
Abstract
Shutdown of translation is a highly conserved response of cells to a severe form of metabolic, thermal, or physical stress. After the metabolic stress induced by transient cerebral ischemia, translational recovery is observed only in cells that withstand the transient interruption of blood supply, implying that restoration of translation critically determines the final outcome. On the other hand, apoptosis is believed to play a role in ischemia-induced cell death. Apoptosis is an active process that is blocked by agents known to suppress protein synthesis. Thus, the question arises whether stress-induced suppression of protein synthesis is protective or toxic for the affected cells. Accepting the notion that endoplasmic reticulum (ER) dysfunction is the mechanism underlying shutdown of translation after transient cerebral ischemia, an attempt may be made to try to solve the protein synthesis paradox by understanding the role of protein synthesis suppression in conditions associated with ER dysfunction. Endoplasmic reticulum dysfunction-induced accumulation of unfolded proteins in the ER lumen is the trigger of two signal transduction pathways: PKR-like ER kinase–induced shutdown of translation to suppress new synthesis of proteins that cannot be correctly folded, and IRE1-induced expression of ER stress genes, a protein synthesis–dependent pathway needed to restore ER functions. Together these comprise the unfolded protein response. They are also induced after transient ischemia, implying a dual effect of protein synthesis suppression, a protective and a pathologic effect during early and prolonged reperfusion.
Keywords
Shutdown of translation is a common response of cells to a severe form of stress (Fig. 1). This stress response is highly conserved from yeast to mammalian cells (Ron and Harding, 2000; Schneider, 2000). It is the initiation step of protein synthesis that is affected. The signal transduction cascade resulting in stress-induced shutdown of translation is initiated by activation of one of the kinases that specifically phosphorylates the alpha subunit of the initiation factor 2 (eIF2α). Phosphorylation of eIF2α leads to eIF2 becoming an inhibitor of eIF2B. The consequent reduction in levels of active eIF2-GTP results in impairment of the initiation process of protein synthesis (Hinnebusch, 2000). This stress response has been investigated in detail in various models of transient cerebral ischemia (Kleihues et al., 1975; Cooper et al., 1977; Bodsch et al., 1985; Thilmann et al., 1986; Burda et al., 1994; DeGracia et al., 1996; Althausen et al., 2001; Mengesdorf et al., 2002a). In the following, transient cerebral ischemia will be considered as a pathologic state of considerable clinical relevance to elucidate the possible significance of shutdown of translation for the final outcome of cells exposed to a severe form of stress.
