
Abstract
The evolution of brain infarction after transient focal cerebral ischemia was studied in mice using multiparametric imaging techniques. One-hour focal cerebral ischemia was induced by occluding the middle cerebral artery using the intraluminal filament technique. Cerebral protein synthesis (CPS) and the regional tissue content of adenosine triphosphate (ATP) were measured after recirculation times from 0 hours to 3 days. The observed changes were correlated with the expression of the mRNAs of hsp-70, c-fos, and junB, as well as the distribution of DNA double-strand breaks, visualized by TUNEL. At the end of 1 hour of ischemia, protein synthesis was suppressed in a larger tissue volume than ATP in accordance with the biochemical differentiation between core and penumbra. Hsp70 mRNA was selectively expressed in the cortical penumbra, whereas c-fos and junB mRNAs were increased both in the lateral part of the penumbra and in the ipsilateral cingulate cortex with normal metabolism. During reperfusion after withdrawal of the intraluminal filament, suppression of CPS persisted except in the most peripheral parts of the middle cerebral artery territory, in which it recovered between 6 hours and 3 days. ATP, in contrast, returned to normal levels within 1 hour but secondarily deteriorated from 3 hours on until, between 1 and 3 days, the ATP-depleted area merged with that of suppressed protein synthesis leading to delayed brain infarction. Hsp70 mRNA, but not c-fos and junB, was strongly expressed during reperfusion, peaking at 3 hours after reperfusion. TUNEL-positive cells were detected from 3 hours on, mainly in areas with secondary ATP depletion. These results stress the importance of an early recovery of CPS for the prevention of ischemic injury and suggest that TUNEL is an unspecific response of delayed brain infarction.
Keywords
In small laboratory animals, occlusion of the middle cerebral artery (MCA) results in cerebral infarction even if the occlusion is reversed as early as 30 minutes after the onset of ischemia (Du et al., 1996). It is widely assumed that injury after transient ischemia is mediated by mechanisms similar to those during permanent vascular occlusion, but this assumption is not self-evident. Permanent ischemia causes a threshold-dependent injury, with primary necrosis at flow rates below the value required for maintaining energy metabolism. Transient ischemia, in contrast, produces only temporary suppression of energy state, provided that the duration of ischemia does not exceed the survival time of the brain. A certain pathophysiologic analogy is conceivable only as regards the periinfarct penumbra, in which energy state is initially preserved until complicating side effects provoke slowly progressive tissue injury. It is therefore surprising that many therapeutic interventions have been documented to improve tissue damage after both transient and permanent brain ischemia (Balkan et al., 1997; Eliasson et al., 1997; Endres et al., 1997; Tokime et al., 1998; Zhang et al., 1995). Also, various genemanipulated mutants in which putative molecular pathways of ischemic injury are knocked out or overexpressed respond in a similar way to both forms of brain ischemia. This raises the question of how far similar biochemical pathways are involved in permanent and transient ischemia, despite the differences in hemodynamic pathophysiology.
A straightforward way to investigate this question is to use multiparametric imaging techniques to evaluate the temporal and regional evolution of cell injury. This ethod includes bioluminescence imaging of ATP to detect energy failure, amino acid autoradiography to image protein synthesis, TUNEL histochemistry to detect DNA fragmentations, and in situ hybridization autoradiograms to image specific genomic expression patterns. In a previous investigation, we used this approach to study the evolution of tissue injury after permanent MCA occlusion in mice, and we observed that the final infarct size is heralded by an early inhibition of protein synthesis but not by any of the other investigated parameters (Hata et al., 2000). This led us to conclude that the mechanisms leading to expansion of brain infarcts are closely associated with metabolic disturbances that are caused by or associated with inhibition of protein synthesis. In the present study, we applied a similar approach to investigate the dynamics of tissue injury after transient MCA occlusion in mice.
METHODS
Experiments were carried out according to the National Institutes of Health guidelines for the care and use of laboratory animals and were approved by the local authorities. Animals were housed under diurnal lighting conditions and allowed access to food and water ad libitum until the day of the experiment. Anesthesia was induced by 1.5% halothane and maintained with 1% halothane in 70% N2O and 30% O2.
Animal preparation
A total of 56 adult male C57Black/6J mice weighing 20 to 25 g were used in this study. One group of animals (n = 21) was used to measure physiologic variables, the other group (n = 35) to assess the dynamics of biochemical alterations. The latter group was divided into six subgroups (n = 5 each), subjected to focal cerebral ischemia for 1 hour, and either killed without reperfusion or reperfused for 1, 3, or 6 hours or 1 or 3 days. In both the 1- and 3-day survival groups, one mouse did not survive until the end of the experiments, reducing the number of animals in these groups to four. Twelve sham-operated animals were used as controls, seven for measuring physiologic variables and five for the biochemical investigation.
Focal cerebral ischemia was produced by occluding the MCA using the intraluminal filament technique (Hata et al., 1998a). After a midline neck incision was made, the left common and external carotid arteries were isolated and ligated. A microvascular clip (FE691; Aesculap, Tuttlingen, Germany) was temporarily placed on the internal carotid artery. An 8–0 nylon monofilament (Ethilon; Ethicon, Norderstedt, Germany) coated with silicon resin (Xantopren; Bayer Dental, Osaka, Japan) was introduced through a small incision into the common carotid artery and advanced 9 mm distal to the carotid bifurcation for occlusion of the MCA. The tip diameter of the thread (0.15 to 0.20 mm) was selected to match the body weight of the animals. One hour after occlusion, the thread was withdrawn to allow reperfusion of the MCA territory. Animals selected for reperfusion times of more than 6 hours were returned to their cages after thread retraction and treated with an intraperitoneal injection of saline with glucose (8%) to keep proper water and glucose balances (0.2 mL, twice a day). Sham operation was performed by inserting the thread into the common carotid artery without advancing it to occlude the MCA.
