
Abstract
Estrogen has been shown to protect against stroke-induced brain damage, yet the mechanism is unknown. During the early hours of stroke, cerebral edema forms as increased transport of Na and Cl from blood into brain occurs across an intact blood–brain barrier (BBB). We showed previously that a luminal BBB Na–K–Cl cotransporter is stimulated by hypoxia and arginine vasopressin (AVP), factors present during cerebral ischemia, and that inhibition of the cotransporter by intravenous bumetanide greatly reduces edema in rats subjected to permanent middle cerebral artery occlusion (MCAO). The present study was conducted to determine whether estrogen protects in stroke at least in part by reducing activity of the BBB cotransporter, thereby decreasing edema formation. Ovariectomized rats were subjected to 210 mins of permanent MCAO after 7-day or 30-min pretreatment with 17 β-estradiol and then brain swelling and 2,3,5-triphenyltetrazolium chloride staining were assessed as measures of brain edema and lesion volume, respectively. Diffusion-weighed imaging was used to monitor permanent MCAO-induced decreases in apparent diffusion coefficient (ADC) values, an index of changes in brain water distribution and mobility. Na–K–Cl cotransporter activity of cerebral microvascular endothelial cells (CMECs) was assessed as bumetanide-sensitive K influx and cotransporter abundance by Western blot analysis after estradiol treatment. Estradiol significantly decreased brain swelling and lesion volume and attenuated the decrease in ADC values during permanent MCAO. Estradiol also abolished CMEC cotransporter stimulation by chemical hypoxia or AVP and decreased cotransporter abundance. These findings support the hypothesis that estrogen attenuates stimulation of BBB Na–K–Cl cotransporter activity, reducing edema formation during stroke.
Introduction
During the early stages of ischemic stroke, cerebral edema forms in the presence of an intact blood–brain barrier (BBB) by a process involving increased uptake of cations and water from blood into brain, with BBB breakdown occurring later, around 4 to 6 h after the onset of ischemia (Kimelberg, 1995; Menzies et al, 1993; Schielke et al, 1991). The BBB is well known to function in electrolyte homeostasis in the healthy brain, secreting up to 30% of brain interstitial fluid (comprised primarily of Na and Cl) (Cserr et al, 1989) and removing K from the brain (by absorption across the BBB from brain to blood) (Keep, 1993). During ischemia, BBB secretion of Na and Cl into the brain appears to be greatly increased, contributing substantially to edema formation (Kimelberg, 1995; Menzies et al, 1993; Schielke et al, 1991). Although little is known of the ion transport pathways that may be involved in this phenomenon, previous studies have suggested that a luminally located Na transport pathway plays a major role, working together with abluminal Na/K pump and Cl efflux pathways to ‘hypersecrete’ Na and Cl into the brain, with osmotically obliged water following (Betz, 1983ab; Betz et al, 1994; Kato et al, 1987; Schielke et al, 1991). At the same time, ischemia stimulates astrocytes to take up Na, Cl, and water from the brain interstitial space, causing swelling of those cells (Bourke et al, 1980; Iadecola, 1999; Kimelberg, 1999). The change in distribution of water and electrolytes during cerebral ischemia has been shown to be a complex process. While astrocyte cell bodies and processes swell quickly during ischemia, it has been shown that many neurons shrink but some neurons swell (Garcia et al, 1993) and extracellular space decreases (Nicholson and Syková, 1998) with the extent of these changes depending on both cerebral location and time after onset of ischemia. Despite this complexity of changes in tissue water distribution, during cerebral ischemia total brain water and Na increase resulting in brain edema that includes swelling of astrocytes together with uptake of water and Na from blood into the brain (Kimelberg, 1995; Menzies et al, 1993; Schielke et al, 1991). We have hypothesized that the BBB Na–K–Cl cotransporter is the luminal Na and Cl pathway by which ischemia promotes the net uptake of Na and water across the intact BBB into the brain during the early hours of stroke. In support of this, our previous studies have shown that a Na–K–Cl cotransporter is present in luminal membranes of BBB endothelial cells in situ (O'Donnell et al, 2004) and that the cotransporter is stimulated by factors present during cerebral ischemia (Foroutan et al, 2005; O'Donnell et al, 2005). In particular, the Na–K–Cl cotransporter of cultured cerebral microvascular endothelial cells (CMECs) is stimulated by arginine vasopressin (AVP), which is released during ischemia from extrahypothalamic neuronal processes terminating on brain microvessels (Jójárt et al, 1984; Landgraf, 1992). In addition, our recent studies have shown that hypoxia, which develops rapidly during brain ischemia because of the low O2 store in the brain, is also a potent stimulator of the CMEC Na–K–Cl cotransporter (Foroutan et al, 2005). Further, we have shown previously that inhibition of the BBB Na–K–Cl cotransporter by intravenous administration of the cotransport inhibitor, bumetanide, reduces edema formation (evaluated as increases in brain water by gravimetry) and 2,3,5-triphenyltetrazolium chloride (TTC)-defined lesion volume in the rat permanent middle cerebral artery occlusion (MCAO) model of stroke (O'Donnell et al, 2004). Results of that study also showed that the MCAO-induced decrease in magnetic resonance diffusion weighted imaging (DWI) apparent diffusion coefficient (ADC) values, a measure of changes in brain water distribution and mobility that occur in ischemia, is also significantly reduced by bumetanide.