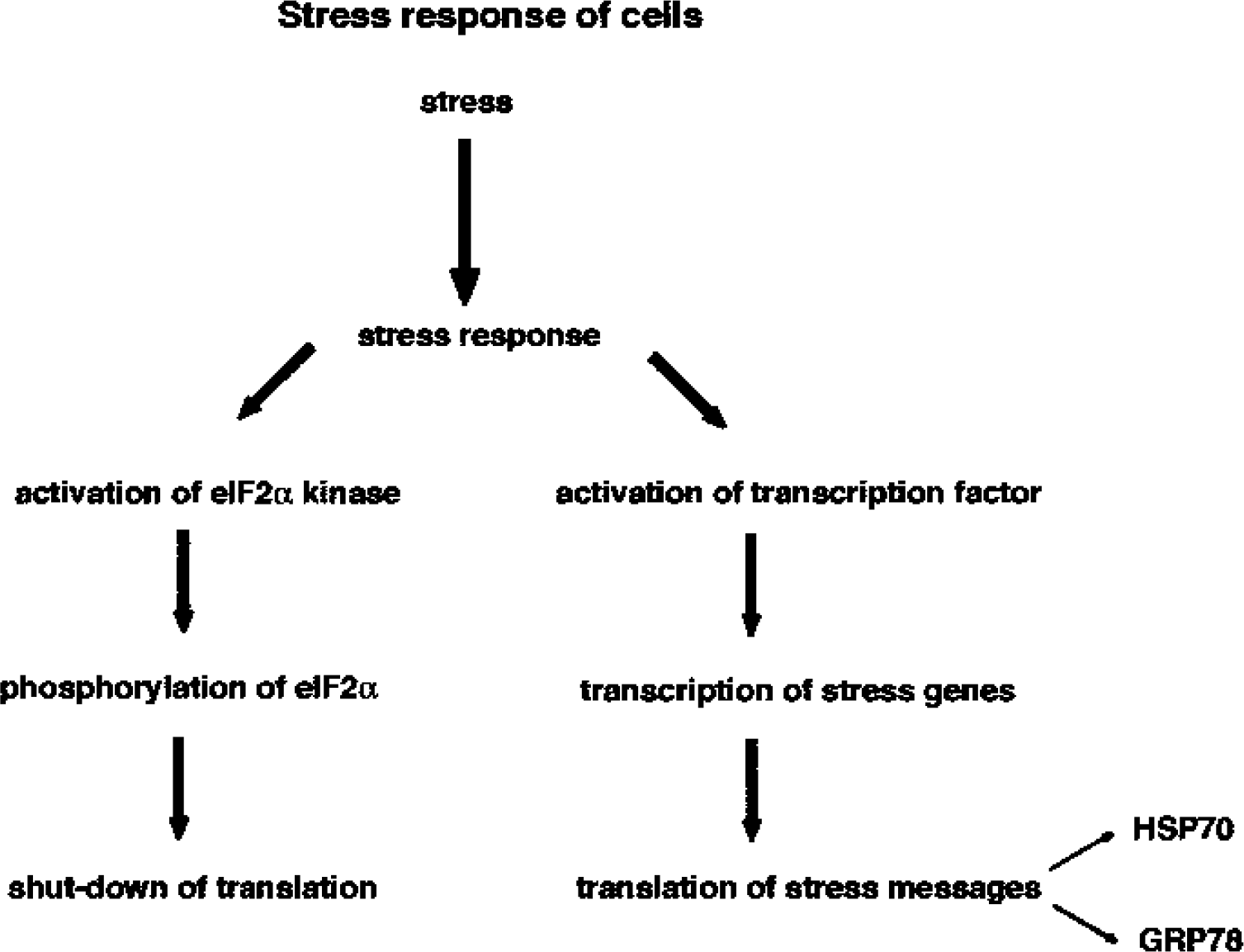
Schematic diagram of the response of cells to a severe form of stress. In cells exposed to a severe form of stress, a highly conserved stress response is triggered. This includes activation of a kinase that specifically phosphorylates the alpha subunit of the eukaryotic initiation factor (eIF2α), resulting in the shutdown of translation. Furthermore, a transcription factor is activated that induces specifically transcription of stress genes. The stress message is then translated into the respective protein. Depending on which subcellular compartment is affected, the genetic response results in the upregulation of protein levels of heat-shock proteins such as HSP70 or glucose-regulated proteins such as GRP78 under conditions associated with impairment of cytoplasmic or endoplasmic reticulum functions, respectively.
After transient cerebral ischemia, protein synthesis is severely suppressed in all cells within brain regions where the blood supply has been critically reduced below the threshold necessary to cover the energy demand of cells (Bodsch et al., 1985; Thilmann et al., 1986). This is a common response of cells to the severe form of stress triggered by a breakdown of energy-producing metabolism after interruption of blood supply. This stress response is elicited in all affected cells, both resistant and vulnerable, irrespective of the final outcome (Bodsch et al., 1985; Thilmann et al., 1986). What distinguishes resistant cells from vulnerable ones is the capability to restore normal rates of protein synthesis. Protein synthesis never recovers in neurons vulnerable to even short periods of ischemia (Bodsch et al., 1985; Thilmann et al., 1986), implying that the inability of vulnerable neurons to reactivate the protein synthesis machinery is associated with induction of cell death. This view is corroborated by the observation that an isolated blocking of protein synthesis is lethal to cells. Shutdown of translation triggered by overactivation of a eIF2α kinase or mutation of eIF2α (S51D), which mimics phosphorylated eIF2α, is sufficient to cause cell death (Srivastava et al., 1998).
