
Abstract
The overall hypothesis that cell death after intracerebral hemorrhage is mediated in part by apoptotic mechanisms was tested. Intracerebral hemorrhage was induced in rats using stereotactic infusions of 0.5 U of collagenase (1-μL volume) into the striatum. After 24 hours, large numbers of TUNEL-positive stained cells with morphologies suggestive of apoptosis were present in the center and periphery of the hemorrhage. Double staining with Nissl and immunocytochemical labeling with antibodies against neuronal nuclei and glial fibrillary acidic protein suggested that these TUNEL-positive cells were mostly neurons and astrocytes. Electrophoresis of hemorrhagic brain extracts showed evidence of DNA laddering into ∼200-bp fragments. Western blots showed cleavage of the cytosolic caspase substrate gelsolin. The density of TUNEL-positive cells at 24 and 48 hours after hemorrhage was significantly reduced by treatment with the broad-spectrum caspase inhibitor zVADfmk. It was unlikely that apoptotic changes were due to neurotoxicity of injected collagenase because TUNEL-positive cells and DNA laddering were also obtained in an alternative model of hemorrhage where autologous blood was infused into the striatum. Furthermore, equivalent doses of collagenase did not induce cell death in primary neuronal cultures. These results provide initial evidence that apoptotic mechanisms may mediate some of the injury in brain after intracerebral hemorrhage.
The prognosis of hemorrhagic lesions in brain is less favorable than that of purely ischemic lesions (Jorgensen et al., 1995). Almost 15% of all strokes are hemorrhagic in nature (Feldman, 1990). It has also been estimated that up to 30% of all ischemic strokes will eventually undergo hemorrhagic transformations (Lyden and Zivin, 1993). More recently, it has been suggested that thrombolytic therapy of ischemic stroke can also lead to serious complications of intracerebral hemorrhage in some patients (ECASS Study Group, 1995; NINDS rt-PA Stroke Study Group, 1995, 1997). Yet, in comparison with ischemia, relatively few experimental investigations have been devoted to studying the pathophysiology of intracerebral hemorrhage.
In the present study, we investigated the hypothesis that brain cell death after intracerebral hemorrhage may be mediated in part by apoptotic mechanisms. This hypothesis was developed based on two lines of reasoning: First, there has been recent evidence that although there is certainly a complex mixture of cell death pathways after cerebral ischemia, apoptotic pathways tend to be more prominent after milder ischemic insults (Bennett et al., 1998; Bonfoco et al., 1995; Du et al., 1996). Review of existing literature reveals that brain tissue adjacent to an intracerebral hemorrhage is affected by relatively mild ischemia (Bullock et al., 1988; Jenkins et al., 1990; Kobari et al., 1988; Ropper and Zervas, 1982; Yang et al., 1994) compared with the severe levels of ischemia present after occlusive strokes (Jones et al., 1981; Katakoa et al., 1987; Lo and Steinberg, 1991; Takagi et al., 1995). Second, a whole host of activated blood components pour into the brain after hemorrhage. It is known that a wide variety of cytokines present in blood and those activated after vascular injury can lead directly into signaling pathways that mediate apoptosis (MacManus and Linnik, 1997; Nagata and Golstein, 1995). Therefore, it is conceivable that mild ischemia together with the extravasation of activated cytokines can lead to brain cell apoptosis after hemorrhage. Here, we report some initial evidence for this phenomenon. Some of these data have been presented in abstract form (Matsushita et al., 1998).
MATERIALS AND METHODS
Collagenase-induced intracerebral hemorrhage in rats
All procedures were performed following an institutionally approved protocol in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Male Sprague-Dawley rats were anesthetized with halothane (1 to 1.2%) under spontaneous respiration and placed into stereotactic frames. Rectal core temperatures were maintained between 37 and 38°C with heat lamps. The skull surface was exposed, a craniotomy was performed, and a 30-gauge needle was lowered into the center of the striatum (stereotactic coordinates from bregma: 0.2 mm posterior, 3 mm lateral, 6 mm depth; Paxinos and Watson, 1982). Collagenase (0.5 U in 1-μL volume; Type IV from Sigma, St. Louis, MO, U.S.A.) was infused via the 30-gauge needle to induce intracerebral hemorrhage. After completion of collagenase infusion, craniotomies were sealed with bone wax, wounds were sutured closed, and rats were allowed to recover. The entire surgical procedure typically lasted 5 to 10 minutes. It is thought that collagenase digests the collagen within the basal lamina surrounding cerebral blood vessels, causing them to rupture and bleed (Rosenberg et al., 1990). This model was first described by Rosenberg and colleagues (Brown et al., 1995; Rosenberg et al., 1990, 1993) and has been extensively used to serve as an animal model for intracerebral hemorrhage (Del Bigio et al., 1996; Elger et al., 1994; Lyden et al., 1997). A total of 35 rats were infused with collagenase: n = 25 rats were used for quantitation of terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL)-positive cells, n = 6 rats were used for DNA analysis with gel electrophoresis, and n = 4 rats were used for western blot analysis of caspase substrate cleavage. Eight rats were infused with 1 μL of artificial CSF into the striatum to serve as sham controls: n = 4 for TUNEL staining and n = 4 for DNA analysis.
