
Abstract
The evolution of brain infarcts during permanent occlusion of the middle cerebral artery (MCA) was studied in mice using multiparametric imaging techniques. Regional protein synthesis and the regional tissue content of ATP were measured on adjacent cryostat sections at increasing intervals after vascular occlusion ranging from 1 hour to 3 days. The observed changes were correlated with the expression of the mRNA of hsp70, c-fos, c-jun, and junB, as well as the distribution of DNA double-strand breaks visualized by terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labelling (TUNEL). One hour after MCA occlusion, the tissue volume with suppressed protein synthesis was distinctly larger than that in which ATP was depleted. With ongoing ischemia time, the ATP-depleted area gradually expanded and, within 1 day, merged with the region of suppressed protein synthesis. Expression of hsp70 mRNA occurred mainly in the penumbra (defined as the region of suppressed protein synthesis but preserved ATP), peaking at 3 hours after vascular occlusion. Expression of the immediate-early genes c-jun, c-fos, and junB increased both in the penumbra and the periinfarct normal tissue already at 1 hour after vascular occlusion, with slightly different regional and temporal patterns for each of these genes. DNA fragmentations were clearly confined to neurons; they appeared after 1 day in the infarct core (defined as the region of suppressed ATP) and never were detected in the penumbra. The late appearance of TUNEL after infarcts had reached their final size and the absence in the penumbra points against a major pathogenetic role of apoptosis. Permanent MCA occlusion in mice thus produces a gradually expanding infarct, the final size of which is heralded by the early inhibition of protein synthesis.
Acute occlusion of a major cerebral artery causes reduction of blood flow, the severity of which increases from the peripheral to the more central parts of the vascular supplying territory (Tamura et al., 1981). According to the threshold concept of cerebral infarction, tissue viability is immediately endangered only in the core of the ischemic territory in which blood supply of oxygen falls below the critical value required for supporting brain energy metabolism (Astrup et al., 1981). At higher flow values corresponding to the periinfarct penumbra, energy metabolism and tissue viability are initially preserved, but with ongoing ischemia, delayed cell death may occur. Noninvasive brain imaging of permanent middle cerebral artery (MCA) occlusion in rats documents that during the initial 6 to 12 hours of vascular occlusion, the infarct core expands into the penumbra and eventually merges with it, leading to the volume expansion of irreversibly injured tissue by as much as 100% (Hoehn-Berlage et al., 1995; Heiss et al., 1995).
Several pathophysiologic mechanisms may contribute to this dramatic progression of injury. Probably the most important single factor is the generation of periinfarct depolarizations (Hossmann, 1996). Such depolarizations lead to the aggravation of the ischemic energy crisis because the reduced hemodynamic capacity of the penumbral circulation precludes adequate coupling of blood supply to the increased oxygen demands of the tissue during restoration of membrane polarization (Back et al., 1994). Other putative mechanisms are inflammation (Becker, 1998; Kogure and Kogure, 1997) or the combination of excitotoxicity with programmed cell death (Leist and Nicotera, 1998; Lipton and Nicotera, 1998; Choi, 1996). The release of excitatory amino acids from the ischemic core is thought to evoke intracellular calcium flooding, which, in turn, leads to the release of cytochrome c and pro-caspase-9 from mitochondria (Krajewski et al., 1999). On entry into the cytosol, cytochrome c binds to the caspase-activating protein Apaf-1, which induces the processing and activation of caspase-9 (Li et al., 1997). This protease cleaves and activates caspase-3, which, in turn, triggers a cascade of events that eventually result in the ordered apoptotic internucleosomal cleavage of DNA (Gorman et al., 1998). Finally, ischemia-induced depletion of calcium from the endoplasmic reticulum causes the inactivation (by phosphorylation) of the eukaryotic initiation factor eIF2α, which, in turn, results in the inhibition of protein synthesis by selective blockade of polypeptide chain initiation (DeGracia et al., 1997; Paschen, 1996).
Although compelling evidence has been provided that each of these pathologic mechanisms is able to damage the brain, their respective contribution to the final ischemic injury is difficult to evaluate. We have, therefore, studied the dynamics of the spontaneous evolution of brain infarction after permanent MCA occlusion by a battery of pictorial assays that allow the evaluation of energy state, protein synthesis, DNA fragmentation, and various genomic expression patterns on adjacent cryostat sections of the same brain. The animal species used was the mouse to provide background information for the further analysis of biochemical interactions in gene-manipulated mutants.
Our findings suggest that the most reliable parameter heralding the expansion of brain infarction is the inhibition of protein synthesis, which correlates better with the final size of ischemic injury than any of the other investigated variables.
MATERIALS AND METHODS
Experiments were carried out according to the National Institutes of Health (NIH) Guidelines for the Care and Use of Laboratory Animals and approved by the local authorities.
Animals were housed under diurnal lighting conditions and allowed access to food and water ad libitum until the day of the experiment. Anesthesia was induced by 1.5% halothane and maintained with 1% halothane in 70% N2O and 30% O2.
Animal preparation
Thirty adult male C57Black/6J mice weighing 20 to 25 g were used. Five groups of animals (n = 5 per group) were subjected to focal cerebral ischemia with the following durations: 1 hour, 3 hours, 6 hours, 1 day, and 3 days. Five shamoperated animals were used as controls. Focal cerebral ischemia was induced by permanent occlusion of the MCA using the intraluminal filament technique (Hata et al., 1998a). After midline neck incision, the left common and external carotid arteries were isolated and ligated. A microvascular clip (FE691; Aesculap, Tuttlingen, Germany) was temporarily placed on the internal carotid artery. An 8-0 nylon monofilament (Ethilon; Ethicon, Norderstedt, Germany) coated with silicon resin (Xantopren; Bayer Dental, Osaka, Japan) was introduced through a small incision into the common carotid artery and advanced 9 mm distal to the carotid bifurcation for occlusion of MCA. Sham-operation was performed by insertion of the thread into the common carotid artery, without advancing it to occlude MCA. Forty-five minutes before killing the animals, L-[4,5-3H]-leucine (150 μCi per animal, specific activity 151 Ci/mmol, Amersham, Braunschweig, Germany) was administered intraperitoneally for evaluating cerebral protein synthesis (CPS) rates. After the predetermined ischemic periods, experiments were terminated by in situ freezing (Mies et al., 1991a). Brains were removed in a cold temperature cabinet (−20°C) and cut into 20-μm thick coronal cryostat sections at −20°C. Sections were mounted on coverslips for ATP-bioluminescence, on object holders for CPS autoradiography, and on 3-aminopropyl triethoxysilane-treated slides for in situ hybridization.