Apoptosis is believed to contribute to neuronal cell death induced by transient cerebral ischemia (for recent reviews see MacManus and Buchan, 2000; Graham and Chen, 2001; Barber et al., 2001; Loetscher et al., 2001; Leker and Shohami, 2002). Typical features of apoptotic cell death have indeed been found after transient ischemia, including degradation of DNA resulting in the typical latter pattern on electrophoresis gels, cytochrome c release from mitochondria, and released cytochrome c–triggered activation of caspases. Apoptosis is a programmed form of cell death that depends on protein synthesis. The ability of cycloheximide or other agents that are known to interfere with protein synthesis to block cell death under experimental conditions is even taken as an indicator that apoptotic mechanisms are involved. Assuming that apoptosis does play a role in neuronal cell death triggered by transient cerebral ischemia, ischemia-induced shutdown of translation may be viewed as a protective response of cells serving to block the genetic program that finally results in cell death.
One way to elucidate the significance of an ischemia-induced shutdown of translation for the affected cells, and to establish whether it contributes to neuronal cell death or protects cells from injury, is to focus on the underlying mechanisms and to understand the role of this process. As will be discussed herein, there is strong evidence that endoplasmic reticulum (ER) dysfunction is involved in the suppression of protein synthesis induced by transient cerebral ischemia. We now need to know why cells exposed to conditions associated with ER dysfunction respond to this pathologic process with a shutdown of translation and whether cells need to react in such a way as to withstand periods of impaired ER function.
ENDOPLASMIC RETICULUM DYSFUNCTION IS RESPONSIBLE FOR ISCHEMIA-INDUCED SHUTDOWN OF TRANSLATION
Transient cerebral ischemia triggers a shutdown of translation at the initiation step, as indicated by the phosphorylation of eIF2α and disaggregation of polyribosomes (Kleihues et al., 1975; Cooper et al., 1977; Burda et al., 1994; DeGracia et al., 1996; Althausen et al., 2001). This is a specific pattern that is also induced in neurons under conditions associated with ER dysfunction (Doutheil et al., 1997; Mengesdorf et al., 2001). We have therefore put forward the hypothesis that transient cerebral ischemia induces ER dysfunction (Paschen, 1996; Paschen and Doutheil, 1999; Paschen and Frandsen, 2001). Four different kinases have been identified that specifically phosphorylate eIF2α, resulting in a shutdown of translation. These are (1) the double-stranded RNA-dependent protein kinase PKR, which is activated after viral infection of cells and also under conditions associated with ER dysfunction (Korth and Katze, 2000; Prostko et al., 1995; Srivastava et al., 1995); (2) the PKR-like ER kinase (PERK) that is specifically activated when ER function is impaired (Harding et al., 1999); (3) the amino-acid regulated kinase GCN2; and (4) the heme-regulated kinase present in erythroid cells. PKR-like ER kinase is the only eIF2α kinase that has been found to be activated after cerebral ischemia (Kumar et al., 2001), indicating that ER dysfunction is indeed responsible for the shutdown of translation induced by transient ischemia. The assumption that transient cerebral ischemia causes impairment of ER function is also supported by the observation that ischemia induces an increase in mRNA levels of genes including grp78, grp94, erp72, gadd153, gadd34, and heme oxygenase-1, the expression of which is specifically activated under conditions associated with ER dysfunction (Wang et al., 1993; Higashi et al., 1994; Lowenstein et al., 1994; Paschen et al., 1994; Gissel et al., 1997; Linden et al., 1998; Paschen et al., 1998a, b ; Doutheil et al., 1999).
The ER is a subcellular compartment playing a pivotal role in important cellular functions. Besides its role in calcium storage and signaling (Garaschuk et al., 1997; Berridge, 1998), the ER lumen is the compartment where the folding and processing of all membrane and secretory proteins takes place. These are strictly calcium-dependent processes that need high calcium activity for correct functioning (Lodish and Kong, 1990; Gosh et al., 1991; Kuznetsov et al., 1992; Lodish et al., 1992). Endoplasmic reticulum calcium activity is therefore several orders of magnitude above that of the cytoplasm (Hofer and Machen, 1993; Chen et al., 1996; Pozzomiller et al., 1997). The need for high ER luminal calcium activity is indicated by the fact that depletion of ER calcium stores is a severe form of stress sufficient to cause cell death (for a comprehensive discussion see Paschen and Doutheil, 1999). Besides a high calcium activity, an oxidative environment appears to be required for the folding of proteins, because these reactions are blocked by compounds with high reducing activity (Kuznetsov et al., 1992). Under conditions associated with ER dysfunction, when the process of protein folding and processing is impaired, unfolded proteins accumulate in the ER lumen. This is the warning signal that activates the unfolded protein response (UPR) (for review see Kaufman, 1999).
