
Abstract
The authors examined the effect of
Apoptosis, which is a cell suicide mechanism under active cell control, is a prominent characteristic of the developing nervous system (Oppenheim et al., 1990). Growing evidence suggests that apoptosis is involved in neuronal cell death after a variety of brain insults such as focal and global ischemia, excitotoxicity, and traumatic brain injury (TBI). Characteristic features, such as cell shrinkage, formation of apoptotic bodies, chromatin condensation, and internucleosomal DNA fragmentation, are seen in animal models of stroke and TBI.
The interleukin-1β converting enzyme (ICE) family is the human homologue of the nematode Caenorhabditis elegans Ced-3 (Yuan and Horvitz, 1990; Ellis et al., 1991; Yuan et al., 1993), and is considered to play a critical role in apoptosis (Nicholson et al., 1995). A growing number of Ced-3-related cysteine proteases, called caspases, have been cloned, and this multigene family can be divided into at least two distinct subfamilies (Alnemri et al., 1996; Thornberry et al., 1997). Members of the family contain a cysteine residue and the highly conserved pentapeptide sequence QACRG at their active sites. Recent in vivo studies have provided evidence of the activation and cleavage of caspase-3 (CPP32) in mice brains after transient focal ischemia (Namura et al., 1998), suggesting the involvement of caspase family proteases in the pathophysiology of cerebral ischemia. In fact, inhibition of caspase family proteases has been reported to decrease the infarct volume after focal ischemia (Loddick et al., 1996; Hara et al., 1997; Endres et al., 1998; Schielke et al., 1998). Also, it has been shown that mRNA levels and the activity of CPP32 are increased after fluid percussion-induced TBI, and that inhibition of CPP32 results in the attenuation of DNA fragmentation (Yakovlev et al., 1997). However, the effect of caspase inhibitors on the lesion volume after TBI has not been examined. In the present study, we examined the effect of the caspase inhibitor, N-benzyl-oxycarbonyl-Val-Ala-Asp(OMe)-fluoromethylketone (z-VAD.FMK) on cold injury-induced brain trauma (CIBT) (Chan et al., 1991). z-VAD.FMK is a relatively nonselective irreversible peptide inhibitor that blocks activity of ICE- or CPP32-like caspases by obstructing the processing of proforms (Thornberry et al., 1994; Cain et al., 1996; Slee et al., 1996).
Cold injury is considered not only to be a classic model for vasogenic brain edema as characterized by increased blood—brain barrier permeability, but also to be a model in which apoptosis as well as necrosis plays a role (Tominaga et al., 1992; Murakami et al., 1997a, 1997b). In the present study, a significant amount of DNA fragmentation was detected as early as 4 hours after CIBT, both by agarose gel electrophoresis and by terminal deoxynucleotidyl transferase-mediated uridine 5'-triphosphate-biotin nick end labeling (TUNEL) staining. Furthermore, the results show that the infarction volumes and the amount of DNA fragmentation in the z-VAD.FMK-treated animals were remarkably reduced compared with those in the vehicle-treated animals. Although further investigation is necessary to elucidate the mechanisms of caspase inhibitors' effects on CIBT, our data suggest that caspase inhibitors could be therapeutic modalities for TBI by blocking apoptosis or necrosis.
MATERIALS AND METHODS
Drug
The caspase inhibitor, z-VAD.FMK, was obtained from Enzyme Systems Products (Livermore, CA, U.S.A.). The compound was dissolved in 0.25% dimethyl sulfoxide (DMSO) in phosphate-buffered saline (PBS). The freshly made solution was used within a day.
CIBT and intracerebroventricular injection
Male CD-1 mice (35 to 40 g, 3 months old) were subjected to CIBT according to the methods of Chan et al. (1991). Experimental animals were anesthetized with chloral hydrate (350 mg/kg) and xylazine (4 mg/kg) intraperitoneally and placed in a stereotactic frame. The scalp was incised on the midline, and the skull was exposed. z-VAD.FMK (100 ng in 0.25% DMSO in PBS) and the vehicle (0.25% DMSO in PBS) were injected intracerebroventricularly (2 µL, bregma: 1.0 mm lateral, 0.2 mm posterior, 3.1 mm deep). Immediately after intracerebroventricular injection, a metal probe 4 mm in diameter, cooled with powdered dry ice, was applied to the surface of the skull by a weight of 100 g for 30 seconds. For the 72-hour model, z-VAD.FMK (100 ng) and the vehicle (0.25% DMSO in PBS) were injected intracerebroventricularly 15 minutes before, and 24 and 48 hours after cold injury.
Histology
The experimental animals were sacrificed at 4, 24, and 72 hours after the CIBT. The brains were removed, frozen in −20°C 2-methyl butane, and stored at −80°C. Coronal sections, 20 µm in thickness, were taken at 500-µm intervals from the anterior side to the posterior side and stained with cresyl violet. The unstained area was measured using an image analysis according to the method of Swanson et al. (1990). The staining image was scanned with a Model GS-700 Imaging Densitometer (Bio-Rad, Hercules, CA, U.S.A.) at a 600-dots per inch resolution. The infarct area was measured by Multi-Analyst version 1.0.2 in 14 brain sections from the anterior tip of the brain. The total infarct volumes were calculated by multiplying the unstained area.