TUNEL assay
Rats used for TUNEL staining and/or immunohistochemistry were killed with an overdose of sodium pentobarbital at 1 or 2 days after collagenase injection. After removal, brains were immediately frozen in liquid N2 vapor (Vonsattel et al., 1995), sectioned in 10-μm thicknesses using a cryostat, and thaw-mounted onto precleaned glass slides. The TUNEL procedure for in situ detection of DNA fragmentation was performed (Gavrieli et al., 1992). In brief, air-dried sections were post-fixed in 4% paraformaldehyde for 15 minutes at room temperature. After washing three times in Tris-HCl (pH 7.7), sections were treated with 2% H2O2 for 10 minutes at room temperature to quench endogenous peroxidase activity. Sections were then incubated with terminal deoxynucleotidyl transferase enzyme solution, containing 25 mmol/L Tris-HCl (pH 6.6), 200 mmol/L potassium cacodylate, 0.25 mg/mL bovine serum albumin, 2.5 mmol/L CoCl2, 40 μmol/L biotinylated-16-dUTP, and 500 U/mL terminal deoxynucleotidyl transferase (Boehringer-Mannheim, Indianapolis, IN, U.S.A.) at 37°C for 1 hour. Sections were dipped in 300 mmol/L NaCl and 30 mmol/L sodium citrate for 15 minutes at room temperature to terminate reactions. After washing three times in Tris-HCl (pH 7.7) and subsequent blocking with phosphate-buffered saline (PBS; pH 7.4) containing 10% normal goat serum and 0.3% Triton X-100, biotinylated-16-dUTP was visualized by the avidin-biotin method with 0.05% 3,3′-diaminobenzidine tetrahydrochloride and 0.005% H2O2. Cell density counts were performed on sections lightly counterstained with Nissl stain. A single axial section through the center of the hemorrhagic lesion was analyzed. Square sampling regions (400 × 400 μm) were used for cell counting. Two sampling regions were randomly placed in the center of the lesion, and eight sampling regions were placed along the periphery. The TUNEL-positive cells were identified and counted. In general, two kinds of TUNEL staining were observed: The first involved diffuse light stains, and the second involved punctate staining with apoptotic-like bodies. According to criteria used by Ben-Ari, Chopp, and others (Charriault-Marlange et al., 1996; Chopp and Li, 1996; Li et al., 1998), we counted only the second type of TUNEL-positive staining. Total counts in these sampling regions were converted into cell densities for quantitation and comparison between treatment groups.
Immunocytochemical labeling for neuronal and astrocytic markers
To identify the cell types involved in the TUNEL-positive staining, mouse anti-neuronal nuclei (anti-NeuN) monoclonal antibody (Chemicon International, Temecula, CA, U.S.A.), and anti-glial fibrillary acidic protein (anti-GFAP) rabbit polyclonal antibody (Sigma) were used. Before immunohistochemistry, air-dried sections were postfixed by 4% paraformaldehyde for 15 minutes at room temperature. After blocking with PBS (pH 7.4) containing 10% normal goat serum, 0.3% Triton X-100, 0.05% NaN3, and 0.3% H2O2 for 1 hour at room temperature, the sections were successively incubated with the primary antibody against NeuN (1:400) or GFAP (1:160), diluted in PBS containing 2% normal goat serum, 0.3% Triton X-100, and 0.05% NaN3 at 4°C overnight. After washing three times in PBS, sections were incubated with Bodipy FL goat anti-mouse IgG-conjugated secondary antibody (1:200; Molecular Probes, Eugene, OR, U.S.A.) for NeuN or Bodipy FL goat anti-rabbit IgG-conjugated antibody (1:200; Molecular Probes) for GFAP for 1 hour at room temperature, respectively. After additional washing in PBS, TUNEL staining was performed as mentioned above, except that Cy3-conjugated streptavidin (Jackson ImmunoResearch Laboratories, West Grove, PA, U.S.A.) was used for the visualization of biotinylated dUTP at a dilution of 1:1,000 for 45 minutes at room temperature. Sections were examined by a Leica DMRB/Bio-Rad MRC 1024 confocal microscope with krypton/argon laser, using excitation/emission wavelengths of 488/522 nm for Bodipy FL and 568/605 nm for Cy3.
DNA electrophoresis
Brain samples (25 mg) were dissected from hemorrhagic and contralateral striatum to extract DNA using the QIA Amp kit (Qiagen, Valencia, CA, U.S.A.) according to manufacturer's instructions. A terminal transferase-dependent [32P]ddATP end-labeling method was used as previously described (Endres et al., 1998; Namura et al., 1998). Five micrograms of DNA was used in the labeling procedure together with 35 ng of 200-bp DNA fragments as internal markers. Electrophoresis was performed on the DNA on a 2% agarose gel and autoradiographed with a 32P standard. For this initial study, absolute levels of DNA fragmentation were not quantified.
Gelsolin cleavage
Western blots were performed using an antibody against the cytosolic caspase substrate gelsolin. Rats (n = 4) were killed 18 to 20 hours after collagenase injection, and brain tissues (50 mg) were removed from hemorrhagic and nonhemorrhagic striatum and homogenized on ice in 10 mmol/L N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid buffer (pH 7.6), containing 42 mmol/L KCl, 5 mmol/L MgCl2, 1 mmol/L ethylenediaminetetraacetate, 1 mmol/L ethyleneglycol-bis-(β-aminoethyl ether)-N,N,N′,N′-tetraacetate, 1 mmol/L dithiothreitol, 1% sodium dodecyl sulfate, 1 mmol/L phenylmethylsulfonyl fluoride, 2 μg/mL pepstatin, 1 μg/mL leupeptin, and 5 μg/mL aprotinin. The protein was denatured in sodium dodecyl sulfate loading buffer (62.5 mmol/L Tris-HCl, pH 6.8, 1% sodium dodecyl sulfate, 10% glycerol, 0.5% 2-mercaptoethanol) at 100°C for 3 minutes, and each sample (5 μg of protein) was loaded on a 4 to 20% Tris-glycine gel (Novex, San Diego, CA, U.S.A.) according to the manufacturer's instructions. After electrophoresis, the protein was transferred to polyvinylidene difluoride membrane (Immobilon-P; Millipore, Bedford, MA, U.S.A.), followed by blocking with 5% nonfat milk in Tris-buffered saline with Tween 20 (10 mmol/L Tris, pH 7.5, 150 mmol/L NaCl, 0.05% Tween 20). The blots were probed at 4°C overnight with the anti-gelsolin rabbit polyclonal antibody (Kothakota et al., 1997), diluted at 1:10,000 in 3% nonfat milk in Tris-buffered saline/Tween 20. The next day, membranes were washed with Tris-buffered saline/Tween three times and then exposed to horseradish peroxidase-conjugated anti-rabbit IgG for 1 hour at room temperature. Proteins of interest were detected using the enhanced chemiluminescence western blotting detection system kit (Amersham, Arlington Heights, IL, U.S.A.). The blots were exposed to Hyperfilm (ECL; Amersham) at room temperature in the darkroom.
Treatment with caspase inhibitor
The broad-spectrum caspase inhibitor zVADfmk was used. In the appropriate treatment groups, rats were stereotactically injected with zVADfmk into the lateral ventricles. Two treatment groups were used: In the first group, zVADfmk (240 ng/kg) was given 10 minutes before hemorrhage was induced with collagenase. Rats were then killed at 24 (n = 5) and 48 (n = 5) hours after hemorrhage, and brains were processed for quantitation of TUNEL cell densities. In a second group, zVADfmk (240 ng/kg) was given 10 minutes before hemorrhage and then a second dose (240 ng/kg) was given again at 24 hours after hemorrhage. These rats were killed at 48 hours (n = 5), and brains were also processed for TUNEL staining. Results from zVADfmk-treated rats were compared with those from untreated controls at either 24 (n = 5) or 48 (n = 5) hours after hemorrhage. Density of TUNEL-positive cells was quantified as described earlier. Comparison across groups was performed with analysis of variance followed by Tukey's Honestly Significant Difference tests.