UNFOLDED PROTEIN RESPONSE
Activation of two ER-resident kinases is required to trigger UPR, the PERK, and IRE1 (Fig. 2). In the physiologic state, activity of PERK and IRE1 is blocked by the 78-kDa glucose-regulated protein (GRP78) binding to both proteins. GRP78 is a chaperone required for the folding of unfolded or misfolded proteins in the ER lumen. Under conditions associated with ER dysfunction, when unfolded proteins accumulate in the ER, GRP78 is required to bind to and help to refold these unfolded proteins. Under these conditions, GRP78 dissociates from PERK and IRE1, resulting in oligomerization and autophosphorylation of both kinases. The main function of UPR is to suppress further synthesis of proteins that cannot be correctly folded by a global shutdown of translation and to activate specifically the expression of genes coding for ER stress proteins. This genetic response is needed to restore GRP78 protein levels sufficiently to refold the unfolded proteins accumulated in the ER lumen, to bind to and hence block PERK and IRE1 activity, and thus to terminate UPR (Kaufman, 1999).
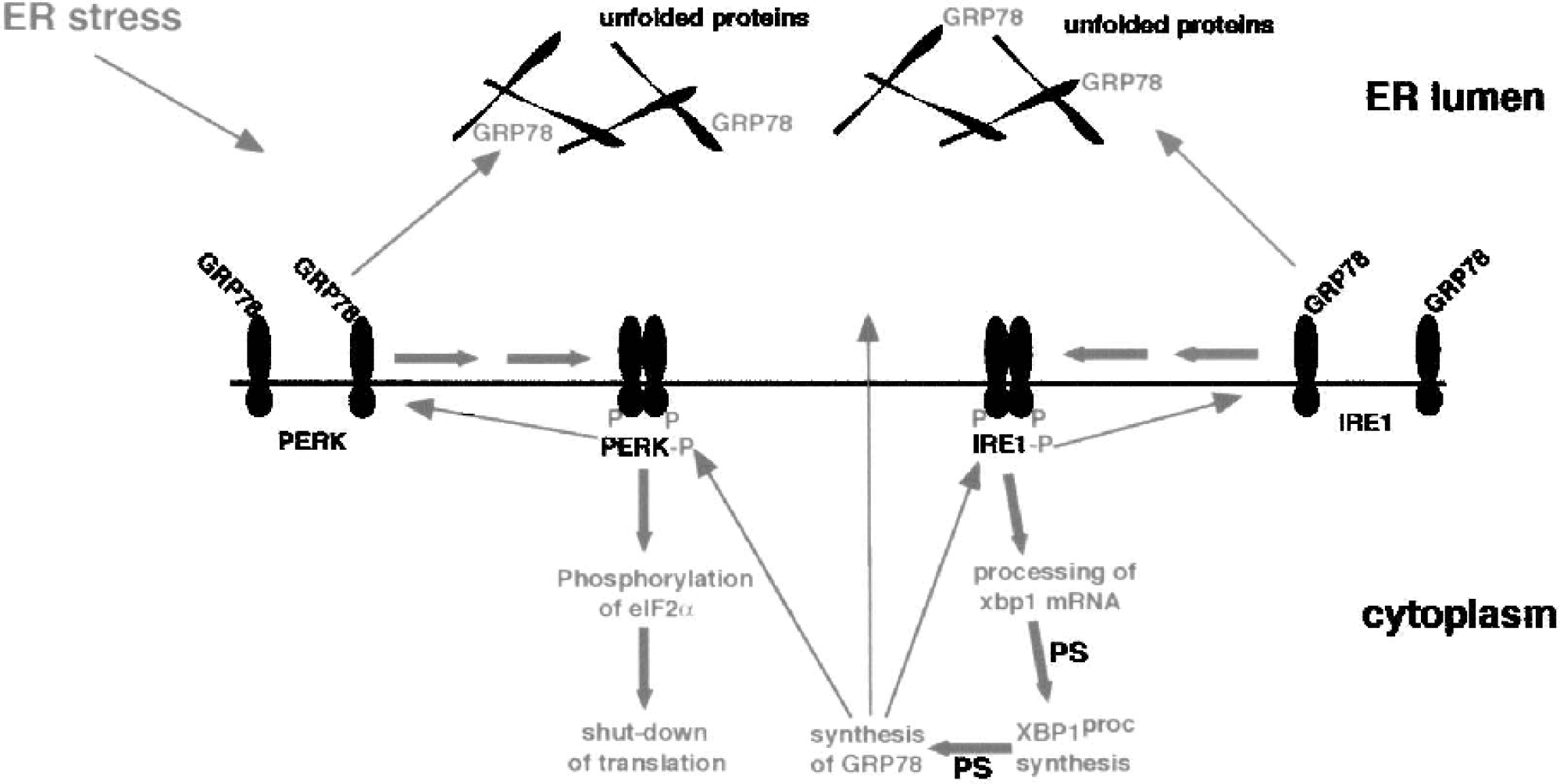
Schematic diagram of activation of the unfolded protein response. In the physiologic state, GRP78 protein is bound to PKR-like ER kinase (PERK) and IRE1. Under conditions associated with ER dysfunction when the processes of protein folding and processing are blocked, unfolded proteins accumulate in the endoplasmic reticulum (ER) lumen. GRP78 protein dissociates from PERK and IRE1 and binds to unfolded proteins to fascilitate the folding reaction. Dissociation of GRP78 from PERK and IRE1 induces oligimerization, phosphorylation, and thus activation. Activated PERK (PERK-P) triggers a shutdown of translation by phosphorylating the eukaryotic initiation factor alpha subunit of the initiation factor 2 (eIF2α), while IRE1 in its activated form IRE1-P cuts out a sequence of 26 bases from xbp1 mRNA. Processed xbp1 mRNA is translated into the processed XBP1 protein (XBP1proc), a highly active transcription