mark et al., 1997a; Blanc et al., 1998) and ion-motive ATPases (Mark et al., 1995, 1997b) in neuronal cultures and synaptosome preparations. Impairment of these transporters results in increased levels of extracellular glutamate, ATP depletion and membrane depolarization, and excessive influx of calcium through N-methyl-
Metabolic and oxidative alterations in neurodegenerative disorders are often not limited to the neuronal populations that ultimately succumb (see Govoni et al., 1996; Connolly, 1998; Schapira et al., 1998 for review). A few studies suggest that abnormalities in ALS are not limited to the spinal cord. For example, positron emission tomography studies of cerebral glucose utilization have documented reduced levels of glucose uptake throughout many regions of cerebral cortex of ALS patients (Ludolph et al., 1992). Levels of glutamate transport proteins are decreased in cerebral cortex of ALS patients (Rothstein et al., 1995), and ALS patients exhibit abnormalities in carbohydrate metabolism (Murai et al., 1983). It is not known whether such abnormalities precede motor neuron degeneration or are secondary phenomena. In the present study we directly tested the hypothesis that abnormalities are present in the brains of Cu/Zn-SOD transgenic mice before onset of motor dysfunction and that such alterations place the neurons “at risk.” We report that synaptic glucose and glutamate transport are impaired in cerebral cortex of the ALS mice and that these abnormalities are associated with increased vulnerability to ischemic injury.
MATERIALS AND METHODS
Mice and transient focal cerebral ischemia protocol
Transgenic mice overexpressing human Cu/Zn-SOD with the ALS-linked G93A mutation (TgN[SODl-G93A]1Gur) were purchased from Jackson Laboratories (Bar Harbor, ME, U.S.A.). Mice were maintained on a B6/SJL background, and transgenic and nontransgenic offspring of crosses of heterozygous mice were used for experiments. Mice were maintained on a 12-hour light: 12-hour dark cycle and were provided free access to food and water. Cu/Zn-SOD mutant mice used in the present study developed progressive lower motor neuron degeneration and associated paralysis beginning from 5 to 7 months of age. All experiments were performed on 3-month-old male mice. The protocol for performing focal cerebral ischemia using a middle cerebral artery occlusion-reperfusion method was similar to that used in our previous studies (Bruce et al., 1996; Keller et al., 1998) and was adapted from Huang et al. (1994). Briefly, mice were anesthetized with 350 mg/kg chloral hydrate. Thermistor probes were inserted into the rectum and temporalis muscles to monitor body and brain temperature, which were maintained at 36 to 37°C by external warming. Blood samples were taken at baseline (10 minutes before occlusion), during ischemia (30 to 35 minutes after thread placement), and 10 minutes after reperfusion for measurements of blood gases and pH level. The left common carotid artery was exposed and the occipital, superior thyroidal, maxillary, and lingual branches of the external carotid artery were coagulated; the pterygopalatine artery was ligated. An incision was made in the external carotid artery and a polylysine-coated suture was advanced into the middle cerebral artery. The thread was left in place for 60 minutes and then removed to allow reperfusion. After surgery mice were returned to their cages and given free access to food and water. Neurologic deficits were assessed 22 hours after ischemia based on a scale from 0 (no deficits) to 3 (severe deficits) as described previously (Huang et al., 1994; Endres et al., 1999). At 24 hours after ischemia the mice were euthanized by anesthesia overdose, and coronal brain sections (1 mm thick) were stained with 1% triphenyltetrazolium chloride. Images of the stained brain slices were captured using a digital camera, and the infarct volume was quantified as described previously (Keller et al., 1998).