Forty-five minutes before the animals were killed, L-[4,5–3H]-leucine (150 μCi/animal, specific activity 151 Ci/mmol; Amersham, Braunschweig, Germany) was administered intraperitoneally to evaluate cerebral protein synthesis (CPS) rates. After the predetermined reperfusion periods, experiments were terminated by in situ freezing (Mies et al., 1991a). Brains were removed in a cold-temperature cabinet (−20°C) and cut into 20-μm-thick coronal cryostat sections at −20°C. Sections were mounted on coverslips for ATP-bioluminescence, on object holders for CPS autoradiography, and on 3-aminopropyl triethoxysilane-treated slides for in situ hybridization (see below).
Regional measurement of ATP and protein synthesis
Pictorial measurements of ATP were carried out using ATP-specific bioluminescence (Kogure and Alonso, 1978). For CPS measurement, brain slices were incubated in 10% trichloroacetic acid to remove labeled free leucine and metabolites other than proteins. Subsequently, slices were exposed for 14 days with 3H standards to tritium-sensitive x-ray film (Hyperfilm 3H; Amersham) for autoradiography of 3H-labeled proteins (Mies et al., 1991b).
Probes for mRNAs
The probe sequences of c-fos (45 mer), junB (45 mer), and hsp70 (30 mer) corresponded to the mouse c–fos gene (bases 290 to 334; Accession No. J00370), the mouse junB gene (bases 1307 to 1351; Accession No. J03236), and the mouse inducible hsp70 gene (bases 1401 to 1430; Accession No. M76613), respectively. Each probe was 3′-end-labeled using terminal deoxynucleotidyl transferase (Gibco BRL, Eggenstein, Germany) and a 30:1 molar ratio of [35S]dATP (1,200 Ci/mmol). Specific activity was greater than 0.5 × 109 dpm/μg.
In situ hybridization
In situ hybridization was performed as described previously (Hata et al., 1998b). Briefly, coronal brain sections (20 μm) were fixed for 15 minutes in 4% paraformaldehyde/phosphate-buffered saline (PBS), pH 7.4. After treatment for 10 minutes with 0.25% acetic anhydride/triethanolamine, sections were dehydrated and incubated in 10 μL hybridization buffer containing 35S-labeled oligonucleotide probe (10 pg/μL), 2 × saline sodium citrate (SSC), 50% formamide, 10% dextran sulfate, 100 μg/mL poly(A), 120 μg/mL heparin, 1 mg/mL herring sperm DNA, 5 mmol/L dithiothreitol, and 1 mg/mL bovine serum albumin, and covered with a coverslip. After overnight hybridization at 42°C, sections were washed twice at 42°C in 2 × SSC/50% formamide for 30 minutes and exposed together with 14C standards to x-ray film (Hyperfilm β-max; Amersham).
Morphometric analysis of ischemia-induced metabolic disturbance and gene expression
Bioluminescence and autoradiographic images were digitized with a CCD camera system and analyzed using National Institutes of Health image software. The volumes of ATP depletion and CPS inhibition were measured using a semiautomated method (Swanson et al., 1990). ATP depletion was defined as a decline to less than 30% of the mean value of the contralateral side. The threshold for CPS inhibition was set to the lowest CPS value of the nonischemic hemisphere, excluding fiber tracts. The areas of ATP depletion and CPS inhibition were measured on each section by subtracting the area of the nonlesioned ipsilateral hemisphere from that of the contralateral hemisphere. The areas of preserved ATP and protein synthesis were outlined and superimposed to demarcate penumbral tissue in which protein synthesis was suppressed but ATP was preserved (Hata et al., 2000). Optical densities of in situ hybridization signals of each mRNA were measured at the level of the caudate-putamen in cortical regions of interest located in the metabolically impaired tissue, as defined by ATP and CPS imaging. Tissue radioactivity (kBq/g) was calibrated using 14C standards. Values were normalized to the radioactivity in the caudate-putamen of the opposite nonischemic hemisphere. Inhomogeneities of the illumination system were eliminated by background shading correction. Film autoradiograms were obtained at two or three different exposure times to ensure that optical densities were within the linear range of the standard curve.
TUNEL
TUNEL was performed as described previously (Wiessner et al., 1996), with minor modifications. Briefly, coronal brain sections were fixed for 15 minutes in ice-cold 4% paraformaldehyde/PBS, pH 7.4. Subsequently, the sections were washed twice in 70% ethanol (1 minute), once in PBS (3 minutes), once in 0.3% hydrogen peroxide/PBS (5 minutes), and then again in PBS (5 minutes). After equilibration for 15 minutes in TDT buffer (100 mmol/L potassium cacodylate, 2 mmol/L cobalt chloride, 0.2 mmol/L dithiothreitol), the buffer was quantitatively removed, sections were incubated in 50 μL TDT-Mix (10 pmol/L biotin-16-dUTP [Boehringer, Mannheim, Germany] and 150 U/mL terminal deoxynucleotidyl transferase [Life Technologies, Eggenstein, Germany]) in TDT buffer, and covered with a coverslip. After incubation for 60 minutes at 37°C, the reaction was terminated by washing the sections for 15 minutes in TB buffer (300 mmol/L sodium chloride, 30 mmol/L sodium citrate). Incorporated biotin was visualized using the avidin–biotin–peroxidase complex method (Vector Laboratories, Burlingame, CA, U.S.A.), as recommended by the supplier. Finally, the sections were dehydrated and embedded in Eukitt (Kindler GmbH, Freiburg, Germany).
Incidence maps of regional alterations
To evaluate the regional reproducibility of TUNEL versus the reduction of CPS and ATP, regional incidence maps were constructed (Hata et al., 2000). The areas of biochemical disturbances were outlined on representative brain sections from each individual experiment and superimposed at two coronal levels, the caudate-putamen and the dorsal hippocampus. Using the image analysis software, the incidence of the metabolic alterations was calculated for each pixel and expressed as a percentage of the number of animals per group.
Temperature control
Rectal temperature was maintained at 36.5°C to 37.0°C by a heating lamp and a heating pad connected to a thermistor (YSI, Yellow Springs, OH, U.S.A.) until 1 hour after reperfusion. After recovering from anesthesia, the animals were maintained in an air-conditioned room at approximately 22°C.
Statistics
All values are given as means ± SD. Differences in metabolic parameters and relative in situ hybridization radioactivity of mRNAs were compared using one-way analysis of variance, followed by Bonferroni's multiple comparison test. P < 0.05 was considered to indicate statistical significance.