factor specifically responsible for the expression of ER stress genes, including grp78. After activation of transcription and translation of grp78, newly synthesized GRP78 protein binds to and inactivates PERK-P and IRE1-P, and helps to fold unfolded proteins accumulated in the lumen of the ER. In the course of the IRE1-P-mediated signal transduction pathway, protein synthesis (PS) is required at two steps: for the translation of processed xbp1 mRNA into the respective protein, and for the translation of grp78 mRNA into the GRP78 protein. Reactions activated under conditions associated with ER dysfunction are indicated in pink, while the recovery process induced by newly synthesized GRP78 protein is indicated in green.
In the course of UPR, PERK must be activated to stop the further accumulation of unfolded proteins that tend to form potentially toxic aggregates. Because, after being activated, PERK phosphorylates eIF2α resulting in suppression of protein synthesis at the initiation step (Fig. 1), we can assume that the ischemia-induced shutdown of translation triggered by activation of PERK is a neuroprotective response serving to protect cells from the accumulation of unfolded proteins. The view that activated PERK-induced suppression of protein synthesis is a protective response is corroborated by the observation that cells in which the gene coding for PERK has been mutated are particularly sensitive to conditions associated with ER dysfunction (Harding et al., 2000). Furthermore, the translational control provided by PERK is also necessary for normal cell functioning even without induction of a severe form of ER stress. This control is particularly important in secretory cells such as the pancreatic β-cells. Pancreatic β-cells have a high rate of synthesis of secretory proteins and therefore require an ER with a high folding and processing capacity. In PERK−/− mice, increased death of β-cells is observed postnatally, and these animals develop progressive diabetes mellitus (Harding et al., 2001; Harding and Ron, 2002).