Synaptosome preparation and experimental treatment
Eight wild-type and 12 PS 1 mutant mice were used for these studies. For each synaptosome preparation, cerebral hemispheres from 2 mice were pooled and homogenized in a solution containing 0.32 mol/L sucrose, 4 μLg/mL pepstatin, 5 μLg/mL aprotinin, 20 μLg/mL trypsin inhibitor, 4 μLg/mL leupeptin, 0.2 mmol/L phenyl-methylsulfonyl fluoride, 2 mmol/L EDTA, 2 mmol/L EGTA, and 20 mmol/L Hepes. Synaptosomes were prepared using a sucrose density gradient protocol described in our previous studies (Keller et al., 1997; Begley et al., 1999). Protein concentrations in synaptosomal preparations were determined (Pierce BCA kit), and the synaptosomes were diluted in Locke's buffer (NaCl, 154 mmol/L; KCl, 5.6 mmol/L; CaCl2, 2.3 mmol/L; MgCl2, 1.0 mmol/L; NaHCO3, 3.6 mmol/L; glucose, 5 mmol/L; Hepes, 5 mmol/L; pH 7.2) to a final concentration of 200 μLg protein/mL. Synaptosomes were then aliquoted into 1.5-mL Eppendorf tubes for glucose and glutamate transport assays or into wells of 96-well plates for measurement of lipid peroxidation. Synaptosomes were either left untreated or were exposed to 10 μmol/L FeSO4.
Measurement of glucose and glutamate transport and lipid peroxidation
The methods for quantifying glutamate and glucose transport in synaptosomes have been described previously (Keller et al., 1997). For the glutamate uptake assay, synaptosomes (200 μLg/tube) were incubated for 7 minutes with [3H]-glutamate (0.1 μCi/mL). The synaptosomes were then pelleted, washed three times in Locke's buffer, and lysed in 200 μL of solution of 1% sodium dodecyl sulfate in phosphate-buffered saline. The lysate was placed in scintillation vials containing Scintiverse, and radioactivity was counted in a Packard 2500TR liquid scintillation counter. For the glucose uptake assay, synaptosomes (200 μg/tube) were subjected to experimental treatments and then washed three times in glucose-free Locke's buffer, and the assay was started by the addition of 1.5 μCi of [3H]-2-deoxyglucose. Seven minutes later the assay was stopped by pelleting the synaptosomes, washing twice with glucose-free Locke's solution, and lysing the synaptosomes in 200 μL of a 1% sodium dodecyl sulfate-phosphate-buffered saline solution. The lysate was placed in scintillation vials containing Scintiverse, and radioactivity was counted in a Packard 2500TR liquid scintillation counter. Results were expressed as counts per minute per milligram of protein. The rates of glucose and glutamate uptake by synaptosomes remained constant under basal conditions throughout the time course of experiments (up to 4 hours). In such assays of glucose and glutamate transport there is often considerable interassay variability in terms of counts per minute of synaptosome-associated radioactivity, whereas intraassay variability is quite low. Because we always included control conditions within an experiment, the experimental values were compared directly to the control value within the experiment. Levels of membrane lipid peroxidation were assessed using a thiobarbituric acid-reactive substances fluorescence-based method described previously (Keller et al., 1998). Values were expressed as a percentage of the value in untreated synaptosomes from wild-type mice.