Temperature control
During surgery and up to 1 hour after MCA occlusion, rectal temperature was kept between 36.5° and 37.0°C using an infrared lamp and a heating pad connected to a thermistorcontrolled heating system (YSI, Yellow Spring, OH, U.S.A.). After recovering from anesthesia, the animals were housed in an air-conditioned room at about 22°C.
Regional measurement of ATP and protein synthesis
Pictorial measurements of ATP were carried out using ATP-specific bioluminescence (Kogure and Alonso, 1978; Paschen et al., 1981). For measurement of CPS, brain slices were incubated in 10% trichloroacetic acid to remove labeled free leucine and metabolites other than proteins. Subsequently, slices were exposed for 14 days with 3H standards to an x-ray film (Hyperfilm 3H, Amersham, Braunschweig, Germany) for autoradiography of 3H-labeled proteins (Mies et al., 1991b).
Probes for mRNA
The probe sequences of c-fos (45 mer), c-jun (45 mer), junB (45 mer), and hsp70 (30 mer) corresponded to the mouse c-fos gene (bases 290 to 334; accession no. J00370), the mouse c-jun gene (bases 1866 to 1910; accession no. J04115), the mouse junB gene (bases 1307 to 1351; accession no. J03236), and the mouse inducible hsp70 gene (bases 1401 to 1430; accession no. M76613), respectively. Each probe was 3′-end-labeled using terminal deoxynucleotidyl transferase (Gibco BRL, Eggenstein, Germany) and a 30:1 molar ratio of [35S]dATP (1200 Ci/mmol). Specific activity was greater than 0.5 × 109 dpm/μg.
In situ hybridization
In situ hybridization was performed as described before (Hata et al., 1998c). Briefly, coronal brain sections (20 μm) were fixed for 15 minutes in 4% paraformaldehyde/phosphate-buffered saline (PBS), pH 7.4. Sections were treated for 10 minutes with 0.25% acetic anhydride/triethanolamine. After dehydration, sections were incubated in 10 μL hybridization buffer containing 35S-labeled oligonucleotide probe (10 pg/μL), 2 × standard sodium citrate, 50% formamide, 10% dextran sulfate, 100 μg/mL poly(A), 120 μg/mL heparin, 1 mg/mL herring sperm DNA, 5 mmol/L dithiothreitol, and 1 mg/mL bovine serum albumin, and covered with a coverslip. After overnight hybridization at 42°C, the sections were washed twice at 42°C in 2 × standard sodium citrate/50% formamide for 30 minutes and exposed together with a 14C standard to an X-ray film (Hyperfilm β-max, Amersham, Braunschweig, Germany).
Morphometric analysis of ischemia-induced metabolic disturbance and gene expression
Bioluminescence and autoradiographic images were digitized with a CCD camera system and analyzed using the NIH image software (Bethesda, MD, U.S.A.). The volumes of ATP depletion and CPS inhibition were estimated according to the semiautomated method described by Swanson et al. (1990). The ATP depletion was defined as the decline to less than 30% of the mean value on the contralateral side. The threshold for CPS inhibition was set to the lowest CPS value of the nonischemic hemisphere excluding fiber tracts. The areas of ATP depletion and CPS inhibition were measured on each section by subtracting the area of the nonlesioned ipsilateral hemisphere from that of the contralateral hemisphere. The areas of preserved ATP and protein synthesis were outlined and superimposed to demarcate penumbral tissue in which protein synthesis was suppressed but ATP was preserved. Optical densities of in situ hybridization signals of each mRNA were measured at the level of caudate-putamen in cortical regions of interest, located in the ischemic core, the metabolic penumbra, and in normal brain tissue, as defined by ATP and CPS imaging. Values were normalized to the signal in caudate-putamen of the opposite nonischemic hemisphere. Inhomogeneities of the illumination system were eliminated by background shading correction. Film autoradiograms were obtained at two or three different exposure times to ensure that optical densities were within the linear range of the standard curve.
Terminal transferase biotinylated-UTP nick-end labeling
As described before (Wiessner et al., 1996), terminal transferase biotinylated-UTP nick end labeling (TUNEL) was performed with minor modifications. Briefly, coronal brain sections were fixed for 15 minutes in ice-cold 4% paraformaldehyde/PBS, pH 7.4. Subsequently, the sections were washed twice in 70% ethanol (1 minute), once in PBS (3 minutes), once in 0.3% hydrogen peroxide/PBS (5 minutes), and then again in PBS (5 minutes). After equilibration for 15 minutes in TDT buffer (100 mmol/L potassium cacodylate, 2 mmol/L cobalt chloride, 0.2 mmol/L dithiothreitol), the buffer was quantitatively removed, sections were incubated in 50 μL of TDT-mix (10 pmol/L biotin-16-dUTP [Boehringer Mannheim, Germany] and 150 U/mL terminal deoxynucleotidyl transferase [Life Technologies, Eggenstein, Germany]) in TDT buffer and covered with a coverslip. After incubation for 60 minutes at 37°C, the reaction was terminated by washing the sections for 15 minutes in TB buffer (300 mmol/L sodium chloride, 30 mmol/L sodium citrate). Incorporated biotin was visualized using the avidin-biotin-peroxidase complex method (Vector Laboratories, Burlingame, CA, U.S.A.) as recommended by the supplier. Finally, the sections were dehydrated and embedded in Eukitt (Kindler GmbH, Freiburg, Germany).
Incidence maps of regional alterations
To evaluate the regional pattern of TUNEL compared with the reduction of CPS and ATP, regional incidence maps were constructed. The areas of biochemical disturbances were outlined on representative brain sections of each individual experiment and superposed at two coronal levels, caudate-putamen and dorsal hippocampus. Using the image analysis software, the incidence of the metabolic alterations was calculated for each pixel and expressed as a percentage of the number of animals in each group.
Statistics
All values are given as means ± SD. Differences in metabolic parameters and optical densities of mRNA were compared using one-way analysis of variance followed by Bonferroni's multiple comparison test. A p value of less than 0.05 was considered to indicate statistical significance.
RESULTS
General physiologic observations
The effects of MCA thread occlusion on cardiovascular and general physiologic parameters have been described before (Hata et al., 1998b). During the initial 3 hours of vascular occlusion, no significant changes in mean arterial blood pressure or heart rate occurred. There also were no significant group differences in arterial PO2, arterial PCO2, or hematocrit values between ischemic and sham-operated animals.