IRE1-mediated activation of the expression of genes coding for ER stress proteins is a complex process that requires protein synthesis at two steps (Fig. 1). After being activated under conditions associated with ER stress, IRE1 is turned into an active endonuclease that specifically cuts out a sequence of 26 bases from the coding region of xbp1 mRNA (Shen et al., 2001; Yoshida et al., 2001; Calfon et al., 2002). This mRNA processing reaction causes a frame-shift of the coding region, and processed xbp1 mRNA is translated into a new protein. In the physiologic state, xbp1 mRNA is translated into a 33-kDa XBP1 protein, levels of which are low in the brain (Paschen et al., 2003). Processed xbp1 mRNA is translated into a 54-kDa protein (XBP1proc) that is much more stable than the 33-kDa protein species. XBP1proc functions as an active transcription factor specific for the expression of genes coding for ER stress proteins, including GRP78.
UNFOLDED PROTEIN RESPONSE IS INDUCED BY TRANSIENT CEREBRAL ISCHEMIA
PKR-like ER kinase is the only eIF2α kinase that has been found to be activated after cerebral ischemia (Kumar et al., 2001), indicating that ER dysfunction is responsible for the shutdown of translation induced by transient ischemia. We have shown recently that processing of xbp1 mRNA is markedly activated after transient global and transient focal cerebral ischemia (Paschen et al., 2003), implying that ischemia also activates the IRE1-mediated signal transduction pathway (Fig. 3a). However, we found no increase but a transient decrease of grp94 mRNA levels (Paschen et al., 2003) and only a very small postischemic increase in grp78 mRNA levels in brain samples in which processed xbp1 mRNA levels had risen about 40-fold (Fig. 3b). This suggests that ischemia-induced shutdown of translation was so severe that it hindered new synthesis of XBP1proc protein. In fact, we found no increase in XBP1proc protein levels during the first 3 h of reperfusion after transient focal cerebral ischemia, i.e., throughout a recirculation period when processing of xbp1 mRNA was maximally activated (Paschen et al., 2003).
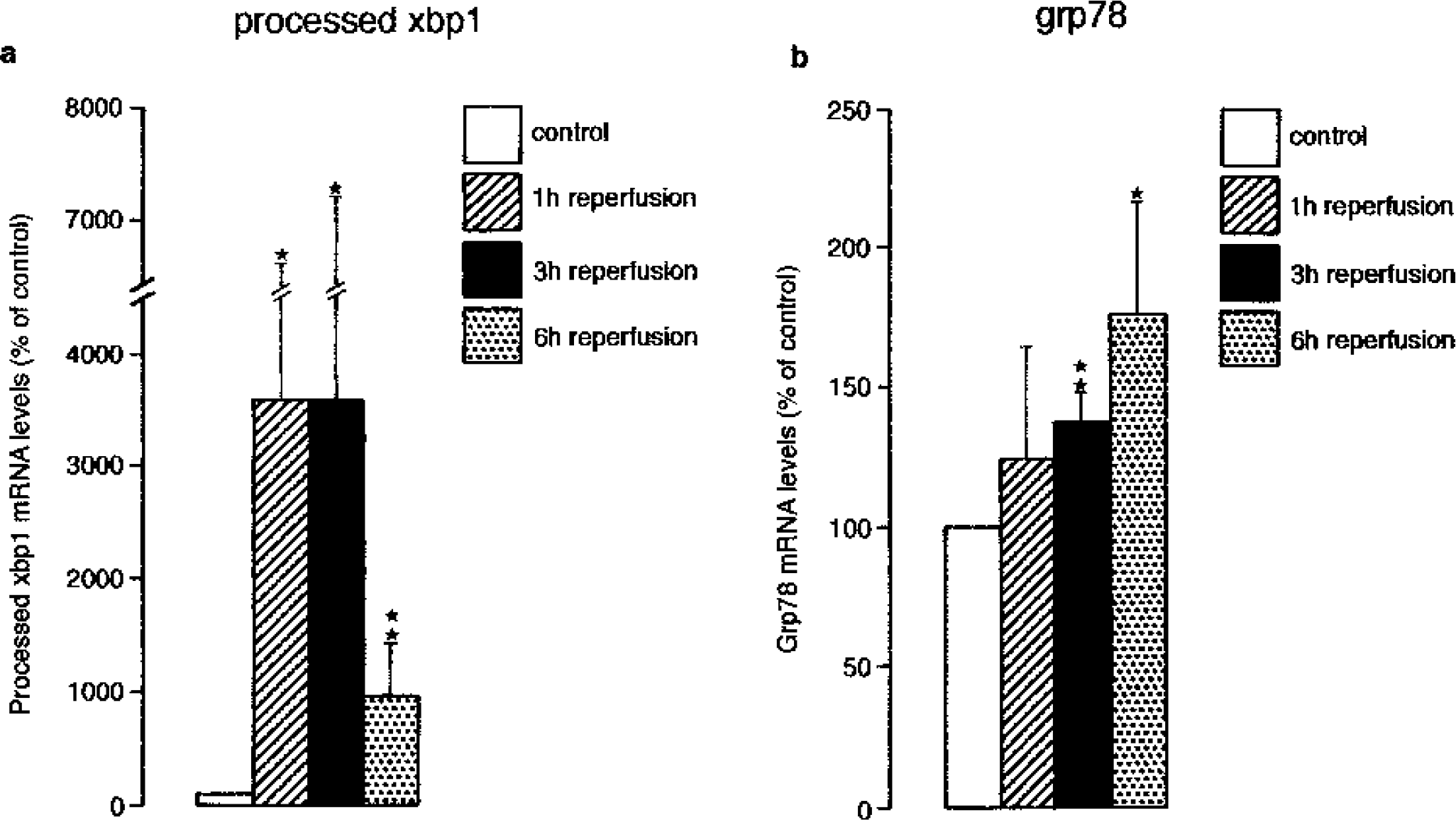