RESULTS
General physiologic parameters
In the mouse, blood sampling for measurement of blood variables can be carried out only once. To study the influence of MCA occlusion on physiologic variables, we therefore used separate groups of animals. As shown in Table 1, arterial pCO2 and blood hematocrit transiently increased during MCA occlusion, but after recirculation all blood variables returned to normal. Blood pressure and heart rate did not change at any time during the experiments.
Physiologic variables before, during, and after 1-hour middle cerebral artery occlusion
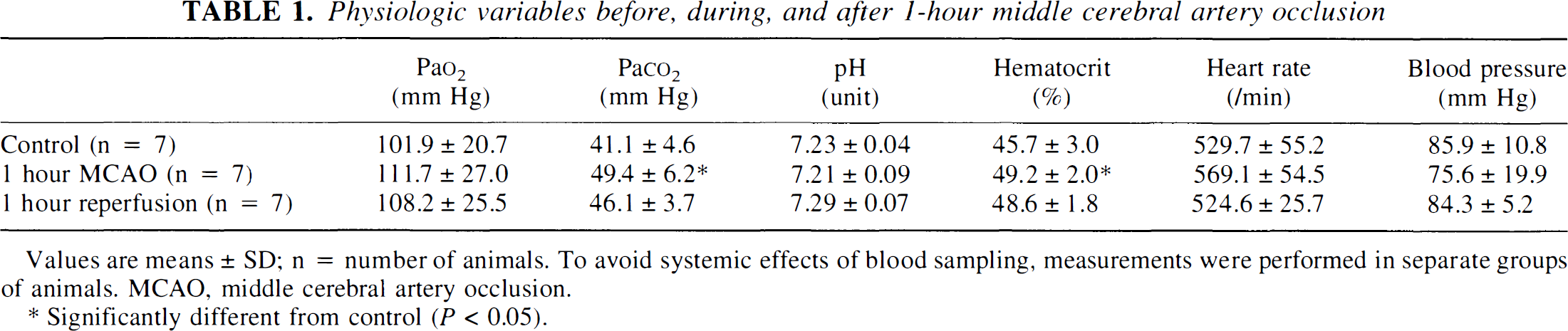
Values are means ± SD; n = number of animals. To avoid systemic effects of blood sampling, measurements were performed in separate groups of animals. MCAO, middle cerebral artery occlusion.
Significantly different from control (p < 0.05).
Neurologic investigations after thread occlusion revealed hemiparesis in all animals but no deaths during the initial 6 hours of reperfusion. In the animals selected for 1 and 3 days of survival, one of five animals spontaneously died during the later phase of reperfusion.
Cerebral metabolism
At the end of one hour of focal ischemia, ATP was depleted in the central parts of the MCA territory—that is, in the frontoparietal cortex, the lateral part of the caudate-putamen, and the piriform cortex (Figs. 1 and 2). Suppression of protein synthesis was present in the same region but extended into the more peripheral parts of the MCA territory. At the level of the caudate-putamen, the ATP-depleted area amounted to 40.1% ± 11.0% and the CPS-inhibited area to 58.9% ± 5.5% of the nonischemic contralateral hemisphere. Comparison with the homotopic areas of the opposite hemisphere revealed significant reductions of both ATP and CPS in the piriform cortex and the somatosensory area of the frontoparietal cortex at the level of the striatum, in the temporal and piriform cortex at the level of the dorsal hippocampus, and in the lateral caudate-putamen (Tables 2 and 3). At the level of the substantia nigra, inhibition of CPS extended further into the temporal and entorhinal cortex.