Regional brain tissue ATP and CPS
In the sham-operated controls, regional ATP content and regional CPS did not differ between both hemispheres (Tables 1 and 2). After MCA occlusion, both ATP and CPS were severely reduced in the ipsilateral hemisphere, but the regional distribution of the changes differed between ATP and CPS on one hand, and with the duration of ischemia on the other (Figs. 1 and 2).
Regional protein synthesis in mouse brain after middle cerebral artery occlusion
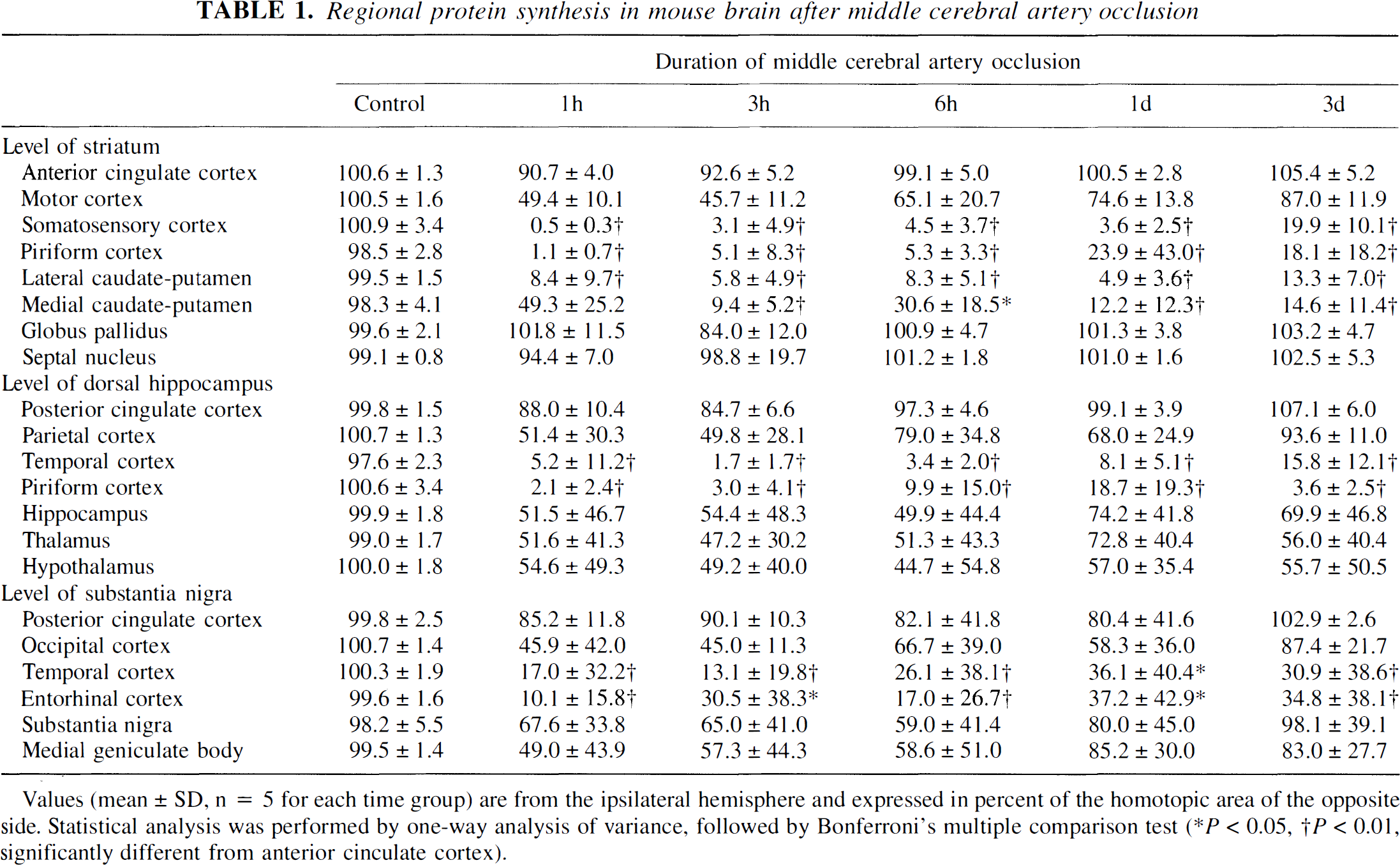
values (mean ± SD, n = 5 for each time group) are from the ipsilateral hemisphere and expressed in percent of the homotopic area of the opposite side. Statistical analysis was performed by one-way analysis of variance, followed by Bonferroni's multiple comparison test (* P < 0.05, † P < 0.01, significantly different from anterior cinculate cortex).