RESULTS AND DISCUSSION
Previous studies have shown that in the same transgenic line of mice used in the present study, mutant Cu/Zn-SOD is expressed in cells throughout the nervous system, although levels of expression are somewhat lower in the brain as compared to the spinal cord (Liu et al., 1998). Our preliminary studies confirmed expression of the transgene in cerebral cortical tissue, wherein levels of overall Cu/Zn-SOD enzyme activity were increased approximately 2- to 3-fold (data not shown). In order to determine whether mutant Cu/Zn-SOD affects the vulnerability of cortical neurons to injury induced by metabolic/excitotoxic insults we subjected nontransgenic (wild-type) mice (n = 10) and Cu/Zn-SOD mutant mice (n = 10) to transient middle cerebral artery occlusion, a model of focal ischemic stroke. There were no significant differences in physiologic parameters (mean arterial pressure; blood
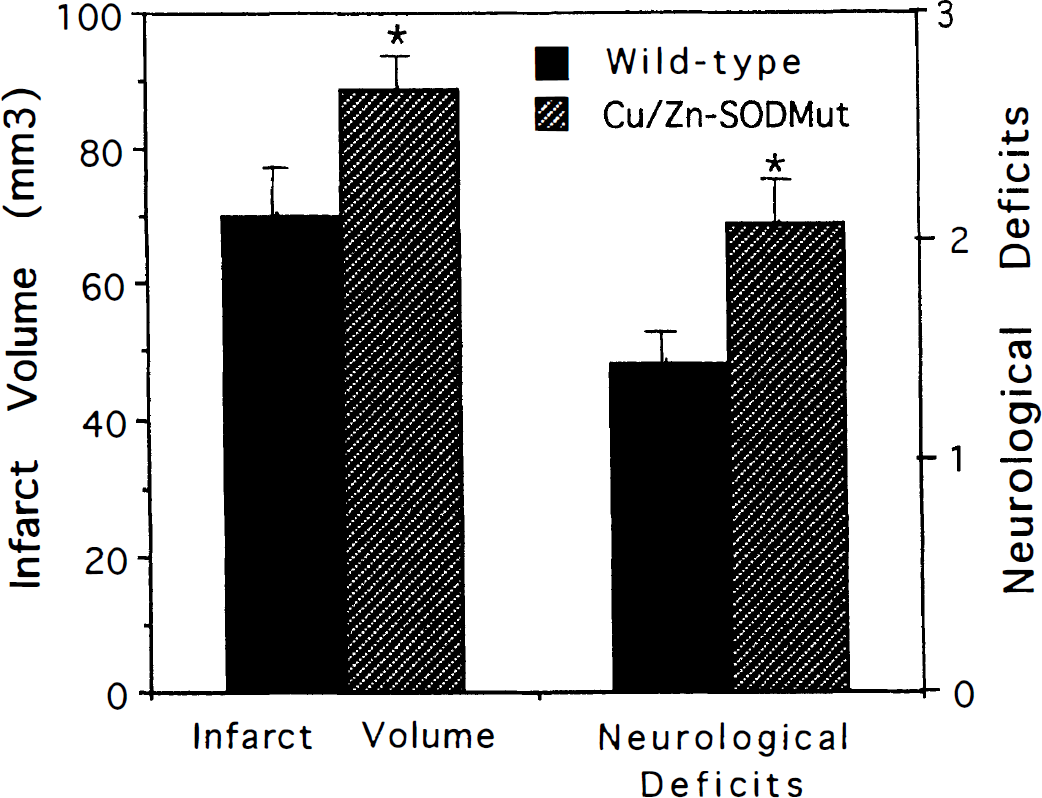
Increased infarct size and behavioral deficits after transient focal cerebral ischemia in copper/zinc superoxide dismutase (Cu/Zn-SOD) mutant mice. Values are the mean and SD (n = 10 mice). *P < 0.01 compared to value for wild-type mice (ANOVA with Scheffe's posthoc tests).
Ischemic neuronal injury involves ATP depletion, oxidative stress, and overactivation of glutamate receptors which results in calcium overload, mitochondrial dysfunction, and either apoptosis or necrosis (see Chan, 1996; Mattson et al., 1999 for review). To elucidate the mechanism(s) responsible for the endangering action of mutant Cu/Zn-SOD toward cortical neurons, we studied cortical synaptosomes from wild-type and Cu/Zn-SOD mutant mice. It was previously reported that levels of membrane lipid peroxidation are increased in spinal cords of ALS patients and Cu/Zn-SOD mutant mice (Hall et al., 1998; Pedersen et al., 1998). Measurement of relative levels of membrane lipid peroxidation, using a thiobarbituric acid-reactive substances assay, revealed a small but significant increase in basal levels of lipid peroxidation in synaptosomes from transgenic mice compared to wild-type mice (Fig. 2A). The magnitude of the increase in lipid peroxidation induced by Fe2+ was also significantly increased in synaptosomes from the transgenic mice, suggesting an increased sensitivity to oxidative stress.