Multiparametric images of cerebral protein synthesis (CPS), tissue ATP content, and hsp70, c-fos, and junB mRNAs of representative brain sections of mice at the level of the caudate-putamen at various reperfusion times after transient middle cerebral artery occlusion for 1 hour. The outlines of preserved ATP and CPS have been superimposed to demarcate the metabolically impaired areas from the normal brain tissue.
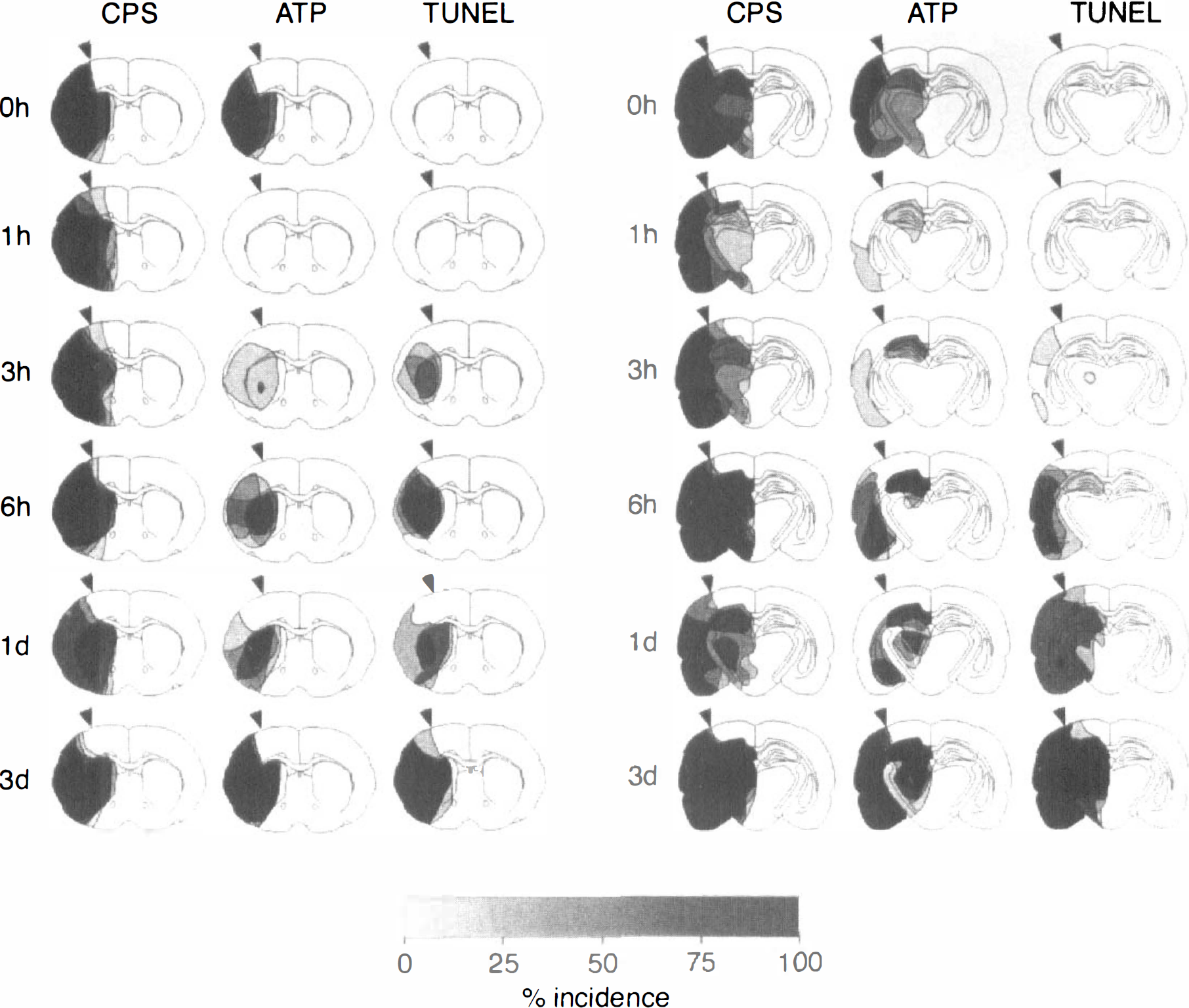
Incidence maps of suppressed protein synthesis (CPS), ATP depletion, and TUNEL-positive cells on coronal sections of the mouse brain at various reperfusion times after transient middle cerebral occlusion for 1 hour. Areas of disturbed metabolism were outlined in four or five animals per time point at the level of the caudate-putamen (left) and the dorsal hippocampus (right) and superimposed to calculate the incidence of alterations as a percentage of the number of animals per group. The demarcation between normal and disturbed protein synthesis in parietal cortex visible at the end of 1 hour of middle cerebral artery occlusion (0 hours reperfusion) was marked by the arrowheads to estimate the evolution or regression of the metabolic lesions at later time points. Note transient recovery of ATP after the beginning of reperfusion and correlation of TUNEL with secondary energy failure.
Regional protein synthesis in mouse brain before and after 1-hour transient middle cerebral artery occlusion
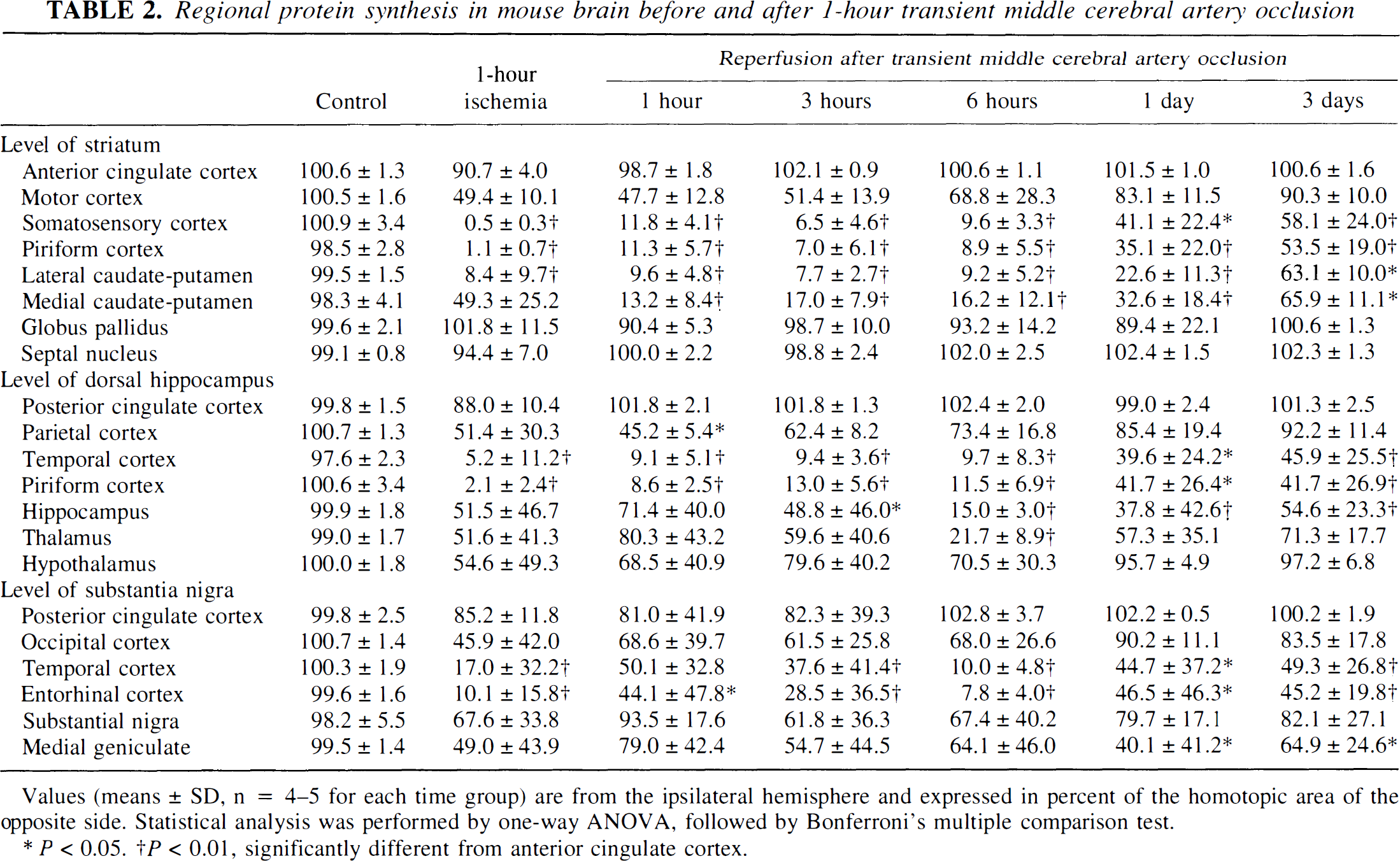
Values (means ± SD, n = 4–5 for each time group) are from the ipsilateral hemisphere and expressed in percent of the homotopic area of the opposite side. Statistical analysis was performed by one-way ANOVA, followed by Bonferroni's multiple comparison test.