Regional ATP content in mouse brain after middle cerebral artery occlusion
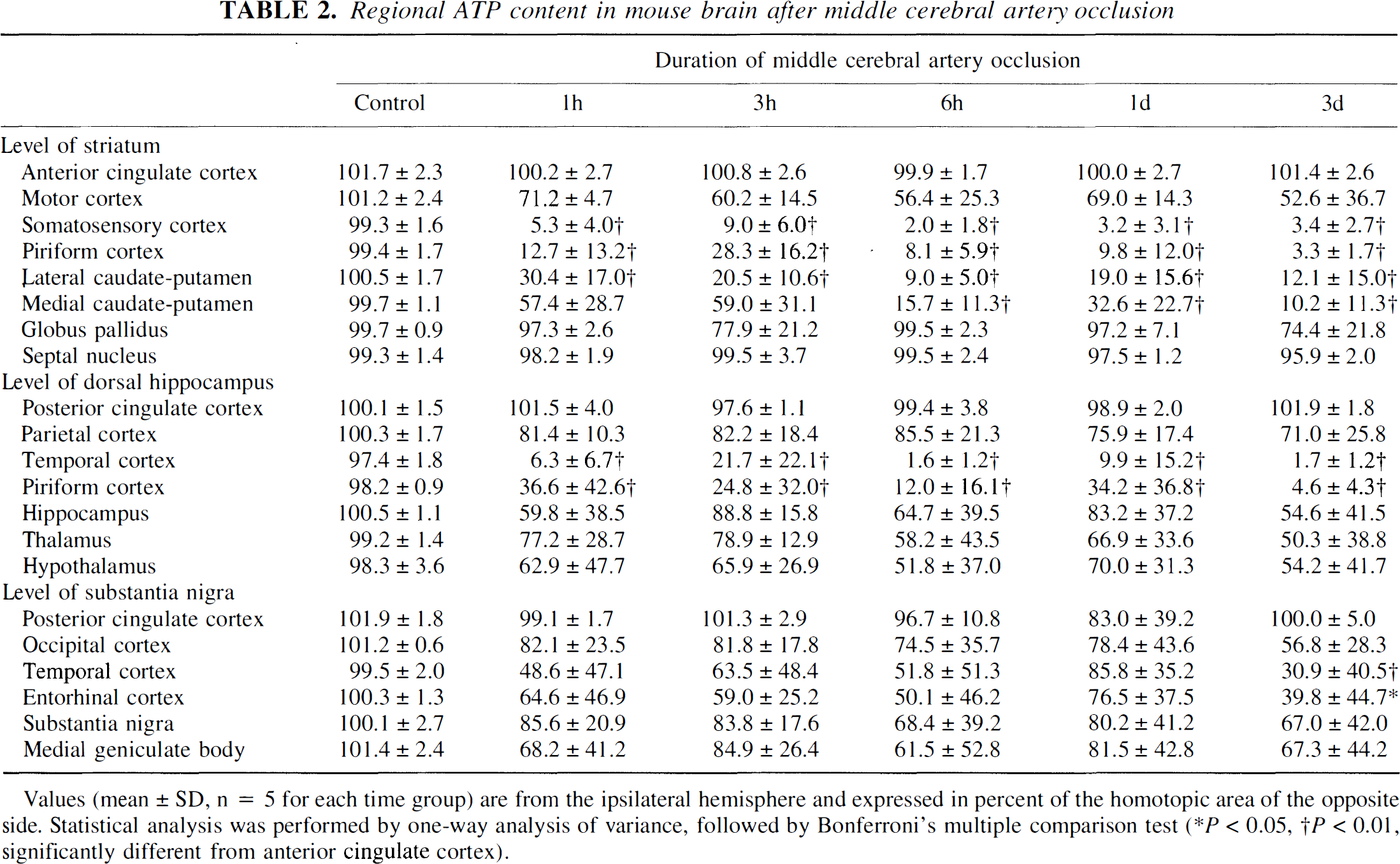
= 5 for each time group) are from the ipsilateral hemisphere and expressed in percent of the homotopic area of the opposite side. Statistical analysis was performed by one-way analysis of variance, followed by Bonferroni's multiple comparison test (* P < 0.05, † P < 0.01, significantly different from anterior cinculate cortex).
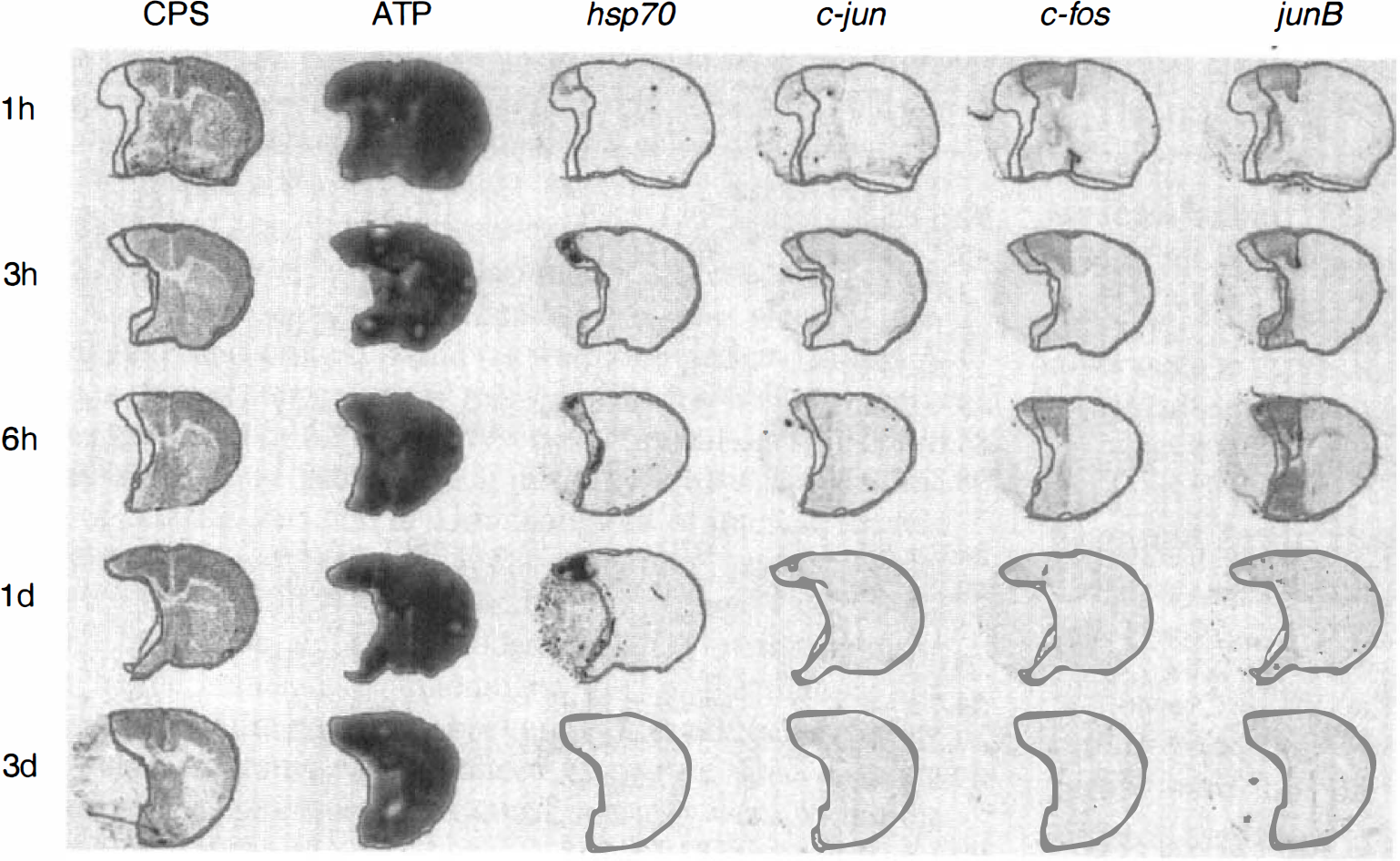
Coronal sections of mouse brain at the level of caudate-putamen at various intervals after the onset of permanent middle cerebral artery (MCA) occlusion. Multiparametric imaging of cerebral protein synthesis (CPS), ATP content, and the expression of hsp70, c-jun, c-fos, and junB mRNA. The outlines of preserved ATP and CPS have been superposed to demarcate the core from the penumbra of the evolving infarct. Notice gradual expansion of infarct core (defined as the ATP-depleted tissue) into the penumbra (defined as the tissue with suppressed CPS but preserved ATP) and differential expression of hsp70 and immediate early genes in the penumbra and the periinfarct normal brain tissue.