Infusion of autologous blood as alternative model
Although the collagenase model is a well established approach to selectively induce intracerebral hemorrhage in animal models, it is not inconceivable that the results obtained in the present study may be due to artifactual effects of collagenase per se rather than reflect the true consequences of hemorrhagic pathophysiology. Therefore, additional experiments were performed using direct infusion of autologous blood into the rat striatum as an alternative model. This approach has been used by many other laboratories (Kobari et al., 1988; Ropper and Zervas, 1982; Wagner et al., 1996). Male Sprague-Dawley rats were anesthetized with halothane (1 to 1.2%) under spontaneous respiration and placed into a rat stereotactic frame as described above. A small craniotomy was performed using the same stereotactic coordinates, and 300 μL of autologous unheparinized blood (from the femoral artery) was infused into the right striatum. These rats were used for the following studies. The first group of rats were killed at 24 (n = 3) and 48 (n = 3) hours after hemorrhage, and extracted brains were processed for TUNEL staining. A second group of rats were killed at 48 hours (n = 2), and the brains were processed for DNA analysis via gel electrophoresis.
In vitro assay of collagenase neurotoxicity
Finally, to further establish that results obtained from the in vivo collagenase-induced hemorrhage models were not due to an artifact of the collagenase per se, we performed studies to examine the direct neurotoxicity of collagenase using in vitro neuronal culture models. Primary cultures of neurons were obtained from newborn (day 0 to 1) rats using standard techniques (Wang et al., 1999). The cortex was dissected in D-Hanks' solution and incubated with 0.125% trypsin for 30 to 40 minutes at 37°C. The cortex was then rinsed in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum at 4°C. Dissociated cortical cells were suspended into Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and plated on 12-well plates coated with poly-L-lysine at a density of 2 × 106 cells/plate. Growth medium was changed every 2 to 3 days. After 4 days in culture, cells were exposed to 5-fluoro-2-deoxyuridine (10 μg/mL) for 48 hours to suppress further glial proliferation. Experiments were finally performed on cultures ranging from 8 to 10 days in culture. It is difficult to translate in vivo doses of collagenase into in vitro concentrations. A reasonable approach in this case was to divide the total amount of collagenase used in vivo (0.5 U) by the resulting volume of the hemorrhagic lesion (typically 30 mm3; unpublished data). This would translate into a concentration of ∼10 U/mL. Therefore, we investigated a range of collagenase concentrations in vitro from 5 to 100 U/mL. All cultures were exposed for 24 hours. Neurotoxicity was quantified with the standard lactate dehydrogenase assay kit (Boehringer-Mannheim) and compared with control cultures not exposed to any collagenase at all. The caveat should be noted, however, that the lactate dehydrogenase release assay used here may not detect more subtle forms of cell injury that do not involve massive membrane damage and intracellular enzyme leakage.
RESULTS
TUNEL-positive cells are present after intracerebral hemorrhage
Rats infused with 0.5 U of collagenase developed large hemorrhages in the striatum that were evident by 24 hours (Fig. 1). The TUNEL staining revealed a high density of positively stained cells within the hemorrhagic lesion itself as well as in the surrounding periphery. Two major TUNEL morphologies were observed: cells that showed diffuse light staining and those that showed deep punctate staining with perinuclear chromatin condensation (Fig. 2A). According to the published criteria of Ben-Ari, Chopp, and co-workers (Charriault-Marlange et al., 1996; Chopp and Li, 1996; Li et al., 1998), only the latter category of TUNEL-positive cells were considered apoptotic and included for quantitation. Sham controls infused with 1 μL of artificial cerebrospinal fluid did not develop hemorrhage, although a few TUNEL-positive cells were sometimes seen adjacent to the needle tract into the striatum. Similar distributions of TUNEL-positive cells were observed in an alternative model of intracerebral hemorrhage whereby 300 μL of autologous blood was infused into the rat striatum (data not shown).
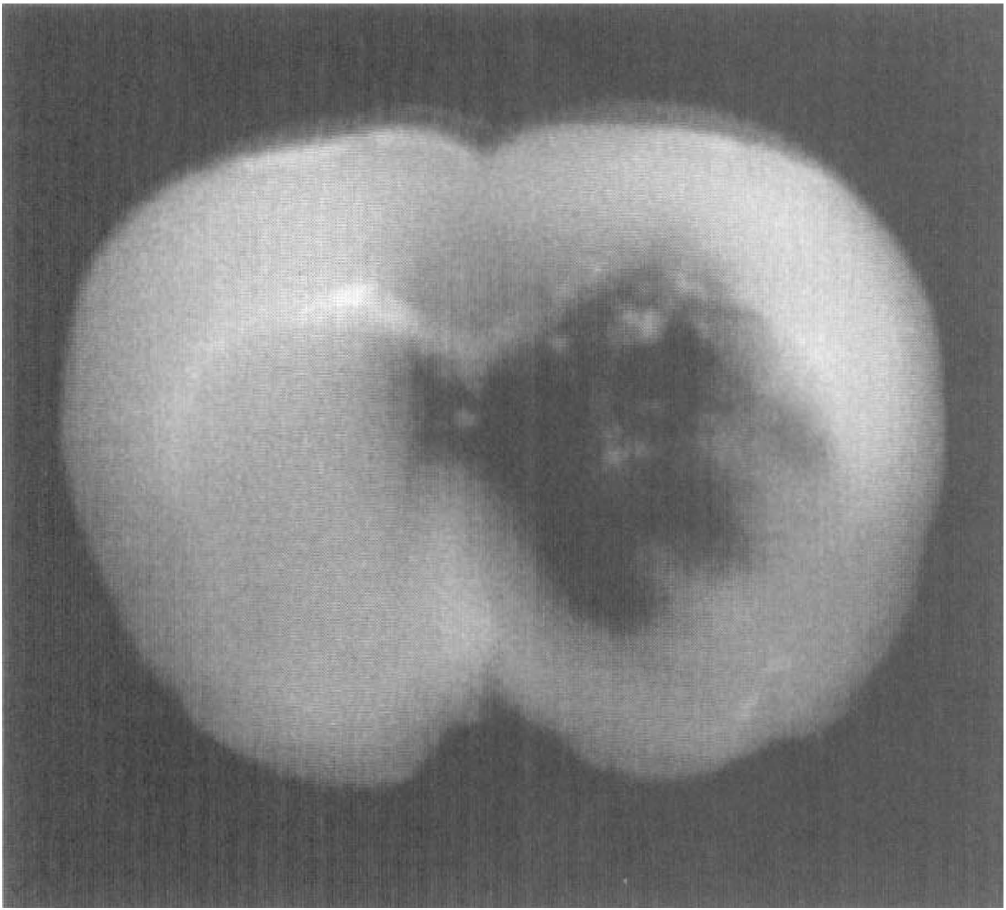
Representative section of rat brain shows the large hemorrhagic lesion induced in rat striatum at 24 hours after collagenase infusion.