P < 0.05.
P < 0.01, significantly different from anterior cingulate cortex.
Regional ATP content in mouse brain before and after 1-hour transient middle cerebral artery occlusion
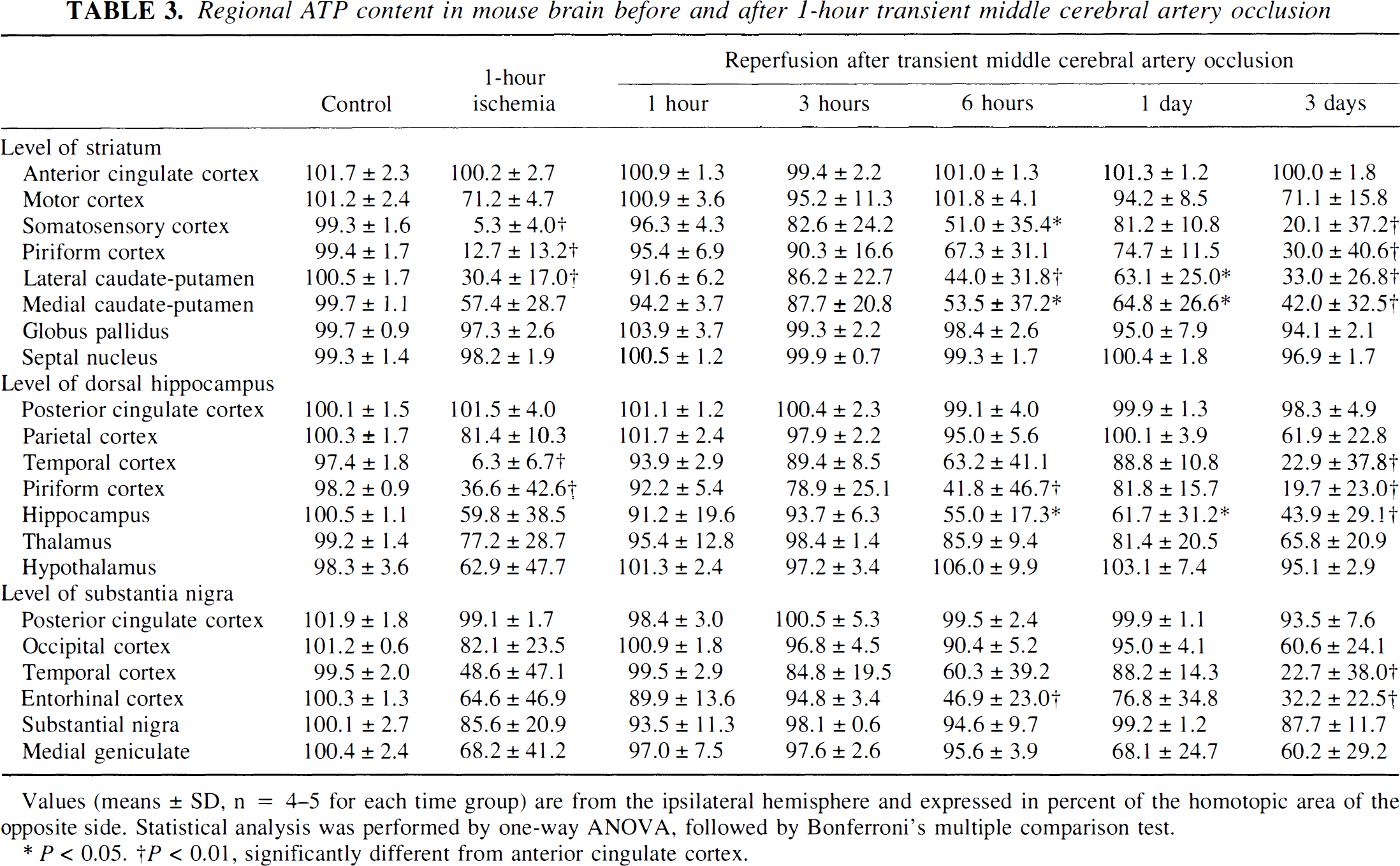
Values (means ± SD, n = 4–5 for each time group) are from the ipsilateral hemisphere and expressed in percent of the homotopic area of the opposite side. Statistical analysis was performed by one-way ANOVA, followed by Bonferroni's multiple comparison test.
P < 0.05.
P < 0.01, significantly different from anterior cingulate cortex.
When the brain was reperfused after 1 hour of MCA occlusion, inhibition of CPS changed little during the initial 6 hours (Fig. 3). At the level of the caudate-putamen, the CPS-inhibited area was 54.1% ± 2.6% at 1 hour, 56.9% ± 6.1% at 3 hours, and 56.6% ± 4.8% at 6 hours after the onset of reperfusion. Later it significantly improved to 48.7% ± 4.5% at 1 day (P < 0.05) and to 37.2% ± 7.3% at 3 days (P < 0.01) after reperfusion.