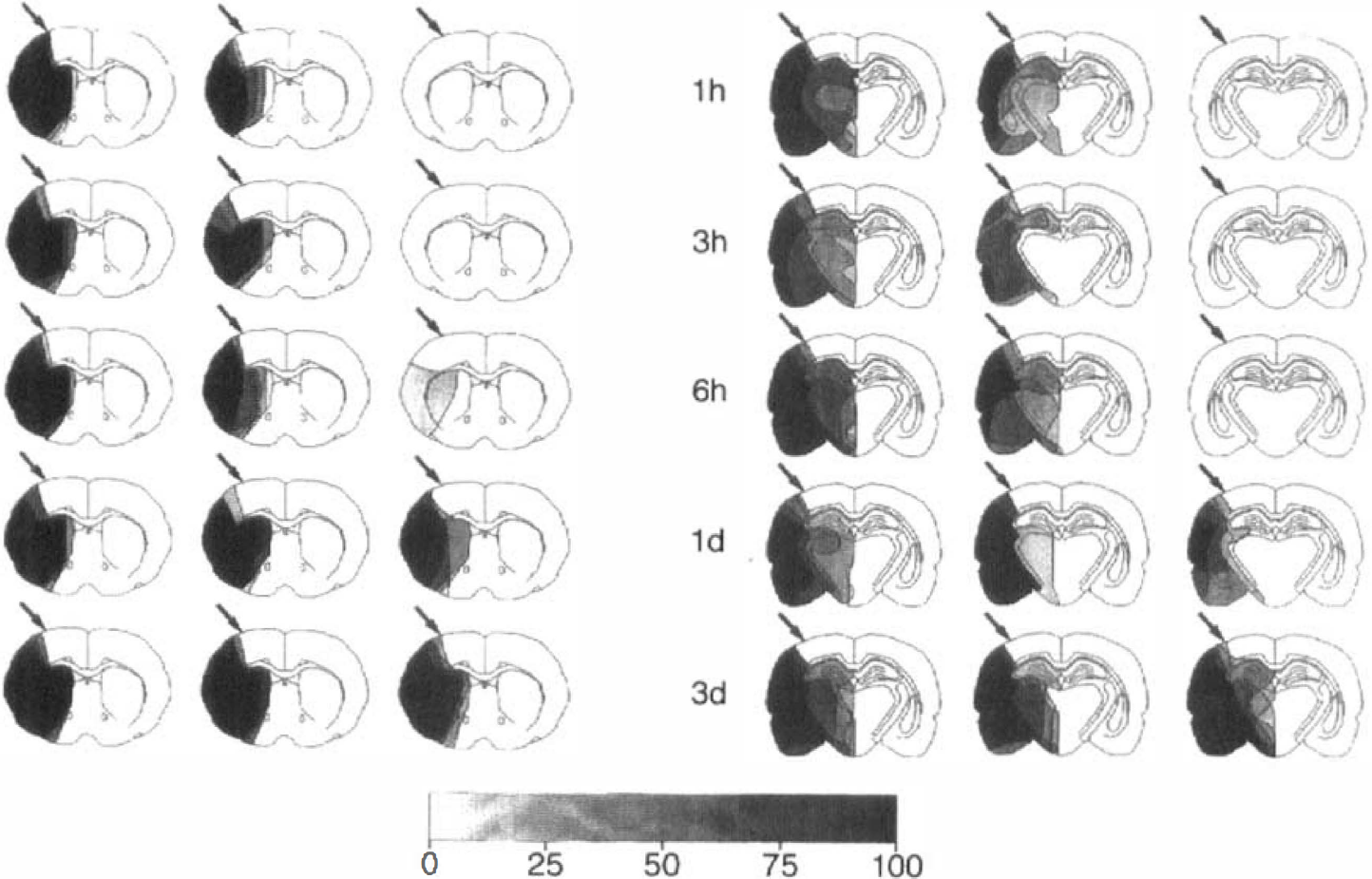
Incidence maps of suppressed protein synthesis (CPS), ATP depletion, and neurons positive for terminal transferase biotinylated-UTP nick end labeling (TUNEL) on coronal sections of the mouse brain at various times after the onset of permanent middle cerebral artery (MCA) occlusion. Areas of disturbed metabolism were outlined in five animals per time point at the level of caudate-putamen (left) and dorsal hippocampus (right) and superposed to calculate the incidence of alterations as the percentage of the number of animals per group. The demarcation between normal and disturbed protein synthesis in parietal cortex visible at 1-hour MCA occlusion was marked by the arrows to estimate the evolution of the metabolic disturbances at later time points. Notice gradual expansion of the ATP-depleted area into but not beyond the area of disturbed protein synthesis, visible after 1 hour, and the delayed appearance of TUNEL within but not outside of the ATP-depleted region. These findings demonstrate that early inhibition of protein synthesis heralds the final manifestation of infarction, and that DNA fragmentation occurs in the core but not in the penumbra of evolving infarcts.
After 1 hour of vascular occlusion, ATP was depleted in the frontoparietal cortex, the lateral part of caudateputamen, and the piriform cortex. Suppression of protein synthesis was present in the same areas, but the changes extended distinctly more into the frontal, medial, and temporo-occipital parts of the MCA territory. With increasing ischemia time, the ATP depleted tissue mass-but not the volume of tissue with inhibition of CPS-gradually expanded until, within 1 day, ATP- and CPS-impaired regions merged (Figs. 1 and 2). On coronal brain sections at the level of caudate-putamen, the ATP-depleted area thus increased from 40.1 ± 11% (means ± SD) of the contralateral hemispheric area at 1 hour of MCA occlusion to 47.4 ± 11.9% after 3 hours, 49.0 ± 13.6% after 6 hours, 56.2 ± 4.7% after 1 day, and 58.2 ± 3.9% after 3 days (Fig. 3). The area of CPS inhibition remained almost constant during this time and amounted to 58.9 ± 5.5% after 1 hour, 59.3 ± 14.9% after 3 hours, 59.6 ± 9.4% after 6 hours, 59.0 ± 5.4% after 1 day, and 60.2 ± 3.8% after 3 days of MCA occlusion. Accordingly, the penumbra-defined as the area of disturbed protein synthesis but preserved energy state-gradually declined from close to 20% of the contralateral hemisphere after 1 hour of ischemia to less than 3% after 1 day.