Estradiol has been shown to be neuroprotective in stroke (Dubal et al, 1988; Hurn and Macrae, 2000). In general, men and postmenopausal women (not taking hormone replacement therapy) exhibit more damage from stroke than do premenopausal women and postmenopausal women taking hormone replacement therapy (Hurn and Macrae, 2000; Zhang et al, 1998). In the MCAO experimental stroke model, estradiol reduces cerebral TTC-defined lesion volume in both male and female rats, whether subjected to ischemia alone (permanent MCAO) (Dubal et al, 1988) or to ischemia/reperfusion (reversible MCAO) (Dubal et al, 1988; Toung et al, 1998; Zhang et al, 1998). Neuroprotection in experimental ischemic stroke has been reported for both acute estradiol treatment (given immediately before or at the start of ischemia) (Rusa et al, 1999; Yang et al, 2000; Zhang et al, 1998) as well as for chronic treatment (given several days before the start of ischemia) (Dubal et al, 1988; Hurn and Macrae, 2000). Also, the estrogen receptor antagonist ICI 182,780 exacerbates MCAO-induced ischemic injury in female mice (Sawada et al, 2000). Estradiol is well recognized for exerting both slow onset effects, occurring over hours, and also very rapid effects, initiated within minutes of exposure (often referred to as ‘genomic’ and ‘nongenomic’ effects, respectively) (Dhandapani and Brann, 2002; Lee and McKewen, 2001; Linford et al, 2000). However, the mechanisms whereby estradiol provides neuroprotection in ischemic stroke are far from clear. A previous study has provided evidence that estradiol reduces DWI-defined lesion volume in rats subjected to ischemia/reperfusion (Shi et al, 2001). Together with our previous finding that the BBB Na–K–Cl cotransporter appears to be an important participant in stroke-induced edema formation (O'Donnell et al, 2004), this suggests the possibility that estradiol may offer neuroprotection in part by acting on BBB endothelial cells to reduce Na–K–Cl cotransporter-mediated cerebral edema formation. The present study was conducted to test this possibility. We show here that treatment of ovariectomized (OVX) rats with 17 β-estradiol (7 days or 30 mins) reduces brain swelling and ischemic injury volume after permanent MCAO. We also show that ischemia-induced early changes in brain water distribution and mobility, assessed as decreases in DWI ADC values, are attenuated by estradiol throughout the first hours of permanent MCAO and that this effect correlates well with estradiol reduction of brain swelling and TTC-defined lesion volume. Further, we show that in cultured CMECs, estradiol reduces Na–K–Cl cotransporter activity stimulated by chemical hypoxia (oligomycin) or by AVP and that estradiol modulates expression of the CMEC Na–K–Cl cotransporter protein in a manner sensitive to the estradiol receptor antagonist ICI 182,780.