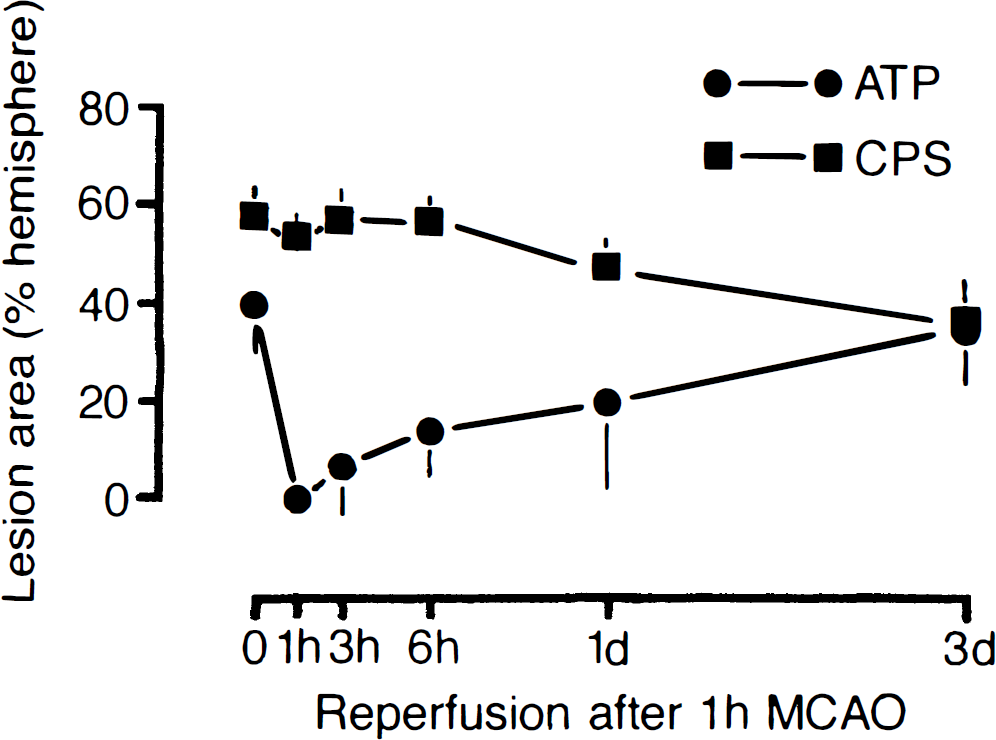
Dynamics of metabolic lesions in mouse brain after transient middle cerebral artery occlusion for 1 hour. The areas of suppressed cerebral protein synthesis (CPS) and of ATP depletion were measured at the level of the caudate-putamen and expressed as a percentage of the opposite hemisphere. Note transient recovery of ATP followed by secondary deterioration.
In contrast to CPS, ATP transiently recovered on reperfusion. After recirculation for 1 hour, ATP was almost completely restored, but with longer periods of reperfusion it again deteriorated. Starting in the central parts of the caudate-putamen, ATP was secondarily suppressed, followed by a gradual expansion of the ATP-depleted region into the more peripheral parts of the MCA territory. The mean area of ATP depletion at the level of the caudate-putamen thus increased from 7.2% ± 10.9% of the nonischemic contralateral hemisphere at 3 hours to 14.2% ± 10.4% at 6 hours, 19.7% ± 17.9% at 1 day, and 34.8% ± 9.2% at 3 days after the onset of reperfusion. At 3 days after the onset of reperfusion, ATP depletion had merged with the area of CPS inhibition, leading to a sharp demarcation of the brain infarct from the normal brain tissue.
The reproducibility of the regional pattern of metabolic alterations was studied by calculating injury incidence maps of disturbed protein synthesis and ATP depletion at two representative coronal levels of the brain, passing through the caudate-putamen and the dorsal hippocampus (see Fig. 2). These maps clearly document that CPS inhibition remained virtually constant from the end of the 1-hour ischemic period up to 6 hours after reperfusion. Thereafter, CPS inhibition recovered in the most peripheral parts of the MCA territory but not in the central regions, where it remained completely suppressed until tissue infarction evolved.
In contrast to CPS, ATP transiently recovered after the beginning of reperfusion. However, starting at 3 hours after ischemia it secondarily deteriorated, first in the dorsal hippocampus and the central parts of the caudate-putamen and later in the more peripheral parts of the MCA territory. After 3 days of recirculation, the region of ATP depletion precisely matched the region of suppressed CPS.
Genomic expressions
Representative in situ hybridization autoradiograms of hsp70, c-fos, and junB mRNAs are shown in Fig. 1. The outlines of preserved ATP and normal CPS were superimposed on adjacent cryostat sections to facilitate the regional allocation of hybridization signals.
At the end of the 1-hour MCA occlusion, expression of hsp70 mRNA was slightly increased in the cortical penumbra (i.e., the region of suppressed CPS but preserved ATP located in the periphery of the MCA territory). In addition c-fos and junB mRNAs also increased in the lateral part of the penumbra but even more so in the normal cortex outside the area of suppressed CPS, extending up to the midline but not into the opposite hemisphere (Fig. 4).
After reperfusion, hsp70 mRNA expression sharply increased throughout the area of suppressed CPS, peaking at 3 hours after the release of vascular occlusion. With longer recirculation times it gradually declined, particularly in the areas of secondary ATP failure; it disappeared after 3 days—that is, when the area of secondary ATP depletion merged with that of CPS suppression. In contrast to hsp70 mRNA, the expression pattern of the immediate-early genes (IEGs) changed little after reperfusion: the signal intensity gradually declined to control levels between 6 hours and 1 day after the onset of reperfusion (see Fig. 4).