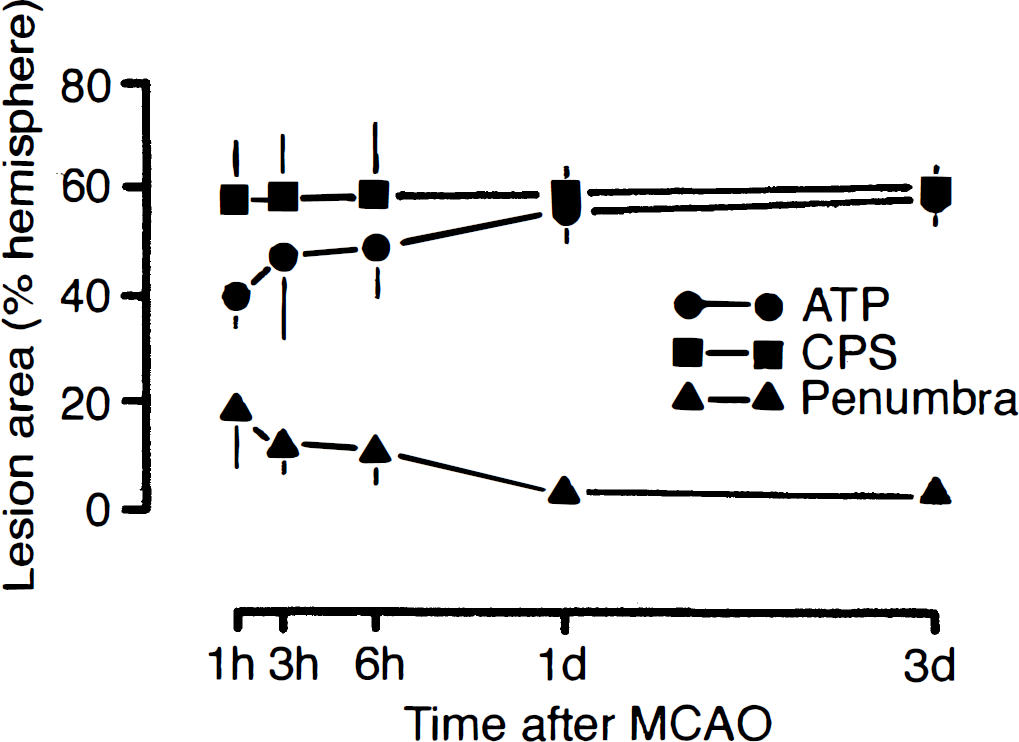
Dynamics of infarct evolution in mouse brain during permanent occlusion of middle cerebral artery. The areas of suppressed cerebral protein synthesis (CPS), ATP depletion, and penumbral tissue (defined as the area of suppressed CPS but preserved ATP) were measured at the level of caudate-putamen and expressed as the percentage of the opposite nonischemic hemisphere. Notice gradual expansion of the ATP-depleted brain tissue into the region of suppressed protein synthesis, indicating growth of the infarct core into the periinfarct penumbra.
Quantitative evaluations of ATP and CPS in different anatomic structures confirmed the progression of metabolic disturbances and further revealed that 3 days after MCA occlusion, ATP reduction was most pronounced in somatosensory, temporal, and piriform cortex, as well as in lateral and medial caudate-putamen (Tables 1 and 2). Reductions also were seen in hippocampus, thalamus, and hypothalamus, but these changes did not reach the level of significance.
To evaluate the reproducibility of the regional extent of metabolic alterations, injury incidence maps were prepared by superposition of the areas of reduced ATP and CPS observed in each individual experiment (Fig. 2). These incidence maps revealed a remarkable reproducibility of the metabolic alterations and confirmed the robustness of early CPS inhibition for predicting the final size of brain infarcts.
Genomic expressions
Representative examples of in situ hybridization autoradiograms of the mRNA of hsp70 and the immediate-early genes (IEGs) c-fos, c-jun, and junB are shown in Fig. 1. Transcription of the stress gene hsp70 was mainly confined to the penumbra, whereas transcription of the IEGs c-jun, c-fos, and junB extended into the normal brain tissue of the ipsilateral hemisphere.
The dynamics of gene expression were studied by measuring the optical densities of the in situ hybridization signals in the infarct core, in penumbral cortex, and in the paramedian, normal cortex of the ipsilateral hemisphere (Fig. 4). Expression of hsp70 mRNA sharply increased in the penumbra, reaching the peak at 3 hours after vascular occlusion. Expression of the IEGs increased from 1 to 6 hours after MCA occlusion, both in the penumbra and the periischemic normal cortex, but the relative distributions were different: the concentrations of c-fos and junB mRNA were highest in the nonischemic normal cortex of the ipsilateral hemisphere, whereas c-jun mRNA was increased more in the penumbra. Interestingly, c-jun mRNA expression remained elevated throughout the observation time of 3 days, whereas all of the other mRNA returned to normal during this time.