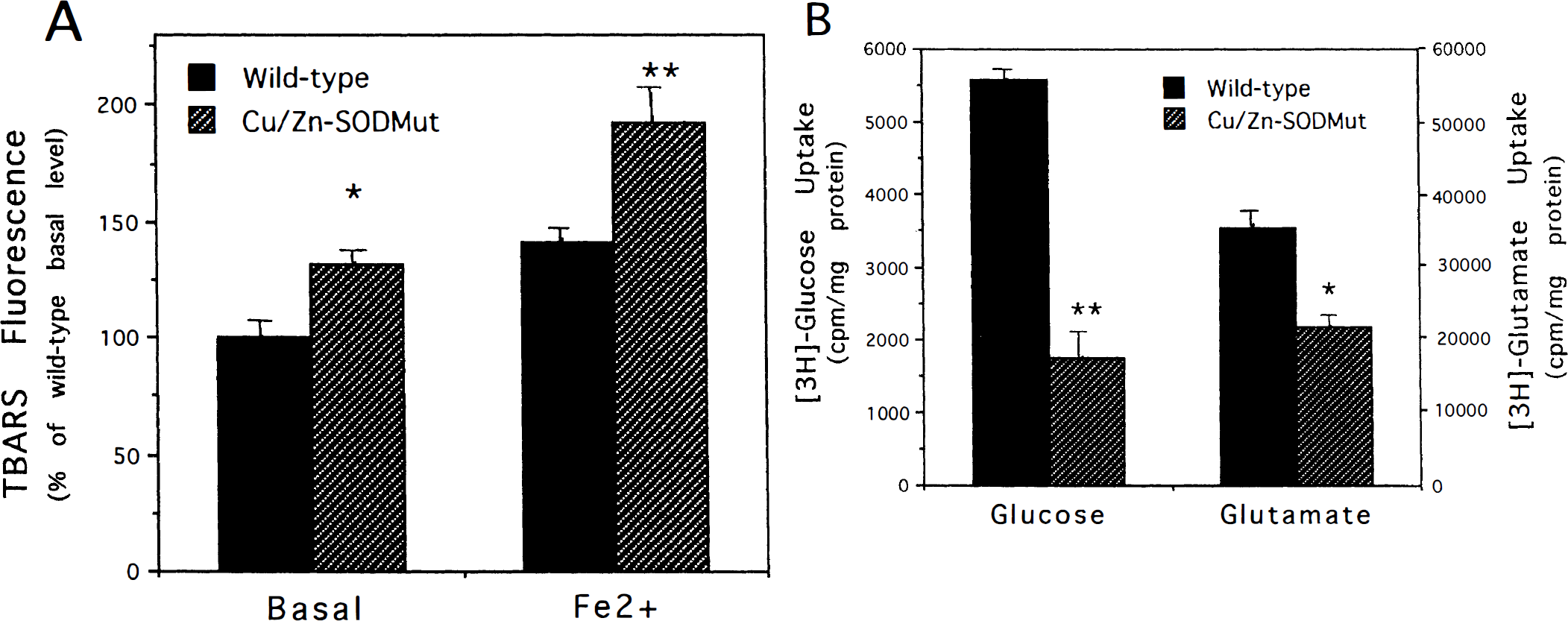
Levels of membrane lipid peroxidation are increased, and glucose and glutamate transport are decreased in cortical synaptosomes from copper/zinc superoxide dismutase (Cu/Zn-SOD) mutant mice. (
Physiologic parameters in nontransgenic (wild-type) mice and Cu/Zn-SOD mutant transgenic mice