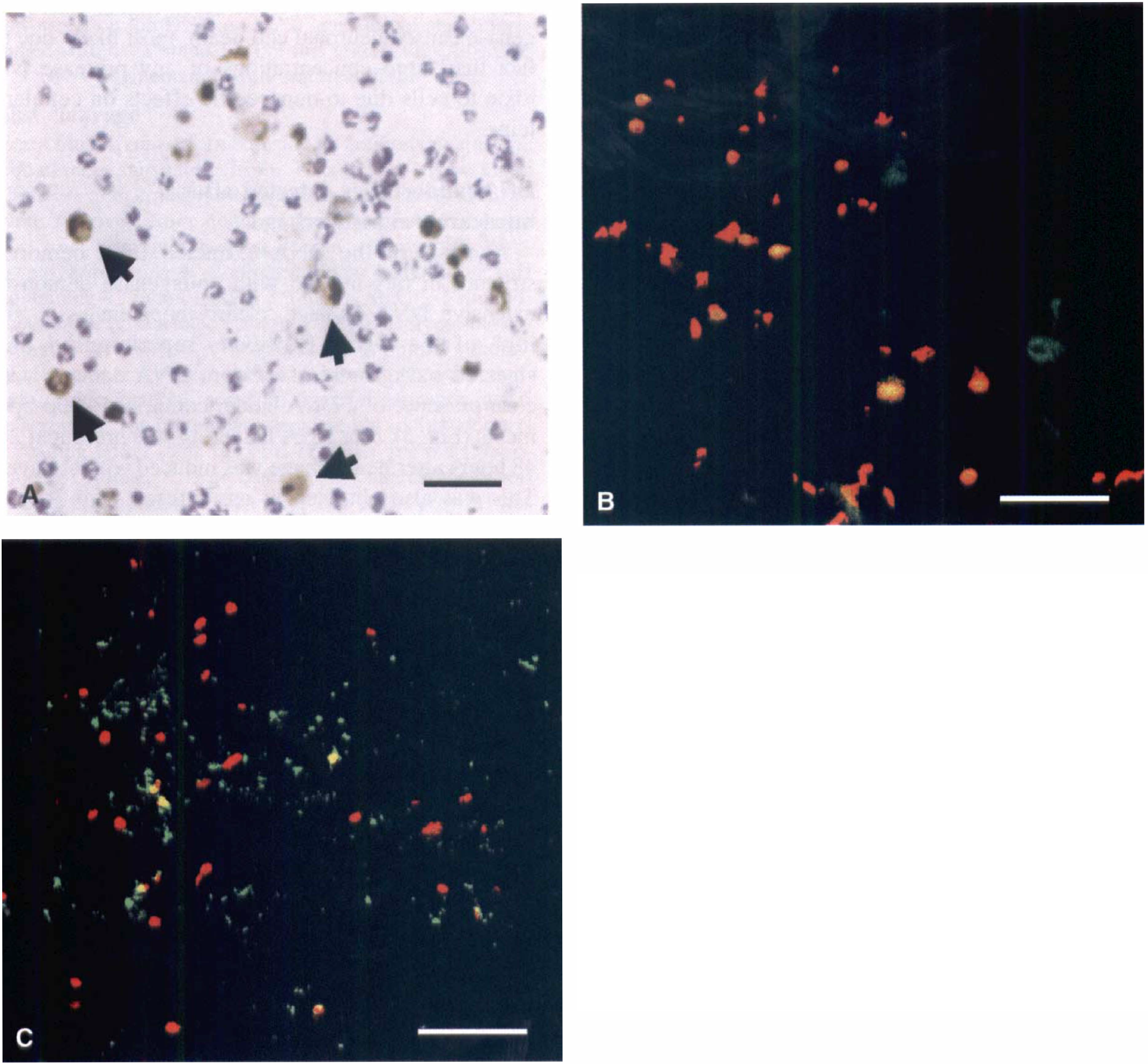
Brain sections that were counterstained with Nissl showed that the TUNEL-positive cells were not likely to be infiltrating polymorphonuclear leukocytes (Fig. 2A). Immunocytochemical studies were conducted with antibodies against neurons (NeuN) and astrocytes (GFAP) to determine the cell types involved. Within the hemorrhagic lesion, overall immunoreactivity against NeuN was somewhat diminished compared with the contralateral striatum, indicating that loss of neuronal nuclear and/or cytoskeletal structure was taking place as the cells died. Immunoreactivity against GFAP also showed signs of fragmented labeling, suggesting the presence of astrocytic degeneration. Nevertheless, fluorescent confocal microscopy showed that the majority of the TUNEL-positive cells were co-localized with these markers for neurons and astrocytes (Figs. 2B and 2C).
To further exclude the possibility that results obtained from the collagenase-induced hemorrhage models were due to an artifact of collagenase neurotoxicity per se instead of true hemorrhagic pathophysiology, we performed in vitro assays using primary neuronal cultures. These experiments showed that collagenase concentrations (10 to 15 U/mL) within the estimated range of those used in our in vivo studies did not cause cell death in vitro (Table 1). However, at much higher doses, collagenase caused neuronal cell death, most likely due to the fact that large concentrations of any protease will be toxic to cells due to nonspecific effects on cellular proteins.