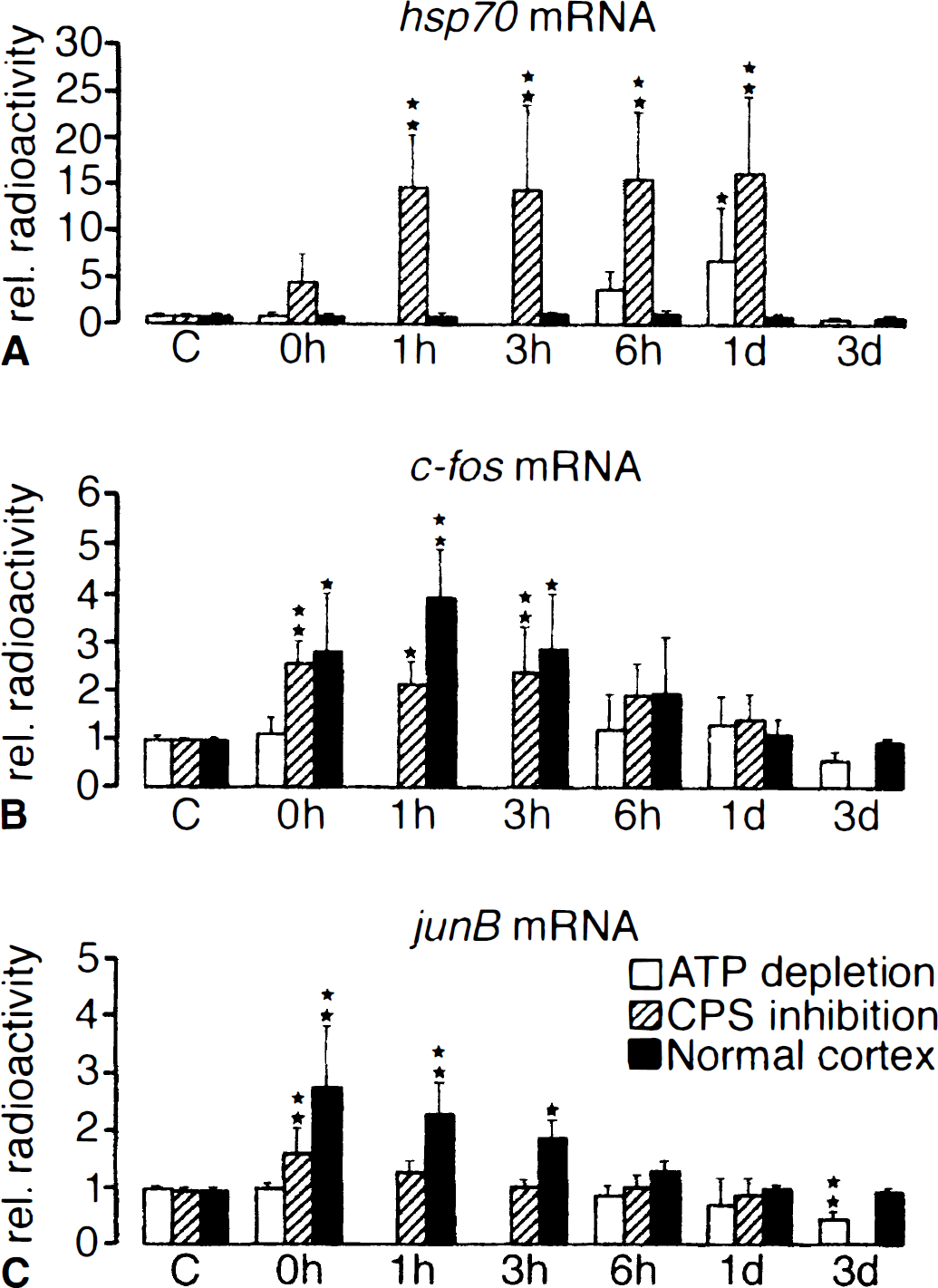
Expression of hsp70, c-fos, and junB mRNAs in the cerebral cortex of mouse at various reperfusion times after transient middle cerebral artery occlusion for 1 hour. Comparison of mRNA levels in the infarct core (area of ATP depletion, white bars), the penumbra (area of suppressed protein synthesis but preserved ATP, hatched bars), and the normal periinfarct cortex (black bars). Note differences in the regional pattern of gene expressions.
TUNEL
Double-strand DNA breaks visualized by TUNEL could first be detected between 3 and 6 hours after the beginning of reperfusion (see Fig. 2). Most of the TUNEL-positive cells appeared to be neurons, but the use of cryostat sections precluded precise identification. The comparison of the distribution pattern of TUNEL-positive cells with the inhibition of CPS revealed an initially central localization that between 1 and 3 days merged with but did not expand beyond the area of CPS suppression. Moreover, TUNEL-positive cells were mainly encountered in areas of secondary ATP depletion, although precise colocalization was not present in all animals (see Fig. 2).
DISCUSSION
The evolution of a delayed type of brain infarction after transient MCA occlusion is in agreement with the results of previous studies (Du et al., 1996; Endres et al., 1998), but the interval between the beginning of recirculation and the manifestation of secondary ATP depletion was much shorter. This could be due to differences in the methodology used to detect delayed infarction and also to differences in the duration of ischemia, the animal species, or the efficacy of postischemic recirculation. The evolution of the various pathobiochemical disturbances will be discussed separately.
CPS suppression after reperfusion
Postischemic CPS suppression is a well-known phenomenon that has been related to the selective block of polypeptide chain initiation (Cooper et al., 1977). The reason for this disturbance is phosphorylation of Ser51 on the alpha subunit of the eukaryotic initiation factor 2 (eIF2-alpha), resulting in the inactivation of this factor (Burda et al., 1998; DeGracia et al., 1999; DeGracia et al., 1997). Paschen and Doutheil (1999) recently suggested that eIF2-alpha inactivation is a stress response of the endoplasmic reticulum triggered by the sharp decline of endoplasmic reticulum calcium activity after the onset of ischemia. This hypothesis is supported by the recent cloning of a novel 150-kDa oxygen-regulated stress protein (ORP150) that localized within endoplasmic reticulum, was upregulated in the ischemic neurons in vivo, and suppressed hypoxia-induced apoptotic cell death in vitro (Matsushita et al., 1998; Ozawa et al., 1999).
In a previous study of transient global ischemia of 1 hour's duration, we described slow but progressive recovery of protein synthesis in most parts of the brain within 24 hours (Bodsch et al., 1986). In the present investigation of focal ischemia, in contrast, recovery of protein synthesis occurred in the peripheral but not in the central parts of the ischemic territory, although energy metabolism was rapidly resumed on recirculation. An important difference between the previous global and the present focal ischemia is the fact that after global ischemia recovery of energy metabolism is permanent, provided that secondary recirculation disturbances can be prevented (Hossmann, 1997). The persistence of CPS inhibition in the central part of the ischemic territory may therefore be secondary to the emerging energy failure, and not the other way around.