Western blot analysis
Protein extraction of the cytosolic fraction was performed as described (Fujimura et al., 1998). Approximately 20 mg of tissue of both the infarction and the contralateral cortex were cut into pieces after 1, 4, and 24 hours after CIBT, homogenized gently by douncing 30 times in a glass tissue grinder (Wheaton, Millville, NJ, U.S.A.) in seven volumes of cold suspension buffer (20 mmol/L HEPES-KOH [pH 7.5], 250 mmol/L sucrose, 10 mmol/L KCl, 1.5 mmol/L MgCl2, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L dithiothreitol) with a protease inhibitor cocktail (0.1 mmol/L phenylmethylsulfonyl fluoride, 2 mg/mL aprotinin, 10 mg/mL leupeptin, 5 mg/mL pepstatin, and 12.5 mg/mL N-acetyl-leu-leu-norleucinal). The homogenates were centrifuged at 4°C at 10,000g for 20 minutes, and then at 100,000g for 60 minutes. Protein concentrations were determined by the Bradford method (Bio-Rad). Electrophoresis and blotting were performed according to the manufacturer's instructions (Novex, San Diego, CA, U.S.A.). The supernatants were diluted and boiled in sodium dodecyl sulfate sample buffer (Novex), loaded on a 10% bis-Tris NuPAGE gel (Novex) in a reducing condition, and transferred onto a Hybond ECL nitrocellulose membrane (Amersham International, Buckinghamshire, England). The primary antibodies were diluted 1:500 for rabbit anti-ICE polyclonal antibody (Upstate Biotechnology, Lake Placid, NY, U.S.A.) and incubated with the membrane. The primary antibody was raised against a peptide corresponding to amino acid residues 129 through 152 of human ICE, which recognizes both the p20 (20 kDa) subunit of reduced ICE isoform and the p45 (45 kDa) proenzyme. The membrane was further incubated with 1:25,000 dilution of horseradish peroxidase-conjugated anti-rabbit immunoglobulin G (Boehringer Mannheim, Indianapolis, IN, U.S.A.), and developed using enhanced chemiluminescence Western blotting detection reagents (Amersham). The membrane was reprobed with 1:10,000 dilution of β-tubulin monoclonal antibody (Amersham) and developed as above. Densitometric analysis was made on the results by using GS-700 imaging densitometer (Bio-Rad) for scanning the film, and Multi-Analyst software (Bio-Rad) for quantifying the results.
In situ labeling of fragmented DNA cells
Brain sections were stained to detect the DNA free 3'-OH ends using an in situ technique, the TUNEL reaction, as described (Kondo et al., 1997). Briefly, frozen brain sections were fixed for 30 minutes in 3.7% formaldehyde in 0.1 mol/L PBS, pH 7.4. The slides were placed in 1× terminal deoxynucleotidyl transferase buffer for 15 minutes, followed by reaction with terminal deoxynucleotidyl transferase enzyme and biotinylated 16-dUTP at room temperature for 60 minutes. The slides were then washed in 2× SSC (150 mmol/L sodium chloride, 15 mmol/L sodium citrate, pH 7.4) for 15 minutes, followed by a washing in PBS two times for 15 minutes. Avidin-biotin horseradish peroxidase solution (ABC kit, Vector Laboratories, Burlingame, CA, U.S.A.) was applied to the sections for 30 minutes, then the slides were washed for 15 minutes with 0.175 mol/L sodium acetate. Staining was visualized using 0.0025% diaminobenzidine and 0.075% H2O2 in PBS with 0.4 mg/mL nickel sulfate. The slides were rinsed with water, stained with methyl green for 10 minutes, dehydrated, and mounted.
DNA fragmentation assay using gel electrophoresis
Thirty to 50 mg of brain tissue from two mice was taken from the lesion and the controls, homogenized in 0.6 mL lysis buffer (0.5% sodium dodecyl sulfate, 10 mmol/L Tris-HCl, 0.1 mol/L EDTA, pH 8.0) containing 1 mg/mL proteinase K (Boehringer Mannheim), and incubated in the same buffer overnight at 55°C. DNA was extracted with phenol and phenol-chloroform-isoamyl alcohol (25:24: 1) and precipitated overnight in 85 mmol/L sodium chloride in 95% ethanol at −20°C. The DNA was washed twice with 75% ethanol, dried under speed vacuum, and resuspended in a 100-µL buffer (10 mmol/L Tris-HCl, 1 mmol/L EDTA, pH 8.0). DNA was incubated at 37°C overnight in proteinase K (1 mg/mL) and reextracted, reprecipitated, and resuspended in 80 µL DNase-free water (Sigma, St. Louis, MO, U.S.A.). The DNA concentration was measured using the dye To-Pro-1 (Molecular Probes, Eugene, OR, U.S.A.). Before electrophoresis, 0.5 µg DNA was incubated with 50 µg/mL DNase-free RNase (Boehringer Mannheim) for 30 minutes at 37°C. Gel electrophoresis for DNA laddering detection was performed according to the manufacturer's instructions (Trevigen, Gaithersburg, MD, U.S.A.). The samples were labeled with dNTP by Klenow enzyme in 1× Klenow buffer for 10 minutes at room temperature, using the TACS apoptotic DNA laddering kit, chemiluminescent method (Trevigen). DNA (0.5 µg), separated in a 1.5% agarose gel (Trevigen), was transferred to a nylon membrane overnight in 10× SSC. The membrane was first incubated with PBS containing 5% nonfat dry milk for 30 minutes, and then Streptavidin-horseradish peroxidase conjugate (Trevigen) diluted 1:2,000 in PBS. Labeled DNA was visualized by incubating the membrane with PeroxyGlow solution (Trevigen). The nylon membrane was exposed to x-ray film for an appropriate amount of darkness.