The IRE1-mediated signal transduction pathway is activated after transient focal cerebral ischemia. Mice were subjected to transient focal cerebral ischemia by unilateral occlusion of the right middle cerebral artery (MCA) using the thread occlusion technique. After 1-h MCA occlusion, brains were reperfused for 1 h to 6 h. Tissue samples were excised from the right and left hemispheres from the territory supplied by the MCA. Total RNA was isolated and reverse-transcribed into cDNA. Ischemia-induced changes in processed xbp1 and grp78 mRNA levels were evaluated by quantitative polymerase chain reaction. Transient focal cerebral ischemia induced a marked increase in processed xbp1 mRNA
In addition to signs of PERK and IRE1 activation induced by transient cerebral ischemia, protein aggregates, the pathologic consequences of disturbed ER functioning, have been observed after transient cerebral ischemia (Hu et al., 2000, 2001). Protein aggregates were found attached to the ER membrane (Hu et al., 2002), implying that they originated from unfolded proteins accumulated in the ER lumen. Formation of potentially toxic protein aggregates is indicative of an excessive formation of unfolded proteins. If the assumption is valid that protein aggregates accumulating after transient cerebral ischemia result from ischemia-induced impairment of ER functioning, insufficient resynthesis of the GRP78 protein required to facilitate the folding process may be seen as the main obstacle to recovery after transient cerebral ischemia. In other words, recovery from ischemia-induced ER dysfunction is blocked in vulnerable neurons by the inability of cells to restore protein synthesis. Preconditioning may directly affect this process. After ischemic preconditioning, suppression of protein synthesis induced by the second period of ischemia was almost absent in the cortex and of considerably shorter duration in the vulnerable hippocampal CA1 subfield (Kato et al., 1995), implying that preconditioning may upregulate GRP78 protein levels far enough for cells to withstand ER dysfunction. This assumption needs to be validated in future experiments.