Cytotoxic effects of collagenase in primary neuron cultures
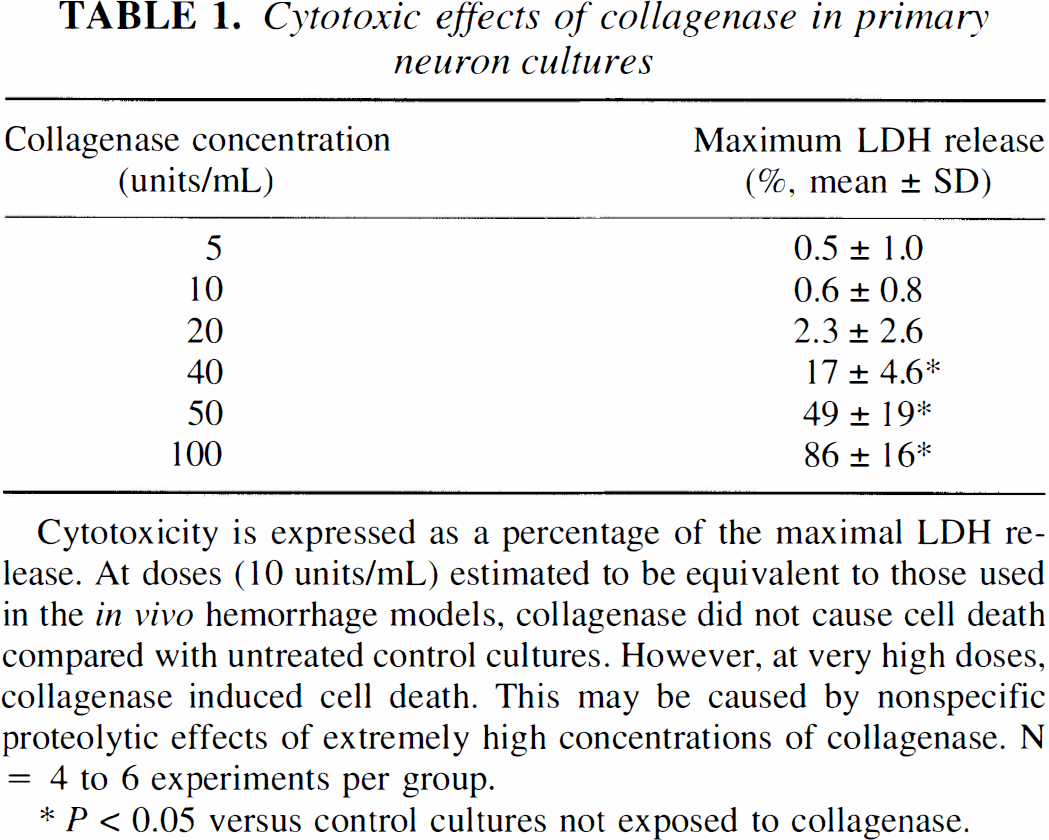
Cytotoxicity is expressed as a percentage of the maximal LDH release. At doses (10 units/mL) estimated to be equivalent to those used in the in vivo hemorrhage models, collagenase did not cause cell death compared with untreated control cultures. However, at very high doses, collagenase induced cell death. This may be caused by nonspecific proteolytic effects of extremely high concentrations of collagenase. N = 4 to 6 experiments per group.
P < 0.05 versus control cultures not exposed to collagenase.
DNA laddering is detected after intracerebral hemorrhage
Analysis of the DNA extracted from hemorrhagic striatum in rats infused with collagenase demonstrated extensive DNA damage, clearly apparent as a “smearing” of the DNA. However, superimposed on this smeared background of random DNA damage was the clear presence of a DNA ladder comprised ∼200-bp fragments (Fig. 3). The DNA laddering was present at 24 and 48 hours after hemorrhage was induced with collagenase. This was also obtained in rats infused with 300 μL of autologous blood as an alternative model of intracerebral hemorrhage (Fig. 3). No DNA damage was observed in contralateral striatum samples. However, very faint traces of DNA laddering were detected in sham-treated rats infused with 1 μL of artificial cerebrospinal fluid, most likely related to perturbations from needle insertion.
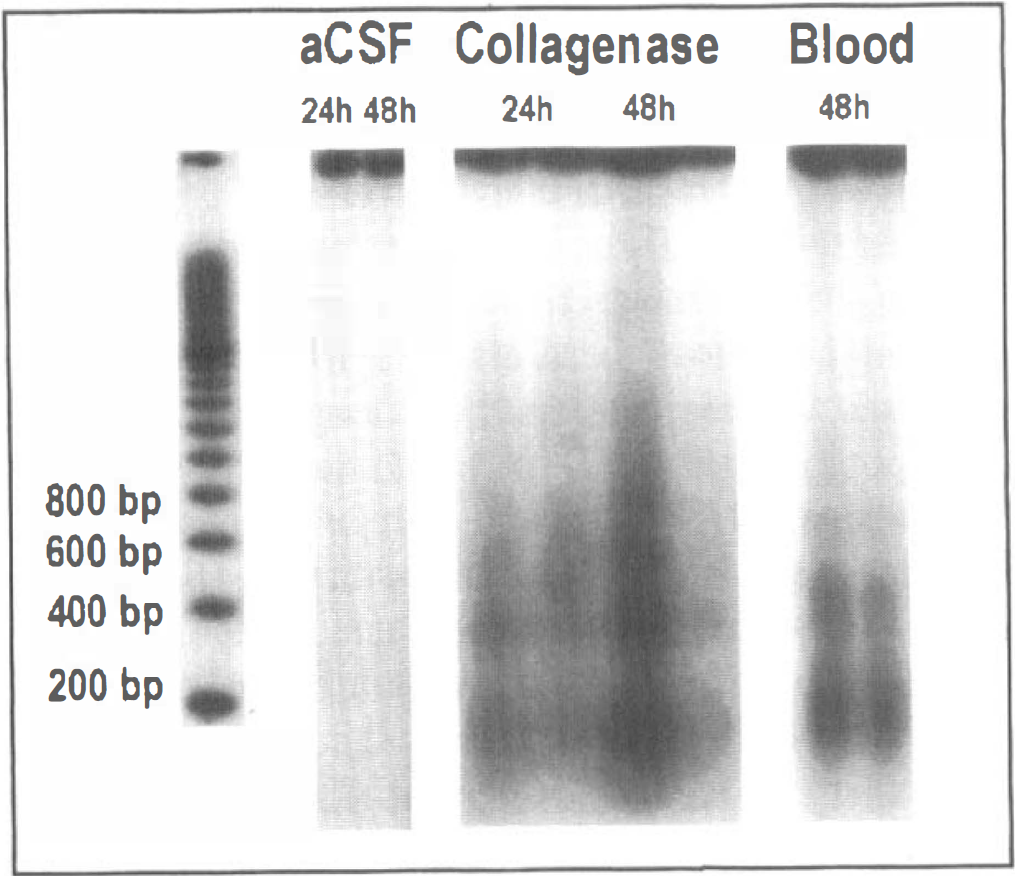
Gel electrophoresis reveals evidence of DNA laddering into ∼200-bp segments after intracerebral hemorrhage induced by collagenase (n = 3 at 24 hours, n = 3 at 48 hours) or autologous blood infusion into rat striatum (n = 2 at 48 hours). Note that DNA ladders were superimposed on a background of DNA smearing, demonstrating that random DNA damage was also present. Representative data from two rats per group are shown here. Sham rats infused with 1 μL of artificial cerebrospinal fluid showed faint traces of DNA laddering most likely related to perturbations from needle insertion into the striatum.