Statistical analysis
Data are presented as the mean ± SD. Statistical comparisons were made by analysis of variance using the software StatView, version 4.0 (Abacus Concepts, Berkeley, CA, U.S.A.). A P < 0.05 was considered statistically significant.
RESULTS
Reduction of CIBT by a caspase inhibitor
To determine the role of the caspase family proteases in CIBT, we examined the effect of a relatively nonselective caspase inhibitor, z-VAD.FMK, which is known to block both ICE-like and CPP32-like caspases. The vehicle alone or with z-VAD.FMK was administered into the cerebral ventricles of mice 15 minutes before and 24 and 48 hours after cold injury. Typical brain sections were stained with cresyl violet at 4, 24, and 72 hours after CIBT (data not shown). The infarction volumes of the z-VAD.FMK-treated animals were apparently reduced compared with those of the vehicle-treated animals. Figure 1 shows the infarction volumes at various times after cold injury. At 4 hours, the infarction volume of the vehicle-treated animals was 4.48 ± 1.43 mm3 (n = 7), and was further increased at 24 (5.72 ± 2.43 mm3, n = 13) and 72 hours (7.46 ± 3.53 mm3, n = 7). When z-VAD.FMK was injected into the ventricle, reduction in infarct volumes was noted at 4 (2.98 ± 1.74 mm3, n = 8) and 24 hours after injury. The brain infarction was approximately 55% smaller (3.15 ± 2.50 mm3, P = 0.025, n = 9) than that of the vehicle-treated animals. Furthermore, at 72 hours, the brain infarction was reduced approximately 85% to 90% (0.92 ± 1.81 mm3, P = 0.0005, n = 8).
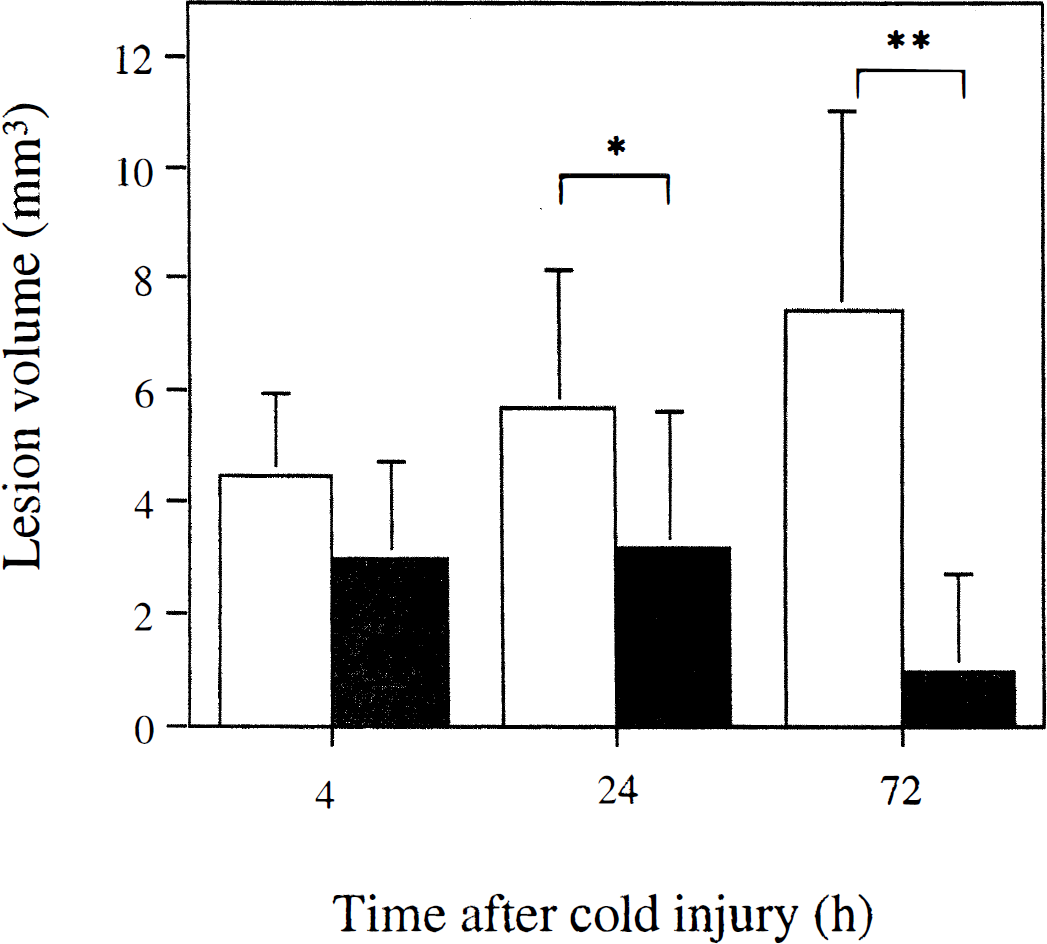
Reduced infarct size in cold-injured brain of z-VAD.FMK- and vehicle-treated mice. The infarct size was analyzed by multiplying the infarct area in the coronal sections stained with cresyl violet, at 4 hours (n = 7 to 8), 24 hours (n = 9 to 13; P < 0.025, compared with vehicle-treated mice), and 72 hours (n = 7 to 8; P < 0.005) after cold injury. Values are mean ± SD, using analysis of variance followed by factorial t test. (□) Vehicle. (■)z-VAD.FMK.