Materials and methods
Induction of Focal Ischemia by Permanent Middle Cerebral Artery Occlusion
This study was conducted in accordance with the Animal Use and Care Guidelines issued by the National Institutes of Health using a protocol approved by the University of California, Davis, Animal Use and Care Committee. Ovariectomizedfemale Sprague–Dawley rats (Charles River Laboratories, Wilmington, MA, USA) weighing 250 to 300 g were used in the study. Animals were anesthetized by intraperitoneal injection of sodium pentobarbital (65 mg/kg body weight). Body temperature was monitored and maintained at 36.8°C to 37.0°C using a rectal probe and an electric heating blanket (during surgery) or water heating pad (during DWI data acquisition) as described previously (O'Donnell et al, 2004). In addition, the left femoral artery was cannulated with PE-50 polyethylene tubing for continuous monitoring of arterial blood pressure (MAP) during surgery and data acquisition. Blood samples were drawn from the descending aorta at the end of the DWI data acquisition period for determination of pH, pCO2, HCO3, hemoglobin, blood glucose, blood urea nitrogen, Na±, K±, and Cl− (I-STAT; Sensor Devices, Waukesha, WI, USA). To induce focal cerebral ischemia, rats were subjected to occlusion of the left middle cerebral artery (MCA) as described by us previously (O'Donnell et al, 2004). Briefly, the left common carotid artery (CCA) was surgically exposed, occipital and thyroid artery branches of the external carotid artery (ECA) and the pterygopalatine artery were ligated. After ligating the ECA distal to the CCA, a 3-0 dermalon filament with a blunted tip was inserted into the ECA and advanced to the origin of the MCA. Occlusion was confirmed by laser Doppler evaluation of cerebral blood flow (CBF) (Moor Instruments, Wilmington, DE, USA) through a craniotomy (approximately 3 mm posterior to bregma and 3 mm lateral to midline). In these studies, MCAO reduced CBF to 26% to 30% of preocclusion flow. For sham surgery, rats were subjected to surgery without occlusion. In our previous studies evaluating the effects of bumetanide on cerebral edema formation in MCAO, we nephrectomized the rats immediately before MCAO to avoid any possible contribution from bumetanide renal effects (O'Donnell et al, 2004). While we found in that study that the same results were obtained regardless of whether the rats were nephrectomized, to optimize comparison of the edema reducing effects of bumetanide with those of estradiol, we chose to nephrectomize the rats in this study. Thus, immediately before MCAO, the rats underwent nephrectomy using standard methods, as described previously (O'Donnell et al, 2004).
Estradiol Pretreatments
For 7-day estradiol pretreatment, silastic capsules containing either 1 mg/mL 17 β-estradiol (0.05 mL) or vehicle (sesame oil) were implanted subcutaneously in OVX female rats. Plasma estradiol levels were then analyzed by radioimmunoassay (RIA) after 7 days treatment at the end of the MCAO DWI experiment period. For acute estradiol pre-treatment, rats were injected with either 17 β-estradiol (0.05 mg in 0.05 mL sesame oil) or vehicle (0.05 mL sesame oil) 30 mins before induction of MCAO and plasma estradiol levels analyzed by RIA at the end of the 210-min MCAO period. RIA analyses of plasma estradiol levels were performed in the UC Davis Clinical Endocrinology Laboratory as described previously (Shille et al, 1979).
Magnetic Resonance Diffusion Weighted Imaging Analysis of Apparent Diffusion Coefficient Values
Immediately on induction of MCAO (or completion of sham surgery) rats were subjected to magnetic resonance diffusion weighted imaging analysis of ADC values as described by us previously (O'Donnell et al, 2004). Briefly, rats were placed on a homemade Plexiglas stage with bite bar and ear clamps, then the stage with rat was positioned in the magnet bore of a 7-T Bruker Biospec MRS/MRI system (Bruker Biospin MRI, Inc., Billerica, MA, USA) and diffusion weighted images acquired, using 2-mm thick axial slices. Apparent diffusion coefficient values were then determined for selected brain regions (three regions in the cortex and one in the striatum) using four gradient strengths (Tatlisumak et al, 1996) and ParaVision 2.1 software (Bruker Biospin GMbH, Rheinstetten, Germany). For each region of interest, ipsilateral (left) and contralateral (right) ADC values were compared and the ratio of left/right ADC values was calculated. Previous studies have shown that a decrease in ADC values provides an index of early changes in the distribution and mobility of water in the brain (Hasegawa et al, 1994; Knight et al, 1994; Liu et al, 2001; Moseley et al, 1990). The ADC value appears to depend on the size of various cellular and extracellular compartments in the brain and the movement of water within those compartments, as well as the permeability and surface area of membranes separating the various compartments (Liu et al, 2001). Thus, the factors that cause the ischemia-induced decrease in ADC are complex and incompletely understood. Despite this, ADC values have been found to decrease as the combined swelling of astrocytic processes and neuronal dendrites increases (evaluated 90 mins after induction of MCAO) (Liu et al, 2001). In this regard, while a decrease in ADC is not a quantitative index of brain cell swelling during cerebral ischemia, it provides valuable information about the time course of these initial changes in water distribution and mobility occurring during cerebral ischemia, which appear to be due at least in part to astrocyte swelling. In the present study, ADC values were evaluated for both the occluded ipsilateral and contralateral hemispheres which provides an internal control and allows for effective real-time assessment of ADC values in the living rat throughout the ischemic period (O'Donnell et al, 2004).