Genomic responses after reperfusion
Correlation analysis of IEGs and brain metabolism revealed that 1 hour after MCA occlusion, junB mRNA was expressed mainly in the periinfarct tissue with normal metabolism. In contrast, c-fos mRNA increased both in the penumbra and the periinfarct tissue. Induction of IEGs has been attributed to the transient increase in cytosolic calcium evoked by the passage of periinfarct spreading depressions (Ginty, 1997; Herdegen and Leah, 1998). A possible explanation for the differential expression pattern of IEGs is the difference of the promoter construction that mediates IEG induction after calcium influx. The expression of c-fos and junB mRNAs vanished between 6 hours and 1 day after the onset of reperfusion, suggesting that periinfarct spreading depressions were not longer generated after this interval (Kristian et al., 1998). Interestingly, the regional and temporal expression profiles of the two IEGs did not correlate with the appearance of DNA fragmentations or the secondary energy failure after reperfusion, indicating that induction of IEGs does not contribute to secondary brain injury.
The expression of hsp70 mRNA after ischemia closely correlated with the inhibition of protein synthesis and was presumably triggered by ischemic denaturation of proteins. Denatured proteins bind to Hsp90, which in turn dissociates from the heat shock factor-1 (HSF-1) (Zou et al., 1998). HSF-1 forms a trimer, is phosphorylated, migrates to the nucleus, and binds to the heat shock element (a sequence in the promoter or regulatory region of the gene), resulting in transcription of hsp70 mRNA (Morimoto et al., 1996). The Hsp70 protein is a chaper-one that restores protein structure and function; therefore, it has been implicated in protection against ischemic injury (Sharp et al., 1999). However, our study and previous studies (Nowak and Jacewicz, 1994; Planas et al., 1997) clearly demonstrate that hsp70 mRNA was expressed in regions with severe inhibition of protein synthesis, precluding translation of the message. This may be one of the reasons why infarction evolved despite massive expression of hsp70 mRNA.
DNA fragmentation after reperfusion
The TUNEL technique has been widely used to detect brain cells that undergo apoptosis after transient ischemia. Our study confirms data from several laboratories that reported abundance of TUNEL-positive cells during recirculation after reversible MCA occlusion (Campagne and Gill, 1996; Charriaut-Marlangue et al., 1996; Fink et al., 1998; Namura et al., 1998). We could not, however, document the preponderance of TUNEL-positive neurons in the inner (Li et al., 1995) or outer (Du et al., 1996) boundary zones of infarct. Indeed, TUNEL-positive cells appeared first in the center of the evolving infarct and then gradually expanded into the more peripheral parts, together with the manifestation of secondary energy failure.
The late onset of TUNEL contrasts with the much earlier appearance of single-strand breaks, which can be detected 1 minute after the beginning of reperfusion (Chen et al., 1997). Obviously, these breaks are initially repaired, as reflected by the massive expression of DNA repair proteins (Li et al., 1997; Tu et al., 1998), but accumulation of unrepaired DNA triggers the mitochondrial release of cytochrome c, which contributes to the activation of caspases as executioners of apoptotic cell death (Green and Reed, 1998). This interpretation is supported by increasing evidence that prevention of either DNA fragmentation or of the associated activation of apoptosis-related pathways markedly reduces infarct size (Eliasson et al., 1997; Endres et al., 1998; Fink et al., 1998; Weisbrot-Lefkowitz et al., 1998).
Postischemic energy failure
The initial homogenous recovery of ATP after 1 hour of transient MCA occlusion is in full agreement with previous studies of global cerebrocirculatory arrest and supports the notion that brain energy metabolism can be resuscitated after this length of ischemia (Behar et al., 1989). Secondary postischemic energy failure after transient focal ischemia was first reported by Folbergrova et al. (1995) after 2 hours of MCA occlusion, but in this study and another similarly designed one (Selman et al., 1999), energy metabolism recovered only partially before secondary deterioration. To the best of our knowledge, the present study is the first demonstration of gradually progressive secondary energy depletion despite full and homogenous primary recovery. This excludes deterioration from complicating side effects of a persisting primary lesion, such as that induced by periinfarct edema.
The mechanisms of secondary energy failure are still poorly understood. Folbergrova et al. (1995) were able to reduce this disturbance with the spin-trapping agent alpha-phenyl-
Another explanation is delayed energy failure caused by mitochondrial dysfunction or DNA repair. Postischemic uptake of calcium into mitochondria is thought to generate oxygen free radicals and to open mitochondrial permeability transition pores, causing disturbances of mitochondrial oxidative phosphorylation (Siesjö et al., 1999). Energy failure may also result from postischemic DNA repair processes. According to the poly(ADP-ribose) polymerase (PARP) suicide concept, oxygen free radicals and nitric oxide formed during ischemic reperfusion mediate DNA strand breaks, which in turn leads to the activation of PARP, a nuclear DNA repair enzyme that functions upstream of p53 to identify DNA strand breaks (de Murcia and Menissier de Murcia, 1994; Lautier et al., 1993). PARP uses oxidized NAD+ to form poly(ADP-ribosyl) nuclear proteins for the repair of DNA breaks. The utilization of NAD+ deprives the tricarboxylic acid cycle of its main electron acceptor and therefore contributes to energy failure (Endres et al., 1997). The close temporal and topical correlation between secondary energy failure and the appearance of TUNEL-positive neurons supports this concept: as long as sufficient amounts of ATP and NAD+ are present, DNA breaks are repaired and TUNEL-positive cells are absent, but with the collapse of this system, TUNEL-visible DNA fragmentations and energy failure concur in unison.
Finally, the possibility of edema formation and microcirculatory disturbances must be considered. In a recent nuclear magnetic resonance study, van Dorsten et al. (1999) observed persistent hypoperfusion after thread withdrawal. According to the threshold concept of brain ischemia, this hypoperfusion could be responsible for the failure of CPS recovery and in consequence for the secondary breakdown of energy metabolism.
In conclusion, the present investigation stresses the importance of an early recovery of CPS for the prevention of ischemic injury and suggests that TUNEL is an unspecific response of delayed brain infarction. The close correlation of secondary energy failure with the region of initial energy breakdown points to complex pathobiochemical interactions that are difficult to understand and that require more detailed investigation.
Footnotes
Acknowledgments
The authors thank Mrs. U. Beckmann, Mrs. U. Gillert, and Mrs. P. Lorenz-Korenkov for excellent technical assistance and Mrs. D. Schewetzky and Mr. A. Grasse for careful preparation of the manuscript.