Activated form of ICE was detected in the control brain by Western blot analysis, which showed moderate reduction after CIBT
To determine whether ICE-like proteases are activated in brain lesions after CIBT, we examined whether ICE cleavage products appear on a Western blot of the brain lesion before and after CIBT using a polyclonal antibody of ICE that detects the cleavage product (p20) and the proenzyme (p45). As demonstrated in Fig. 2, the activated form of ICE (p20) was observed constitutively in the control brain (Fig. 2, lane 1, upper panel), which showed moderate reduction at 4 hours (79.8% optical density, compared with control) and 24 hours (27.4% optical density, compared with control) after CIBT on the ipsilateral side (Fig. 2, upper panel). On the other hand, p20 level was sustained on the contralateral side after CIBT (Fig. 2). The proform of ICE (p45) was seen as having dense characteristic 45-kDa bands before and after CIBT (Fig. 2, upper panel). There was no difference in p-tubulin protein expression among the lanes, suggesting that the amount of loaded proteins was consistent (lower panel).
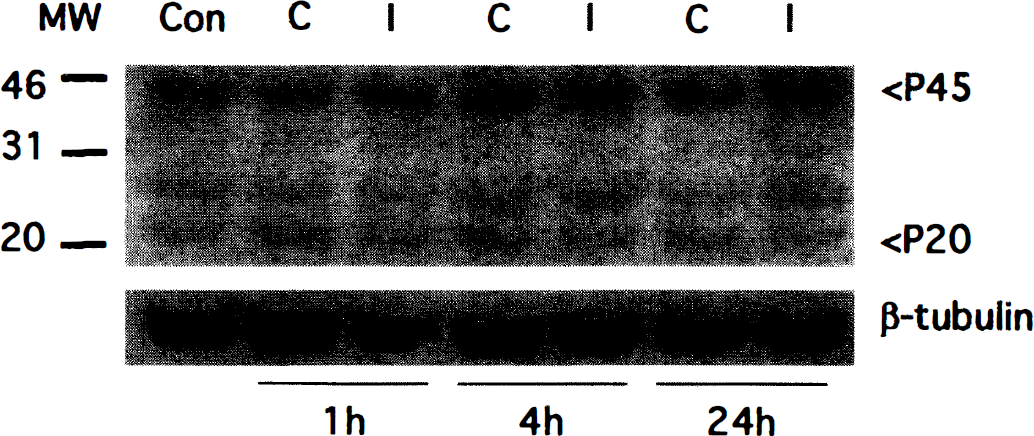
Western blot analysis before and after CIBT using polyclonal antibody of ICE. An activated form of ICE is shown as a characteristic band of p20 in the control brain (lane 1, upper panel), which was sustained after CIBT on both the ipsilateral and contralateral sides (lanes 2 through 7, upper panel). The proform of ICE was also shown as dense characteristic p45 bands before and after CIBT. β-tubulin was used as an internal control (lower panel).
In situ detection of DNA fragmentation by TUNEL staining
To assess the potential role of caspases in trauma-mediated apoptosis, we examined the ability of the caspase inhibitor z-VAD.FMK to effect cold injury-induced DNA fragmentation. Apoptosis induction on tissue sections from injured mice brains was first examined by the TUNEL staining technique. TUNEL-positive cells were observed especially at the center of the lesion 4 hours after the injury (Fig. 3A), and in the peripheral area 24 hours (Fig. 3C) after the injury. At 72 hours, TUNEL-positive cells were detected mainly in the peripheral area (Fig. 3E), and a number of accumulated necrotic cells were observed in the center of the lesion (Fig. 3E). In the lesions of the z-VAD.FMK-treated mice brains, a marked decrease in TUNEL-positive cells was observed (Fig. 3B) compared with those of the vehicle-treated mice brains (Fig. 3A) at 4 hours in the entire injured area. At 24 and 72 hours after injury, more necrotic cells were observed only in the core of the lesion (Fig. 3D and 3F), and a lesser amount of TUNEL-positive cells was observed in the peripheral lesion. To quantify the DNA-fragmented cells after cold injury, the percentage of TUNEL-positive cells in the total of methyl green-stained cells was counted at 4, 24, and 72 hours after cold injury (Fig. 4A—C). TUNEL-positive cells with shrunken morphology, condensed nuclei, and formation of apoptotic bodies were detected in the ipsilateral injured cortex (Fig. 4D) as early as 4 hours after injury (Fig. 4A). These cells are compatible with those in the apoptotic cell death process previously described (Murakami et al., 1997b). Besides these typical TUNEL-positive cells, slightly stained cells that had diffuse nuclear and cytoplasmic staining were also observed (Fig. 4E). These cells are consistent with the necrotic cells described in a previous report (Murakami et al., 1997b). Only densely labeled cells are considered to be TUNEL-positive cells in this study. No apoptotic or necrotic cells were observed in the contralateral cortex (Fig. 4F). Three regions, as defined previously (Murakami et al., 1997b), were used to assess the distribution of TUNEL-positive cells as shown in the schema in Fig. 4. At 4 hours, the percentage of TUNEL-positive cells in all the regions was significantly reduced in the z-VAD.FMK-treated animals compared with the vehicle-treated animals (Fig. 5A, 5B, and 5C, P < 0.03, n = 4). Moreover, at 24 hours, the percentage of TUNEL-positive cells was significantly reduced in the peripheral area (Figs. 3C and 4B, P < 0.01, n = 4), but not in the center of the lesion (Fig. 4A, P = 0.086, n = 4), where more necrotic cells were observed. No significant inhibition of the percentage of TUNEL-positive cells was detected in the lesion at 72 hours (Fig. 4A, 4B, and 4C, n = 4). The number of TUNEL-positive cells in the peripheral area (region C) at 24 hours was significantly reduced in the lesion of the z-VAD.FMK-treated mice (416.0 ± 169.3/mm2, n = 4) compared with the number in the vehicle-treated mice (732.0 ± 186.3/mm2, P = 0.046, n = 4).