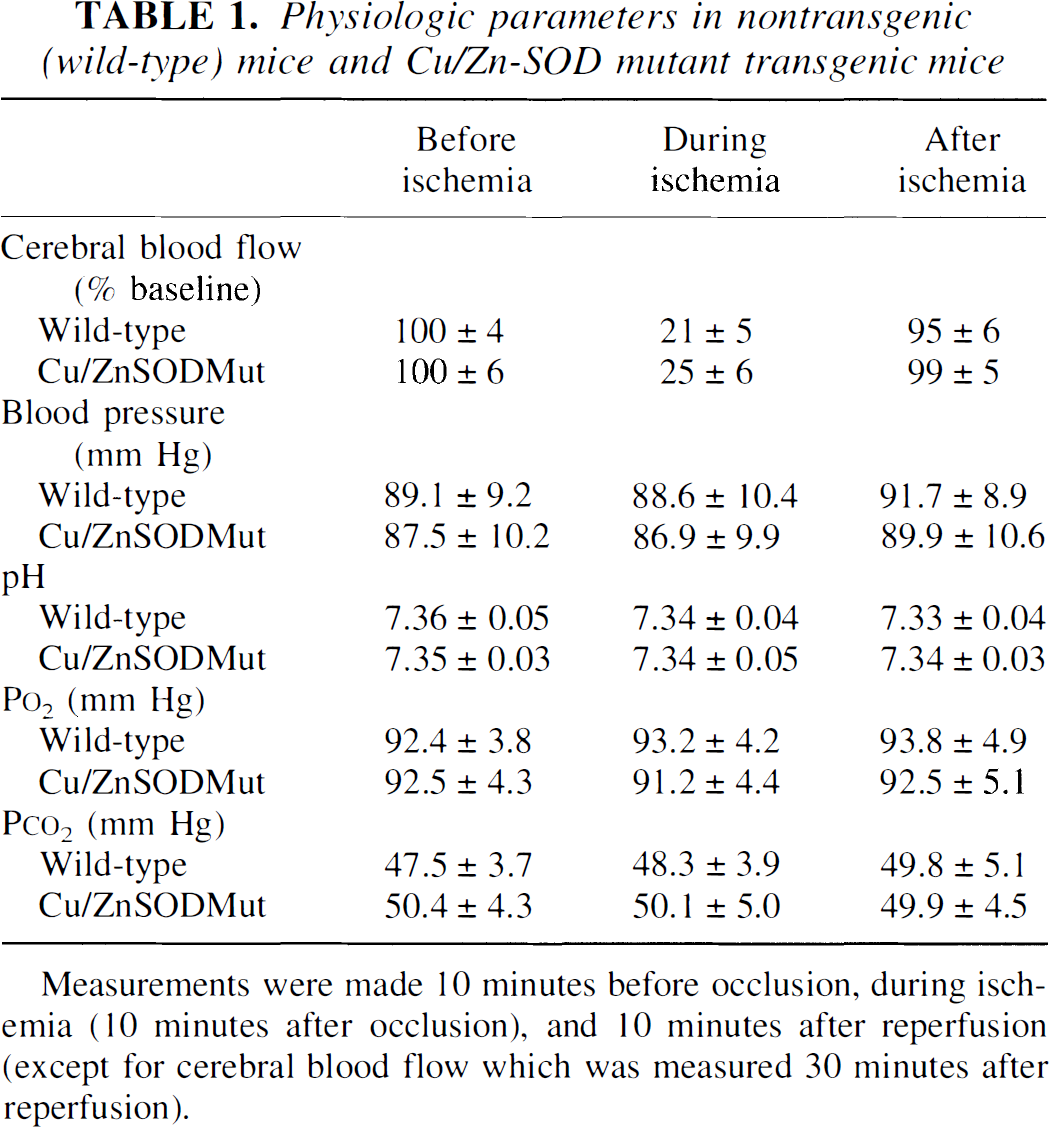
Measurements were made 10 minutes before occlusion, during ischemia (10 minutes after occlusion), and 10 minutes after reperfusion (except for cerebral blood flow which was measured 30 minutes after reperfusion).