SYNTHESIS
Transient cerebral ischemia causes impairment of ER functioning, which in turn triggers shutdown of translation. Endoplasmic reticulum dysfunction results in accumulation of unfolded proteins that tend to form toxic aggregates. To withstand this pathologic process, affected cells need to activate the entire UPR comprising PERK-induced shutdown of translation and the protein synthesis–dependent IRE1-mediated pathway that triggers expression of genes coding for ER stress proteins, including GRP78. Thus, the PERK- and IRE1-controlled signal transduction pathways both need to be activated to restore normal ER functioning. Activation of PERK is necessary to block pathologic processes triggered in the course of ER dysfunction, whereas activation of IRE1 is required to restore normal ER functioning. A more severe suppression of translation, brought about by pharmacologic intervention designed to interfere with apoptotic mechanisms, would reinforce the action of activated PERK by more pronounced blocking of further synthesis of proteins that cannot be folded. It would also suppress potentially toxic processes started in the course of programmed cell death that are dependent on protein synthesis. On the other hand, after transient cerebral ischemia, translation is already so heavily suppressed in vulnerable neurons that new synthesis of XBP1proc and GRP78 protein is blocked and restoration of normal ER functioning is hindered. It is therefore unlikely that further pharmacologic suppression of protein synthesis would help to prevent the process culminating in neuronal cell death in those pathologic states of the brain where ER dysfunction plays a central role. In fact, the protein synthesis machinery needs to be reactivated far enough to permits new synthesis of neuroprotective proteins including XBP1proc and GRP78 that are required to restore normal ER functioning. If this restoration process is blocked, cells are trapped in a self-activating pathologic process (Fig. 4). Protein aggregates are formed (Hu et al., 2001) that block proteasomal function (Bence et al., 2001). The ubiquitin/proteasomal pathway has indeed been found to be impaired after transient cerebral ischemia (Keller et al., 2000; Hu et al., 2001; Asai et al., 2002; Mengesdorf et al., 2002b), and impairment of proteasomal functioning causes further ER stress in neurons (Mengesdorf et al., 2002b; Paschen et al., 2003). The same scenario most probably takes place in degenerative diseases that, like transient cerebral ischemia, are characterized by dysfunctioning of the ER and the ubiqutin/proteasomal pathway and by the formation of toxic protein aggregates (Chung et al., 2001) (Fig. 4).
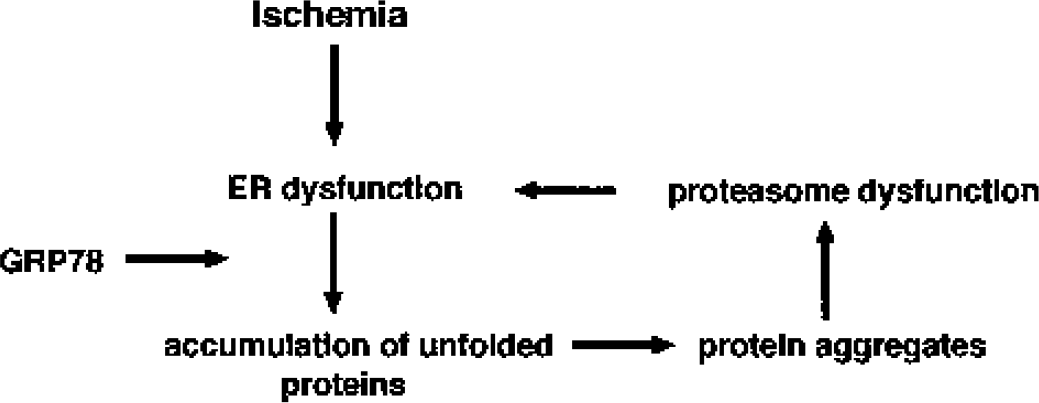
Schematic diagram of the self-aggravating pathologic process in which vulnerable neurons may be trapped after transient cerebral ischemia. Ischemia induces endoplasmic reticulum (ER) dysfunction, resulting in accumulation of unfolded proteins in the ER lumen. When the unfolded protein response cannot be activated to provide new GRP78 protein sufficient to fold these unfolded proteins, potentially toxic protein aggregates may be formed. Protein aggregates block proteasomal function, and impaired proteasomal function causes further ER stress. Cells can escape this pathologic cycle only when the protein synthesis machinery is sufficiently reactivated for the synthesis of enough new GRP78 protein to fold the unfolded proteins and thus to permit normal ER functioning.