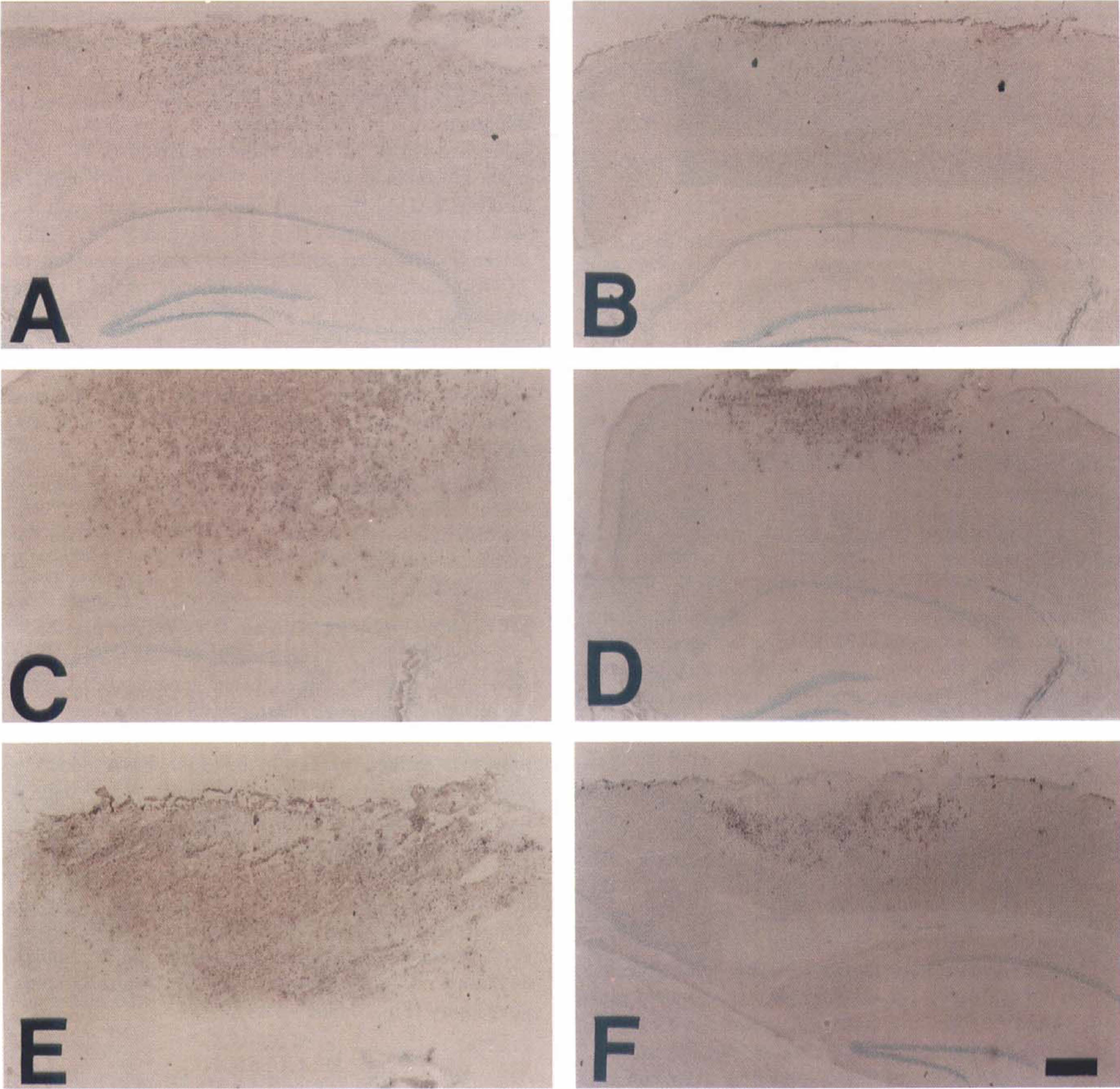
Distribution of DNA fragmentation in the brains of vehicle-treated (
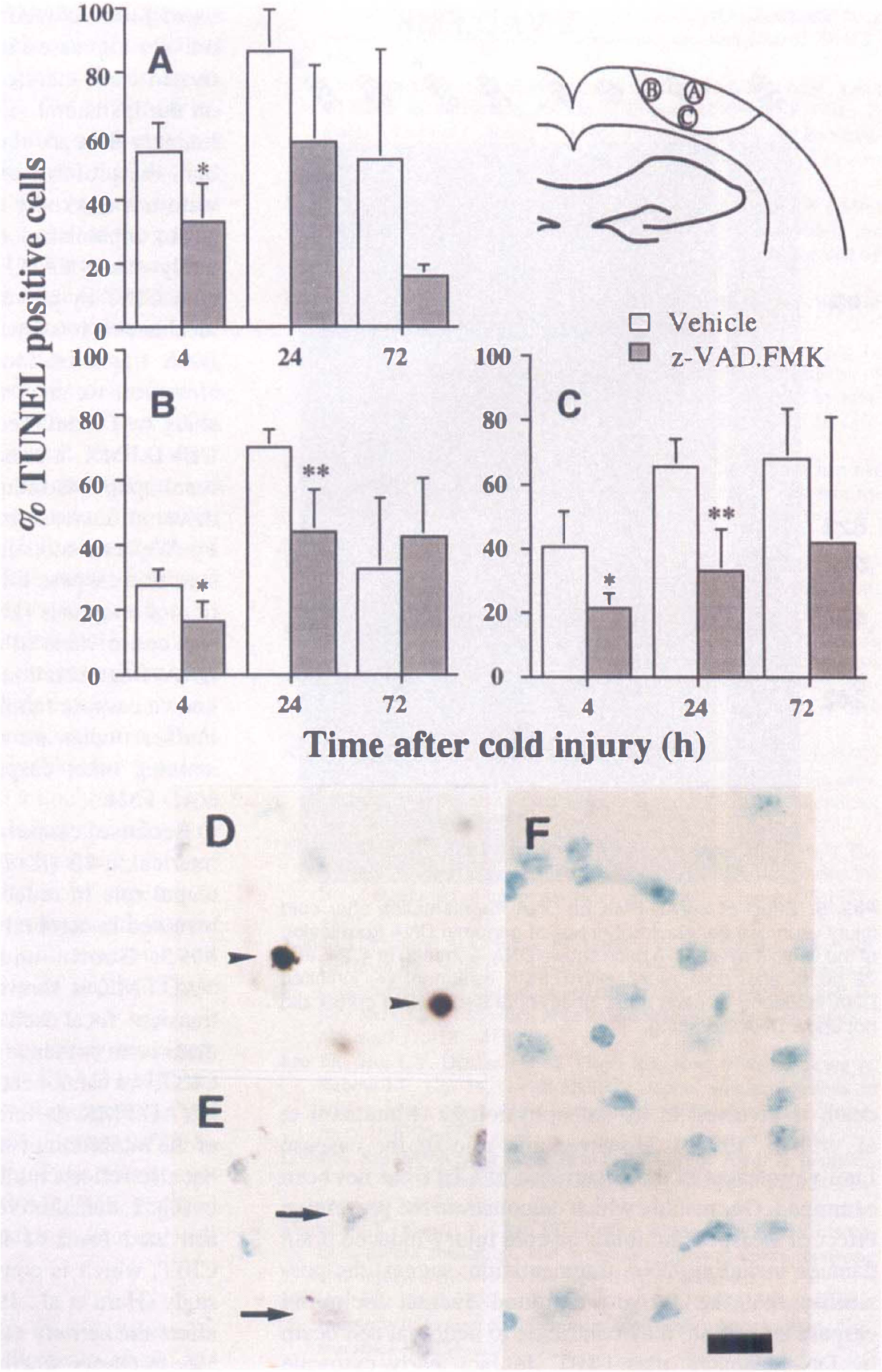
Temporal profile of TUNEL-positive cells in the infarct area of vehicle-treated animals (
z-VAD.FMK inhibits genomic DNA fragmentation
To confirm that TUNEL staining reflects nucleosomal DNA fragmentation, we extracted genomic DNA from the lesion and the contralateral brain homogenates, and analyzed them by gel electrophoresis using a chemiluminescent method (Fig. 5). In the lesion, DNA laddering was first observed at 4 hours after injury (Fig. 5, lane 4 h–), and became more apparent at 24 (Fig. 5, lane 24 h–) and 72 hours (Fig. 5, lane 72 h–) after cold injury. z-VAD.FMK, administered intracerebroventricularly before cold injury, inhibited DNA laddering at 4 (Fig. 5, lane 4 h+) and 24 hours after injury (Fig. 5, lane 24 h+). At 72 hours, DNA laddering was further inhibited when additional z-VAD.FMK was injected at 24 and 48 hours after injury (Fig. 5, lane 72 h+).
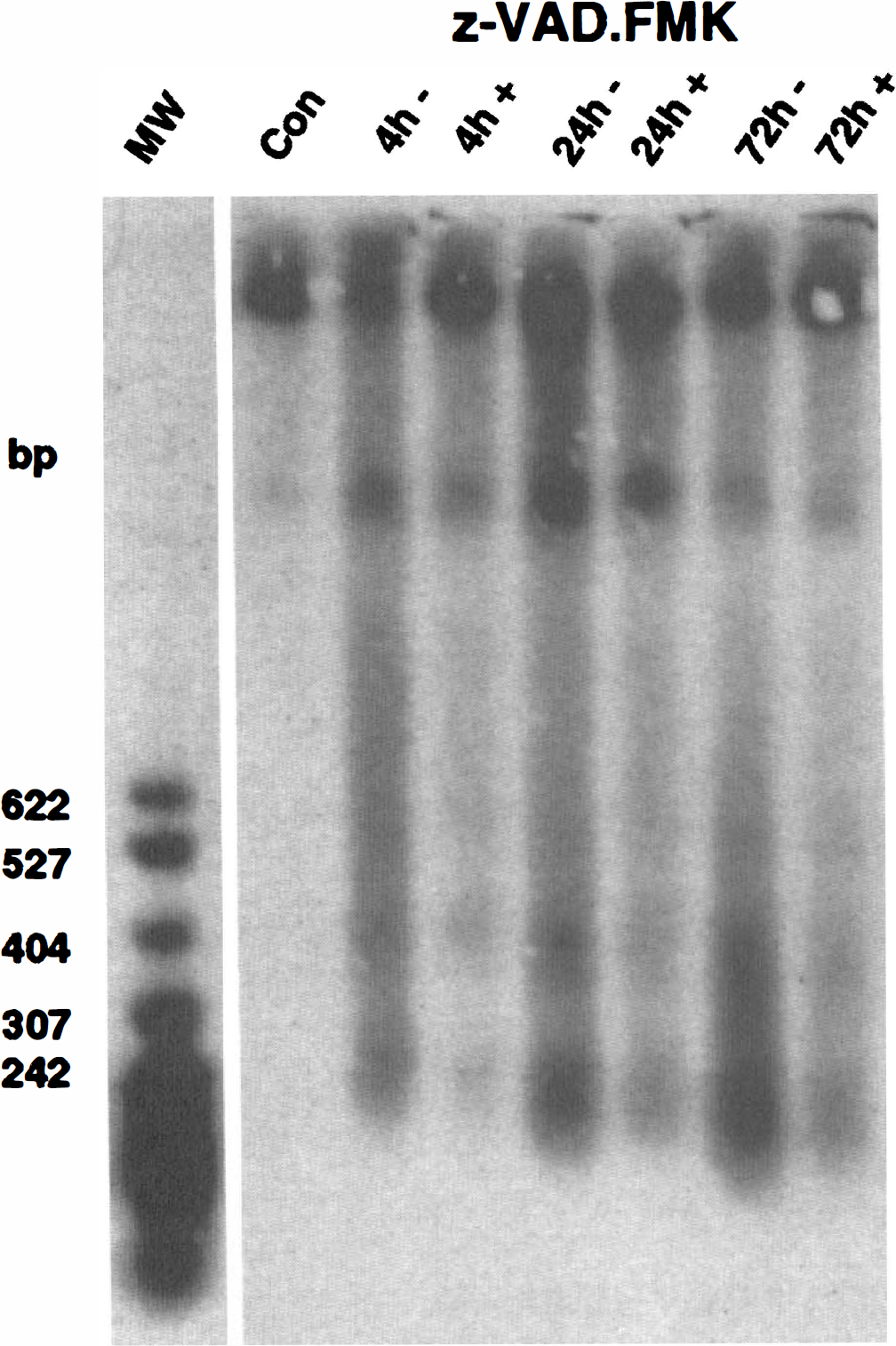
Effect of z-VAD.FMK on DNA fragmentation after cold injury. Agarose gel electrophoresis of genomic DNA from lesion of the vehicle-treated (–) mice shows DNA laddering at 4, 24, and 72 hours after cold injury. z-VAD.FMK treatment (+) inhibited DNA laddering at each time. Sham-operated mouse cortex did not show DNA laddering.
DISCUSSION
We demonstrate in the present study that a cysteine protease inhibitor of the caspase family, z-VAD.FMK, remarkably reduced the amount of DNA fragmentation and infarct volume after CIBT. As shown in the Results, a marked decrease of DNA fragmentation in the