2,3,5-Triphenyltetrazolium chloride Assessment of Lesion Size
Rats were euthanized immediately after DWI data acquisition (210 mins after induction of MCAO). The brain was quickly removed and sectioned into 2 mm thick slices starting at the frontal pole using a Brain Matrix Slicer (Vibratome Co., St Louis, MO, USA). Slices were immersed in 2% TTC (Sigma Aldrich Corp., St Louis, MO, USA) in a Petri dish and incubated at 37°C for 20 mins. TTC, a water soluble salt, is reduced by mitochondrial dehydrogenases to formazan, a red, lipid-soluble compound that turns normal tissue deep red (Bederson et al, 1986; Rich et al, 2001). Thus, reduced TTC staining identifies regions of diminished mitochondrial function in the ischemic tissue. To assess lesion volume, TTC-stained slices were scanned using an Epson Perfection 1200U scanner (Epson America, Inc., Long Beach, CA, USA) and Adobe Photoshop software (Adobe, San Jose, CA, USA) and analyzed by Image-J analysis software (public domain software developed at NIH, and available on the Internet at http://rsb.info.nih.gov/nihimage/). Lesion volume was determined as the percent of the total ipsilateral hemispheric volume as described previously (Swanson et al, 1990; O'Donnell et al, 2004). Briefly, volume of the lesion was calculated as:
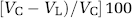
where VC is the volume of the control hemisphere and VL is the volume of nonlesioned tissue in the lesioned hemisphere.
Cerebral Microvascular Endothelial Cell Culture
Bovine CMECs were maintained on collagen type I- and fibronectin-coated tissue culture flasks in Eagle's minimal essential medium (EMEM) supplemented with L-gluta-mine, 5% fetal bovine serum (FBS) and 2 ng/mL fibroblast growth factor (FGF) (complete EMEM). The growth medium was changed to a 50:50 mixture of complete EMEM plus either C6 glial cell-conditioned medium (Figures 4A and 4B) or astrocyte conditioned medium (Figures 5–6) 24 to 48 h before using the CMECs in an assay as described previously (O'Donnell et al, 1995; Sun et al, 1995). We have found these two types of astroglial cell-conditioned media to be equally effective for studies of CMEC Na–K–Cl cotransport (O'Donnell, 1993; Sun et al, 1995). Cultured CMECs were obtained from Cell Systems Inc. (Kirkland, WA, USA) and C6 glial cells were from the American Type Culture Collection (Rockville, MD, USA). Primary cultures of astrocytes were isolated from the cerebral cortex of neonatal Sprague–Dawley rats and maintained on collagen-coated flasks in EMEM supplemented with L-glutamine, 5% FBS and 5% horse serum (O'Donnell et al, 1995; Sun et al, 1995). Bovine CMEC were used only up to passage 8. During these passages, we have not observed any changes in CMEC morphology, Na–K–Cl cotransporter or Na/K pump activities, total ion fluxes, expression of cotransporter protein, or responsiveness of the cotransporter to agents of interest (e.g., arginine vasopressin, hypoxia, and estradiol).