Caspase substrate gelsolin is cleaved in hemorrhagic brain
Brain homogenates derived from hemorrhagic and contralateral striatum were subjected to western blot analysis using antibodies against the cytosolic caspase substrate gelsolin. Uncleaved gelsolin (86 kDa) was present in all samples tested. At 18 to 20 hours after hemorrhage was induced by intrastriatal collagenase injection, cleavage of gelsolin (48 kDa) was observed (Fig. 4). Contralateral striatal homogenates did not show cleavage of this caspase substrate.
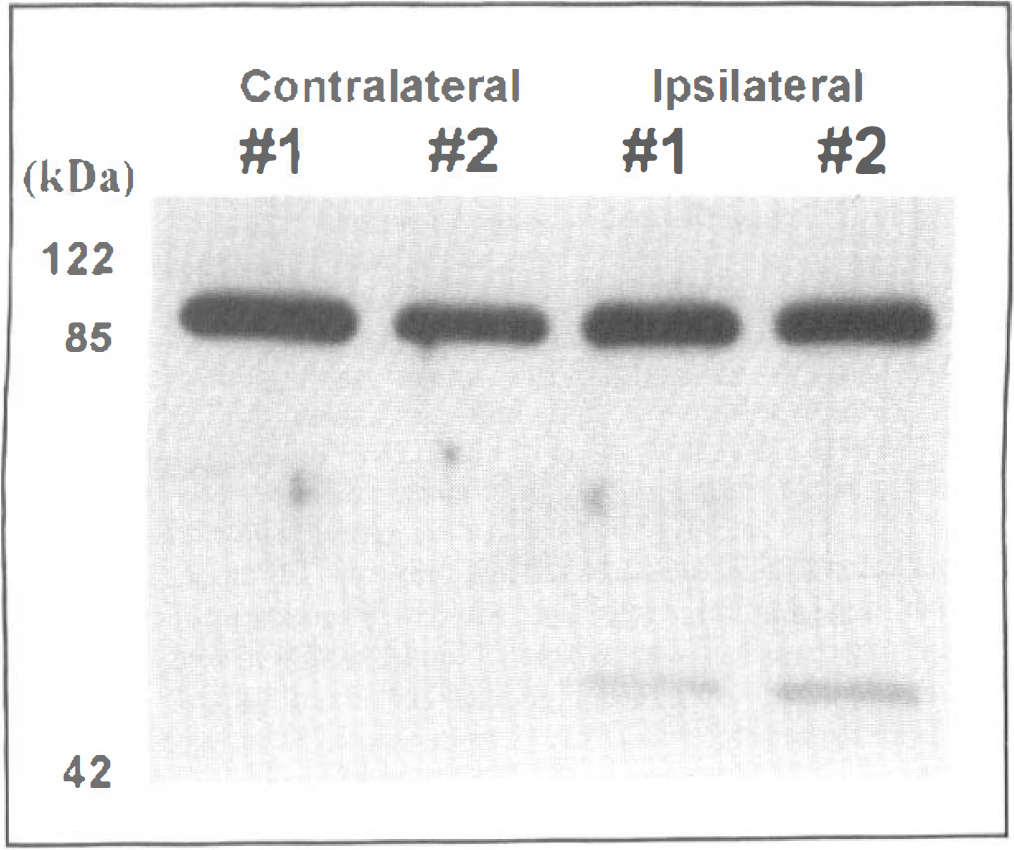
Western blots demonstrate cleavage of the caspase substrate gelsolin at 18 to 20 hours after intracerebral hemorrhage was induced with infusion of collagenase into rat striatum (n = 4). Uncleaved gelsolin bands (86 kDa) were present in both hemorrhagic and contralateral samples. Cleaved gelsolin bands (48 kDa) were present only in hemorrhagic samples. Representative data from two rats are shown here.
TUNEL-positive cell density after hemorrhage is reduced by caspase inhibitors
Untreated rats subjected to collagenase-induced striatal hemorrhage were compared with rats treated with the broad-spectrum caspase inhibitor zVADfmk. In rats treated with a single dose of zVADfmk prior to hemorrhage, the density of apoptotic-like TUNEL-positive cells was significantly reduced at 24 hours in the center as well as the periphery of the hemorrhagic lesion (Fig. 5). This effect was lost by 48 hours, by which time there were no significant differences in TUNEL density between treated and untreated rats (Fig. 5). In rats treated with two doses of zVADfmk (before hemorrhage and 24 hours after hemorrhage), a significant reduction of TUNEL-positive cell density in the periphery of the lesion persisted at 48 hours (Fig. 5). However, no significant reduction of TUNEL-positive cell density was achieved in the hemorrhagic center (Fig. 5).