z-VAD.FMK-treated animals was detected as early as 4 hours after cold injury using both TUNEL staining (Fig. 4) and DNA gel electrophoresis (Fig. 5), and there was no significant difference in infarction volume between the z-VAD.FMK-treated and the vehicle-treated animals (Fig. 1). The reduction of TUNEL-positive cells was more remarkable in the peripheral lesion, as seen in region C in Fig. 4, than in the core lesion of the infarction. At 24 hours after cold injury, the infarct volumes significantly decreased in the z-VAD.FMK-treated animals and further decreased at 72 hours, but did not decrease in the vehicle-treated animals. These data, which provide the first evidence that the reduction of DNA fragmentation precedes the decrease of infarct volume in z-VAD.FMK-treated animals, suggest that z-VAD.FMK could prevent cold injury-induced brain damage by blocking neuronal cell death by DNA damage. Furthermore, we observed a more significant decrease of infarct volume at 72 hours after injury than at 24 hours, and our preliminary data show that preadministration of z-VAD.FMK alone is not enough to reduce the infarct volume at 72 hours (data not shown). Therefore, z-VAD.FMK is likely to have a lengthy therapeutic window from the initiation of the CIBT. Although we administered the agent by intracerebroventricular injection, our results suggest the possibility that z-VAD.FMK could be a therapeutic modality for TBI.