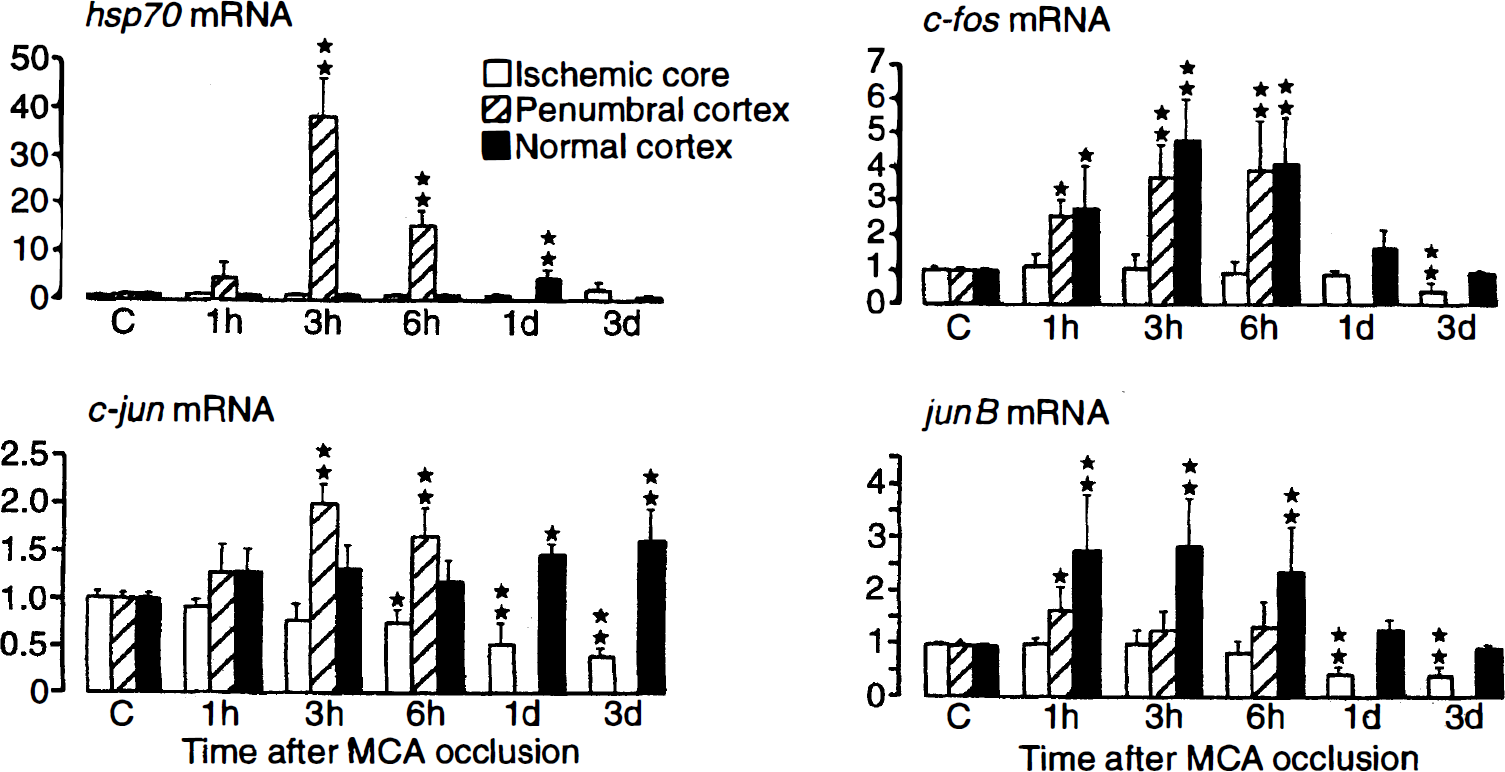
Expression of hsp70, c-fos, c-jun, and junB mRNA in infarct core, penumbra, and normal periinfarct tissue at various times after the onset of permanent middle cerebral artery occlusion. Regions of interest were defined as shown in Fig. 1. Notice differential expression of stress and immediate-early genes in and outside the penumbra.
Terminal transferase biotinylated-UTP nick end labeling
After MCA occlusion, DNA fragmentations, as visualized by TUNEL, were clearly confined to neurons and appeared much later than the changes in ATP and CPS (Fig. 2). The number of TUNEL-positive neurons was highest in the central parts of the ischemic territory, and at no time point could TUNEL be detected in the penumbral regions. On the contrary, TUNEL became prominent at 1 day after MCA occlusion, that is, at a time at which the penumbra had disappeared and brain infarcts had already reached their final size. This excludes any significant contribution of TUNEL-visible DNA fragmentation to infarct expansion.
DISCUSSION
Our investigation is the first detailed pictorial description of the biochemical dynamics of infarct evolution in a murine model of focal brain ischemia. The data provide precise information on the topical relation between core and penumbra, on the time and sequela of penumbral survival, and on the relative contributions of various basic ischemic pathophysiologic pathways to infarct expansion. Using these data, the effects of targeted mutations can be compared easily with the spontaneous evolution of infarcts, allowing the differentiation between disease-related and pathophysiologically irrelevant accompaniments of ischemic injury.
Obviously, the general validity of the data is limited by the model of focal ischemia used here and the selected mouse strain. Ischemia was produced by permanent MCA thread occlusion, which results in large infarcts that affect the entire MCA supplying territory (Hata et al., 1998). Although this technique is the least invasive and the most widely used procedure for MCA occlusion in mice, the results may not be representative for other forms of focal ischemia, notably not for less severe or reversible ischemic impacts. The other limitation is the selection of the C57Black/6 mouse strain. Previous experiments have revealed that the size of infarction after MCA occlusion varies markedly in different mouse strains (Connolly et al., 1996; Maeda et al., 1998). These differences are mainly caused by anatomical variations of the circle of Willis (Fujii et al., 1997) and the angioarchitecture of the MCA supplying territory (Maeda et al., 1998) but variations in molecular ischemic susceptibility also have been described (Schauwecker and Steward, 1997). Strain differences are of importance when transgenic mutants are used because undefined influences from the genetic background of the parent strains may override the influences induced by targeted mutation. The current data can, therefore, be used as reference values only for the C57Black/6J but not for other mouse strains.
Considering these limitations, the main conclusions from our investigation are as follows: (1) brain infarcts produced in mice by permanent MCA occlusion expand and approach their final size by 1 day after occlusion; (2) the final size of infarcts is precisely predicted by the initial suppression of CPS; and (3) DNA fragmentations, as visualized by TUNEL, do not contribute to infarct expansion.
Expansion of infarcts after MCA occlusion has been described before (Hoehn-Berlage et al., 1995; Heiss et al., 1995) and has been associated with the generation of periinfarct spreading depolarizations (Mies et al., 1993a). There are good arguments that this also is true for the current mouse model. Spreading depolarizations evoke a sharp increase in the expression of IEGs with a half-life of about 1 hour (Gass et al., 1992; Hermann et al., 1999). The upregulation of IEGs throughout the time of infarct expansion (i.e. for at least 6 hours) strongly suggests that depolarizations were repeatedly generated during this time.
Depolarizations are thought to increase infarct volume by causing massive activations of the ion exchange pumps (Hossmann, 1996). Since blood flow-and hence oxygen supply-is reduced in the periinfarct surrounding, the increased energy requirements of the ion exchange pumps are not coupled to a parallel increase in oxidative phosphorylation, leading to secondary energy failure. In fact, in rats, the number of spreading depressions correlated linearly with the infarct size (Mies et al., 1993a), and the temporal coincidence of spreading depolarizations with secondary stimulation of anaerobic glycolysis and infarct expansion could be documented by spectroscopical and diffusion-weighted magnetic resonance imaging (Gyngell et al., 1995; Busch et al., 1996). The demonstration of a parallel reduction of the number of spreading depolarizations and infarct size in nitric oxide synthase mutant mice (Shimizu-Sasamata et al., 1998) strongly suggests that such a correlation also exists in the murine model of MCA occlusion.