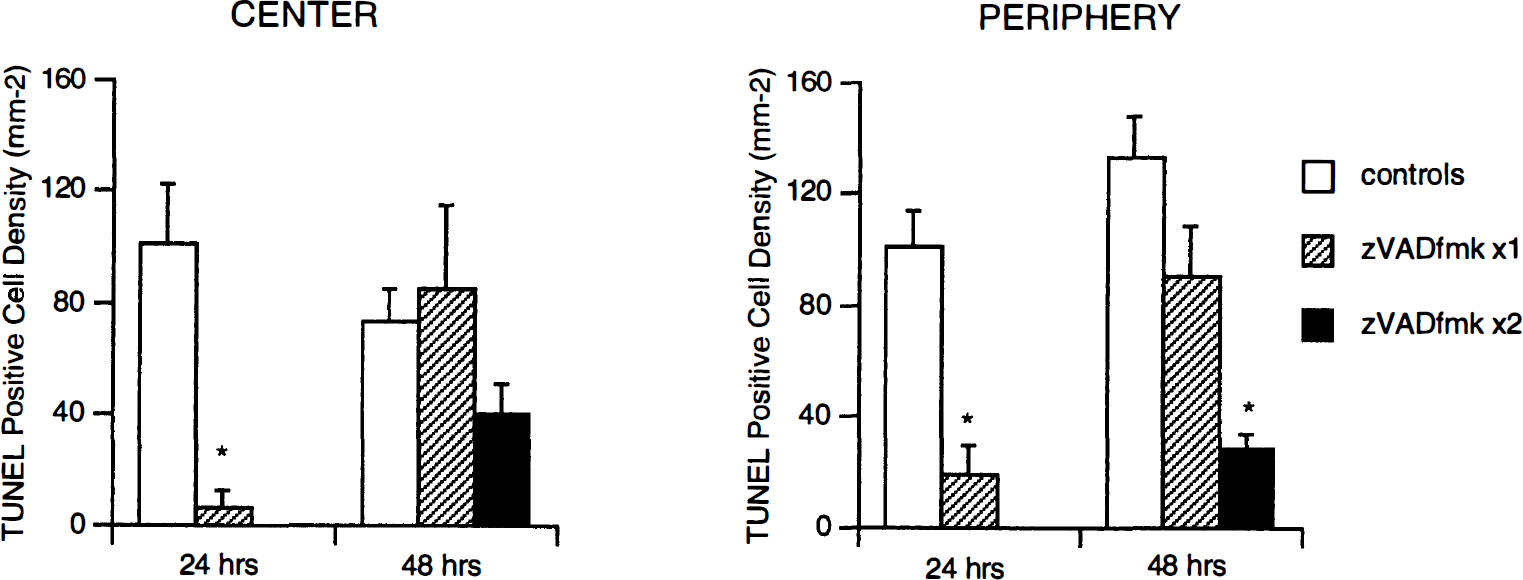
Density of TUNEL-positive stained cells in the center and periphery of hemorrhagic lesions induced by collagenase infusion into rat striatum (mean + SD). A single dose of zVADfmk (240 ng/kg) significantly reduced TUNEL-positive cell densities in both center and periphery. By 48 hours, this effect was lost. When two doses of zVADfmk (240 ng/kg per dose, 10 minutes before hemorrhage and 24 hours after hemorrhage) were administered, TUNEL-positive cell densities in the hemorrhagic periphery were reduced. However, no statistically significant reductions were achieved in the center of the hemorrhage. n = 5 per group, *P < 0.01 versus controls.
DISCUSSION
In this study using rat models of intracerebral hemorrhage, we have presented initial evidence suggesting that apoptosis may contribute to brain damage after intracerebral hemorrhage. After injection of collagenase into rat striatum to cause hemorrhage, (1) there were significant numbers of TUNEL-positive cells within and around the hemorrhagic lesion, (2) TUNEL staining co-localized with immunocytochemical labeling for neurons (NeuN) and astrocytes (GFAP), (3) DNA fragmentation occurred in ∼200-bp ladders as shown by gel electrophoresis, (4) the caspase substrate gelsolin was cleaved, and (5) the density of TUNEL-positive cells was significantly reduced by treatment with the caspase inhibitor zVADfmk. Evidence of DNA fragmentation (TUNEL-positive staining and DNA laddering) was obtained after unheparinized arterial blood was infused into rat striatum, suggesting that direct collagenase toxicity was not the major cause of apoptotic changes. Furthermore, equivalent concentrations of collagenase did not kill cultured primary neurons.
After cerebral injury, distinctions between apoptosis versus necrosis may be less prominent than a continuous spectrum of cellular damage that combines characteristics of both cell death processes (Choi, 1996; Chopp and Li, 1996; Martin et al., 1998; Portera-Cailliau et al., 1997). Positive TUNEL staining can occur in both apoptotic as well as necrotic cells and cannot be used as an unequivocal marker for an active cell death process. However, several investigators have demonstrated that morphological characteristics of the TUNEL-positive cells can be used to partially distinguish apoptosis from necrosis (Charriault-Marlange et al., 1996; Chopp and Li, 1996; Li et al., 1998). In the present study, TUNEL staining indeed showed a mixture of morphologies suggestive of necrosis (diffuse staining) and apoptosis (deep punctate staining with chromatin condensation). Only the latter type of cells were counted in our quantitative analysis. Overall, evidence for apoptosis in our hemorrhage models is based not only on TUNEL-positive cells but also on the presence of DNA laddering, cleavage of caspase substrates, and response to caspase inhibitors.
Necrotic cell death obviously plays a role in our models of intracerebral hemorrhage. In addition to laddered DNA, random DNA cleavage was also present on agarose gel electrophoresis. In fact, DNA fragmentation in various types of cerebral injury may not be accurately described as classic apoptosis according to the original criteria defined in nonneuronal systems (Wylie et al., 1980). For example, some investigators have observed that the nicked ends of the DNA fragments are ragged rather than blunt, as would be normally expected in apoptotic endonuclease-mediated degradation (MacManus et al., 1999; MacManus et al., 1995). It has been suggested that perhaps the term “caspase-mediated cell death” may be a more technically accurate description (Barinaga, 1998). We did not examine in detail the nature of the DNA nicked ends in the present study. Therefore, it is possible that “caspase-mediated cell death” might also be the more accurate description of our findings rather than classic apoptosis.