It has been reported that at least some members of the caspase (ICE/Ced-3) family are essential components of an evolutionarily conserved pathway of apoptosis (Thornberry, 1997). Recent studies have demonstrated a central role for mitochondrial proteins (e.g., cytochrome c and Bcl-2) in the activation of CPP32 (Liu et al., 1996; Kluck et al., 1997; Yang et al., 1997). At the very beginning of the apoptotic event, cytochrome c is released from mitochondria to the cytosol (Liu et al., 1996), where it activates CPP32 by interacting with the protein Apaf-1, which shares its homology with Ced-4 (Zou et al., 1997). Also, we have shown in vivo that the release of cytochrome c from mitochondria to the cytoplasm, which precedes the peak of DNA fragmentation, occurs after ischemia/reperfusion in rats (Fujimura et al., 1998). It is also reported that the activation and cleavage of CPP32 contribute to apoptosis after transient ischemia in mice (Namura et al., 1998), and that CPP32 mRNA and its activity are increased after fluid percussion-induced traumatic injury (Yakovlev et al., 1997). Regarding CIBT, we have demonstrated that apoptotic neuronal cell death is involved in its pathophysiology (Murakami et al., 1997a, 1997b). However, the role of the caspase family proteases in the occurrence of CIBT has not been examined. Our results, which demonstrate the preventive effect of a caspase inhibitor on cold injury-induced brain damage including DNA fragmentation, suggest the possibility that the above-mentioned events, including caspase activation, may contribute to neuronal cell death by DNA damage after CIBT. In fact, early cytosolic redistribution of cytochrome c from mitochondria was detected after cold injury (Y. Morita-Fujimura, unpublished data). Further evaluation is warranted to elucidate the detailed molecular mechanism of the occurrence of DNA fragmentation after CIBT.