We had previously shown that glucose and glutamate transport in cortical synaptosomes are impaired after exposure to insults that induce membrane lipid peroxidation (Keller et al., 1997a, b ). Because impairment of glucose and glutamate transport would be expected to render neurons vulnerable to excitotoxicity, we measured levels of glucose and glutamate transport in synaptosomes from nontransgenic and transgenic mice. There was a striking 70% reduction in the level of glucose transport in synaptosomes from Cu/Zn-SOD mutant mice compared to wild-type mice (Fig. 2B). The level of glutamate transport was also significantly reduced by approximately 40% in synaptosomes from Cu/Zn-SOD mutant mice (Fig. 2B). In light of these findings and because we observed no differences in cerebral vascular anatomy and because cerebral blood flow was similar in mutant and nontransgenic mice (before, during, and after ischemia), we conclude that the increased infarct size after focal ischemia-reperfusion in the the Cu/Zn-SOD mutant mice results from an adverse effect of the mutant enzyme in synaptic terminals of neurons. However, a contribution of indirect effects of the mutant enzyme on the vasculature or glial cells cannot be ruled out.
Previous studies have provided evidence that levels of oxidative stress are increased in cerebral cortex of ALS patients (Ferrante et al., 1997a), and that levels of lipid peroxidation and protein nitration are increased in cerebral cortical tissue from transgenic mice expressing an ALS-linked Cu/Zn-SOD mutation (Ferrante et al., 1997b). The latter studies involved analyses of brain tissue taken from end-stage ALS patients and Cu/Zn-SOD mutant mice. Our studies, performed on brain tissue taken several months before the onset of symptomatic motor dysfunction, suggest that cortical neurons are subjected to increased oxidative stress during the early stages of the disease process. Measurements of lipid peroxidation in spinal cord tissue of Cu/Zn-SOD mutant mice (Hall et al., 1998) and motor neurons in cultures established from embryonic Cu/Zn-SOD mutant mice (Kruman et al., 1999) also indicate that lipid peroxidation occurs before motor neuron degeneration. We found striking deficits in glucose and glutamate transport in cortical synaptosomes from the Cu/Zn-SOD mutant mice suggesting that the neurons are subjected to metabolic and excitotoxic stress. Although the mechanism whereby mutant Cu/Zn-SOD causes impaired glucose and glutamate transport is unknown, it might result from increased oxidative stress. Studies of hippocampal and cortical cell cultures and synaptosomes (Keller et al., 1997a, b ; Mark et al., 1997a, b ; Blanc et al., 1998) and motor neuron-like cells (Pedersen et al., 1999) have shown that membrane lipid peroxidation results in impairment of glucose and glutamate transporters. In addition, mutant Cu/Zn-SOD was reported to inactivate the glutamate transporter GLT-1 (Trotti et al., 1999). The beneficial effect of antioxidants, such as vitamin E that act by suppressing lipid peroxidation in ALS mice (Gurney et al., 1996), is consistent with a role for oxidative impairment of glucose and glutamate transport in the pathogenesis of ALS. Our findings may also provide an explanation for the widespread decrease in glucose utilization (Ludolph et al., 1992) and the decreased levels of glutamate transporter protein GLT-1 (Rothstein et al., 1995) in cerebral cortex of ALS patients.
The demonstration of impaired glucose and glutamate transport in cortical synaptosomes from Cu/Zn-SOD mutant mice provides the first direct evidence for synaptic dysfunction in ALS. Our findings support the excitotoxicity hypothesis of ALS in that impaired glucose and glutamate transport in synaptic terminals would predispose neurons to excitotoxicity.
Footnotes
Acknowledgments
The authors thank W. A. Pedersen and H. Zhu for technical assistance.