Here, we used caspase inhibition with zVADfmk as a method of obtaining pharmacologic evidence for the presence of apoptosis in our models. When treated with a single dose of zVADfmk prior to hemorrhage, the density of TUNEL-positive cells was significantly reduced at 24 hours compared with untreated controls. This effect was lost by 48 hours. A second dose of zVADfmk (given at 24 hours after hemorrhage) was necessary to reduce TUNEL-positive cell density at 48 hours. The need for multiple zVADfmk doses suggests that additional caspase may be processed and activated in brain during the 1 to 2 days after hemorrhage, or unlike cerebral ischemia models, zVADfmk may have been inactivated by some components present in extravasated blood. The density of TUNEL-positive cells at 48 hours did not significantly decrease in the center of the hemorrhage. Three possibilities may account for this result: First, damage may have been too severe in the center and the dose of zVADfmk may not have been high enough. Second, it is possible that the hemorrhagic mass lesion had grown to the extent that hydrostatic pressures made it difficult for zVADfmk to effectively penetrate the center of the lesion at 48 hours. Finally, there may have been insufficient statistical power in our experiments to detect small reductions in TUNEL-positive cell density. Nevertheless, it is possible that an ongoing increase in TUNEL-positive cells may contribute to lesion expansion as the hemorrhage matures so that caspase inhibition may ameliorate secondary processes of cell death. An active process of TUNEL-positive cell death and lesion expansion has been previously demonstrated in a mouse trauma model of cold injury (Murakami et al., 1997). At the present time, it is unclear which caspases are involved as zVADfmk is a wide-spectrum inhibitor or whether caspase inhibition produces any benefit in terms of actually reducing cerebral injury. Experiments are ongoing in our laboratory to determine whether caspase inhibition improves neuronal and/or glial survival, reduces the size of the hemorrhagic lesion, or alters clinical outcome in terms of neurologic function.
Two models of intracerebral hemorrhage were used in the present study. In the first model, vessel rupture and bleeding into brain parenchyma were caused by disruption of the basal lamina via collagenase injection. In the second model, unheparinized arterial blood was directly infused to simulate intracerebral hemorrhage. The advantage of the first model is that it yields reproducible spheroid-shaped lesions and duplicates the primary vascular trauma present in hemorrhage (Brown et al., 1995; Rosenberg et al., 1990, 1993). Some of the results may have been due to direct neurotoxic effects of collagenase per se. However, this seems unlikely because exposure to equivalent concentrations of collagenase in primary cultured neurons did not induce cell death in vitro, and similar markers of DNA damage were also found in the blood infusion models of hemorrhage. Interestingly, recent studies now show that up-regulation of endogenous matrix metalloproteinases such as collagenase may be involved in the development of edema and hemorrhagic transformations after cerebral ischemia (Heo et al., 1999; Mun-Bryce and Rosenberg, 1998; Romanic et al., 1998).
Results from this study provided evidence that apoptosis may be involved in the pathophysiology of cell death after intracerebral hemorrhage. However, the triggers that are responsible for initiating the apoptotic cascade after hemorrhage remain to be defined. Parenchymal ischemia that accompanies hemorrhage (Bullock et al., 1988; Jenkins et al., 1990; Kobari et al., 1988; Ropper and Zervas, 1982; Yang et al., 1994) is one possibility because mild cerebral ischemia has been shown to lead to apoptosis (Bennett et al., 1998; Du et al., 1996; Endres et al., 1998; Fink et al., 1998). Another possibility may involve the indirect effects of trauma due to the mass lesion of the hemorrhage itself. Evidence of apoptosis has also been found in other models of cerebral trauma (Kaya et al., 1999; Rink et al., 1995; Yakovlev et al., 1997), and similar mechanisms may be involved here.
As previously stated, a major reason that led us to hypothesize a role for apoptosis in intracerebral hemorrhage is the fact that activated blood components may induce high levels of extracellular cytokine activity. Signaling via various cytokines has long been associated with apoptosis (MacManus and Linnik, 1997; Nagata and Golstein, 1995). Although there will likely be a multitude of potential cytokines/factors that may be activated and/or released after intracerebral hemorrhage, several of these may be considered to be leading candidates for this role.
The first two candidates may be the classic apoptotic signaling molecules FasL and tumor necrosis factor. A major pathway for apoptotic signaling occurs via the Fas and tumor necrosis factor family of cell surface receptors (Nagata and Golstein, 1995). FasL can be released in soluble form from activated lymphocytes (Nagata and Golstein, 1995), and Fas mRNA is significantly up-regulated in neurons and glia after cerebral ischemia (Matsuyama et al., 1994, 1995). Up-regulation of tumor necrosis factor and tumor necrosis factor receptor is widespread after cerebral ischemia (Botchkina et al., 1997; Liu et al., 1994) and vascular trauma (Feuerstein et al., 1994). A recent study has shown that tumor necrosis factor levels are indeed elevated in brain after intracerebral hemorrhage in a pig model (Hua et al., 1998). Hence, it is possible that Fas and tumor necrosis factor may play critical roles in mediating cellular apoptosis after intracerebral hemorrhage.
A third candidate may be thrombin, which induces apoptosis in neurons and astrocytes in vitro(Donovan et al., 1997). Brain prothrombin levels have not been quantified. However, plasma levels are high (1 to 5 μmol/L), so that upon hemorrhage, large amounts of prothrombin can be released into brain locally. Exposure to only nanomolar concentrations of thrombin for 24 hours leads to apoptosis (Donovan et al., 1997). Therefore, the high levels of thrombin present after intracerebral hemorrhage should easily be within the proapoptotic range.
Iron/heme, which is obviously abundant in blood, is another possible trigger for apoptosis after intracerebral hemorrhage. Amplified processes of injury in cerebral hemorrhage may be due to iron/heme-catalyzed oxidative stress and lipid peroxidation (Koeppen et al., 1995). Oxidative stress and lipid peroxidation have been shown to participate in the signaling pathways for apoptosis (Bondy, 1993; Chan, 1996; Keller et al., 1998; Murphy et al., 1999). Addition of iron induces apoptosis in neuronal cell cultures (Keller et al., 1998; Kruman et al., 1997), and a recent study has shown that transgenic mice overexpressing heme oxygenase 1 are protected against focal cerebral ischemia (Panahian et al., 1999). Hence, locally elevated iron/heme concentrations after intracerebral hemorrhage may be especially conducive to apoptosis. Finally, high concentrations of glutamate in extravasated blood or efflux of glutamate due to parenchymal ischemia may initiate apoptotic processes after hemorrhage. It has been shown that signaling via various glutamate receptors including N-methyl-D-aspartate and kainate receptors can induce apoptosis in neurons (Qin et al., 1996; Simonian et al., 1996).
In summary, the data presented here provide some initial evidence that apoptotic cell death may be present after intracerebral hemorrhage. Additional studies will be required to further dissect the precise pathways that mediate posthemorrhagic cell death. The clinical relevance of these results will depend on whether the reduction in TUNEL-positive cell density after caspase inhibition is accompanied by actual improvements in brain cell survival and/or neurologic function in these animal models.
Footnotes
Abbreviations used
Acknowledgements
The authors thank Dr. David Kwiatkowski for generously supplying the gelsolin antibody.