z-VAD.FMK is known as a broad-spectrum inhibitor for caspases that inhibits at least two caspases: CPP32 and caspase-1 (ICE) (Thornberry et al., 1994; Slee et al., 1996). Therefore, in the present study we examined ICE protein expression before and after CIBT by Western blot analysis (Fig. 2). The results showed that an activated form, as well as the proform, of ICE was constitutively expressed in the normal brain, whereas the activated form manifested moderate reduction after CIBT on the ipsilateral side (Fig. 2). As for CPP32, our preliminary data are showing the constitutive expression of both the proform and activated form of CPP32, which were not markedly affected by CIBT (Morita-Fujimura et al., unpublished data). From these findings, it is unlikely that z-VAD.FMK decreases DNA fragmentation after CIBT by preventing ICE or CPP32 activation. The mechanism by which z-VAD.FMK reduces CIBT and DNA fragmentation in the current study is not clear. However, we may find some clue in a recent in vitro study by D'Mello et al. (1998). They demonstrated that DEVD.FMK, a selective CPP32 inhibitor, inhibits neuronal apoptosis induced by K+ deprivation but failed to detect an increase in processing or activation of CPP32 by Western blot analysis, suggesting that a DEVD-sensitive caspase other than CPP32 mediates the induction of apoptosis (D'Mello et al., 1998). Therefore, it is also conceivable in our study that z-VAD.FMK reduces DNA fragmentation after CIBT by preventing an unknown caspase family protease sensitive to zVAD.FMK. Further studies are warranted to clarify this issue by examining other caspases that could be inhibited by z-VAD.FMK.
Because caspase-1 is reported to process pro-interleukin-1β (IL-1β) to IL-1β, zVAD.FMK has a potential role in reducing IL-1β, which is reported to be involved in cerebral ischemia and TBI (Yamasaki et al., 1995; Gourin and Shackford, 1997). In fact, z-VAD.FMK is shown to prevent IL-1β production after transient focal ischemia (Hara et al., 1997). Although there is no evidence that IL-1β activation plays a role in CIBT, we cannot exclude the possibility that the effect of z-VAD.FMK on infarction volumes is not only because of the inhibition of neuronal cell death by DNA damage, but also reflects inhibition of IL-1β activation. As shown in Fig. 2, constitutive expression of both the proform and activated form of ICE was detected before and after CIBT, which is consistent with a recent focal ischemia study (Hara et al., 1997). Therefore, z-VAD.FMK could affect the activity of IL-1β. Further evaluation is necessary to clarify whether IL-1β is activated by cold injury and whether it can be inhibited by z-VAD.FMK. Nevertheless, z-VAD.FMK is likely to prevent cold injury-induced brain damage mainly, if not exclusively, by blocking neuronal cell death by DNA damage, because our results provide the evidence that a marked reduction of DNA fragmentation, especially in the peripheral lesion, precedes the decrease of infarct volume in z-VAD.FMK-treated animals.
We have reported that reactive oxygen species, superoxide in particular, are involved in neuronal cell death, including apoptosis and necrosis, in the alteration of blood—brain barrier permeability and in the pathogenesis of brain edema after focal cerebral ischemia (Kinouchi et al., 1991; Chan et al., 1995; Chan, 1996; Kondo et al., 1997) and CIBT (Chan et al., 1991). The extent of vasogenic edema and infarct volume after cold injury is significantly decreased in transgenic mice that overexpress copper-zinc superoxide dismutase, suggesting that superoxide radicals play an important role in the pathogenesis of CIBT (Chan et al., 1991), Furthermore, we have shown that DNA fragmentation after focal cerebral ischemia is markedly increased in superoxide dismutase-1 knockout mice compared with wild-type mice, again suggesting that superoxide radicals contribute to neuronal cell death by DNA damage in vivo (Chan, 1996; Kondo et al., 1997). Although it is still unclear how superoxide radicals increase DNA fragmentation and whether superoxide radicals play some role in neuronal cell death by DNA damage after CIBT, an early mitochondrial event is likely to be one of the possibilities that explain the correlation between reactive oxygen species and neuronal cell death by double-strand DNA damage. Because mitochondria are known as the site where oxygen free radicals, especially superoxide anions, are produced during an insult such as cerebral ischemia (Piantadosi and Zhang, 1996), it is conceivable that overproduction of reactive oxygen species in mitochondria could cause mitochondrial dysfunction, including the release of intermembrane proteins such as cytochrome c. It is of great interest to examine the correlation of superoxide radical production with cytochrome c release, subsequent caspase family protease activation, and neuronal cell death by DNA damage after a variety of insults. These studies are taking place in our laboratory.
In conclusion, it has been shown that the cysteine protease inhibitor of the caspase family, z-VAD.FMK, remarkably reduced the amount of DNA fragmentation and infarct volume after CIBT, and that the decrease of DNA fragmentation preceded the reduction of the infarct volume. These results indicate that z-VAD.FMK reduced neuronal damage after CIBT by blocking neuronal cell death by DNA damage.
Footnotes
Acknowledgments
The authors thank Cheryl Christensen for editorial assistance and Liza Reola, Bernard Calagui, and Dr. Sylvia F. Chen for technical assistance.