A remarkable finding of the current study was the precise correspondence of the area of suppressed CPS, visible at 1 hour after vascular occlusion, with the final infarct size observed 3 days later. Moreover, a precise topical correlation existed between the expression of hsp70 mRNA and the penumbra, defined as the region of suppressed CPS but preserved energy state. These findings raise pertinent questions concerning the functional significance of hsp70 mRNA expression on one hand and on the relationship between CPS and infarct expansion on the other.
Penumbral hsp70 expression is a well-known phenomenon, which has been interpreted as a protective mechanism against stress-associated misfoldings of proteins (Kinouchi et al., 1993; Nowak and Jacewicz, 1994). In fact, the Hsp70 protein is a molecular chaperone, and sublethal stress or gene transfer therapy, which increase the expression of this protein, induce tolerance against a later, otherwise fatal ischemic exposure (Chen et al., 1996; Yenari et al., 1998). In the present investigation, hsp70 mRNA expression did not convey such protection: the penumbra underwent delayed cell death although hsp70 mRNA increased, and the more peripheral parts of the MCA supplying territory survived although hsp70 mRNA expression was absent. The failure of Hsp-induced tissue protection probably results from the fact that in the penumbra Hsp70 is transcribed but not translated, as the protein synthesizing machinery is inhibited (Planas et al., 1997). In fact, measurements of both hsp70 mRNA and its protein product revealed that the message was not translated before 24 hours, that is, after the infarct had already expanded to its maximum size (Kinouchi et al., 1993). Synthesis of the Hsp70 protein, if any, would, therefore, come too late to convey neuroprotection.
The pathophysiologically more important alteration for the understanding of infarct expansion is the inhibition of protein synthesis. The precise topical coincidence between this disturbance and the final infarct size suggests that both the metabolic disturbance and the manifestation of ischemic injury, are linked to the same pathophysiologic mechanism. It is unlikely that the inhibition of de novo protein synthesis leads to cell death per se because most of the penumbral tissue infarcts within 6 hours, whereas pharmacologic or hypothermic inhibition of protein synthesis can be survived for much longer times (Lobner and Choi, 1996; Frerichs et al., 1998). However, it is conceivable that the hypoxic episodes induced by the passage of spreading depressions result first in the disturbance of protein synthesis and, after several repetitions, in irreversible breakdown of energy metabolism. This interpretation is supported by the observation that inhibition of the propagation of periinfarct depolarizations by glutomate antagonists prevents penumbral CPS inhibition (Kohno et al., 1994; Mies et al., 1993b). The alternative interpretation that the depolarization-induced ion shifts are responsible for the metabolic disturbance can be refuted because protein synthesis was not impaired peripherally to the penumbra, although periinfarct depolarizations spread over the whole hemisphere. Inhibition of protein synthesis thus marks the region in which periinfarct depolarizations provoke relative tissue hypoxia which, in turn, would be the ultimate cause of infarct expansion.
Hypoxia also could explain the unexpected observation that DNA fragmentation could not be associated with infarct expansion at any of the investigated time points. This finding clearly is at variance with several previous investigations that report on TUNEL-positive neurons in the transition zone between infarct and normal brain tissue (Guegan et al., 1996; Linnik et al., 1995). It is also difficult to reconcile with the previously reported downregulation of apoptosis-preventing and upregulation of apoptosis-promoting genes (Isenmann et al., 1998; Matsushita et al., 1998), or the therapeutic efficiency of drugs that specifically inhibit the activity of apoptosis-associated enzymes, such as interleukin 1-beta converting enzyme or caspase-3 (Loddick et al., 1996; Hara et al., 1997). On the other hand, focal ischemia induces not only apoptosis-promoting but also apoptosis-inhibiting genes (Gillardon et al., 1999), and the morphologic features of injured neurons in the periphery of the infarct are different from those undergoing classic apoptosis (Campagne and Gill, 1996). There are indications that apoptosis may be of greater relevance for delayed injury after transient focal ischemia (Du et al., 1996; Fink et al., 1998; Namura et al., 1998) than for infarct expansion after permanent vascular occlusion. However, it is also conceivable that the TUNEL-detectable DNA fragmentations are late events in a multifactorial apoptosis-promoting pathway, which follow rather than precede the here-described breakdown of energy state (Petito et al., 1997). A possible scenario for this pathophysiologic pathway could be the activation by DNA single-strand breaks of poly(ADP-ribose)polymerase, which may result in NAD depletion and energy failure before DNA double-strand fragmentations become visible by TUNEL (Endres et al., 1997; Eliasson et al., 1997).
Finally, we demonstrated differential expression of IEGs such as c-fos, c-jun, and junB mRNA after MCA occlusion. Correlation analysis of IEG expression and brain metabolism reveals that c-jun mRNA is expressed mainly in the penumbra, junB mRNA is expressed mainly in the periinfarct area with normal metabolism, and c-fos mRNA is expressed in both areas. The reason why IEGs are expressed differentially remains unknown. Induction of IEGs have been attributed to the transient increase in cytosolic calcium evoked by the passage of periinfarct spreading depression. A possible explanation for the differential expression is the difference of promotor construction, which mediates IEG induction after calcium influx (Ginty, 1997). In fact, c-fos possesses one serum-responsive element (SRE) and three cAMP-responsive elements (CREs), c-jun possesses one SRE and no CRE, and junB possesses two SREs and two CREs (Herdegen and Leah, 1998). It also has been reported that in mutant mice that lack A and D isoforms of CRE binding protein, c-fos mRNA expression was significantly attenuated in the periinfarct surrounding (Hata et al., 1998a). These findings and their relevance for infarct evolution are only partly understood and require further investigation.
In conclusion, our study demonstrates that permanent MCA occlusion in mice produces a gradually expanding infarct, the final size of which is heralded by the initial inhibition of protein synthesis. The pictorial biochemical-genomic analysis of the dynamics of spontaneous infarct evolution provides detailed data that can be used in the future to evaluate the pathophysiologic importance of gene disruption or pharmacologic interventions. Such experiments will become of increasing interest for the dissection of complex interactions leading to brain infarction.
Footnotes
Abbreviations used
Acknowledgements
The authors thank Mrs. U. Beckmann, Mrs. U. Gillert, and Mrs. P. Lorenz-Korenkov for excellent technical assistance, and Mrs. D. Schewetzky for the careful preparation of the manuscript.