K Influx Assays
Na–K–Cl cotransport activity was assessed as bumetanide-sensitive K influx, using 86Rb as a tracer for K as described by us previously (O'Donnell et al, 2005, 1995). For these studies, CMEC monolayers cultured on multiwell plates (24- or 96-well cluster plates) were pretreated with 17 β-estradiol, oligomycin, and/or AVP as described in figure legends. Na–K–Cl cotransporter activity was then assessed by incubating the cells for 5 mins in medium containing these agents plus 0 or 10 μmol/L bumetanide, then incubating the cells for another 5 mins in identical medium also containing 86Rb. The medium used for pretreatment/assay medium used was a N-2-hydroxyethylpiperazine-N-2-ethanesulfonic acid (HEPES)-buffered medium (pH 7.4) containing (in mmol/L): 144 Na, 147 Cl, 5.8 K, 1.2 Ca, 4.2 HCO3, 0.4 HPO4, 0.4 H2PO4, 0.4 Mg, 0.4 SO4, 5.6 glucose, and 20 HEPES. To terminate the assay, cluster plate wells were aspirated and rapidly washed in ice-cold 0.1 mol/L MgCl2. Cells were then extracted in 0.2% sodium dodecyl sulfate (SDS) for 86Rb quantitation by liquid scintillation analysis (Tri-Carb 2500 TR liquid scintillation counter) and protein determination by bicinchoninic acid (BCA) assay (Pierce, Rockford, IL, USA). In these assays, Rb was used to assess K uptake because Rb quantitatively substitutes for K and 86Rb has a half-life of days compared with hours for 42K (O'Donnell, 1989). Bumetanide-sensitive K influx was determined as the difference between K uptake rates observed in the absence versus the presence of bumetanide, with K uptake calculated as the slope of an uptake versus time plot as described previously (O'Donnell, 1989). In previous studies, we have found that Na–K–Cl cotransporter activity accounts for ∼50% of total K influx, with Na/K pump activity (ouabain-sensitive K influx) accounting for ∼ 40% of total K influx with the remaining 10% of total K influx mediated by a bumetanide- and ouabain-insensitive K ‘leak’ flux (Foroutan et al, 2005).
Gel Electrophoresis and Western Blot Analysis
Western blot analysis of Na–K–Cl cotransporter protein abundance was performed as described by us previously (O'Donnell et al, 1995; Sun et al, 1995; Yerby et al, 1997) with some modifications, after exposure of CMECs (grown on 24-well cluster plates) to 17 β-estradiol for the doses and times described in figure legends. Briefly, lysates were prepared by washing wells with ice-cold PBS containing 5 mmol/L EDTA (PBS/EDTA) plus protease inhibitors (Roche Diagnostic Complete protease inhibitor cocktail tablet, containing chymotrypsin, thermolysin, papain, pronase, pancreatic extract and trypsin). CMECs were then lysed in PBS/EDTA containing 1% SLS plus protease inhibitors. Samples of each lysate preparation were analyzed in triplicate for protein content (BCA method) to ensure equal loading of membrane protein into each gel lane. Lysate samples and molecular weight markers (BioRad, Hercules, CA, USA) were denatured in SDS reducing buffer containing dithiothreitol (DTT, Invitrogen NuPage, Carlsbad, CA, USA) and heated to 70°C for 100 min, then used immediately for gel electrophoresis. Protein samples were then electrophoretically separated on 7.5% Tris-Glycine gels (Cambrex PAGEr Gold Precast, Rockland, ME, USA; Bio-Rad Mini-PROTEAN II, Hercules, CA, USA) and the resolved proteins were electrophoretically transferred to polyvinylidene fluoride membranes using a Bio-Rad Trans-Blot apparatus. The blots were incubated in 7.5% nonfat dry milk/PBS-Tween for 2 h at room temperature. Subsequently, blots were incubated with T4 monoclonal antibody, which recognizes the Na–K–Cl cotransporter protein (Lytle et al, 1995), rinsed 5 times with PBS-Tween, and then exposed to secondary antibody (Horseradish peroxidase-conjugated goat anti-mouse IgG). After five washes to remove unbound secondary antibody, bound antibody was visualized using the enhanced chemiluminescence assay (ECL, Amersham Biosciences, Little Chalfont Buckinghamshire, England) with a Fuji Film LAS-3000 Imaging System (Medford, UK). Image Quant software (Molecular Dynamics, Sunnyvale, CA, USA) was used to quantitate band density. For these Western blot assays, β-actin was used as a loading control. However, the same results were obtained whether cotransporter protein bands were normalized to the β-actin bands.
Materials
Bumetanide was from ICN Biomedicals (Costa Mesa, CA, USA) and 86Rb was purchased from Dupont NEN (Boston, MA, USA). (Arg8)-vasopressin (AVP) was obtained from Peninsula Laboratories (a division of Bachem, San Carlos, CA, USA). 17 β-Estradiol and oligomycin were obtained from Sigma Chemical Co. (St Louis, MO, USA) and ICI 182,780 was from Tocris Cookson Inc. (Ellisville, MO, USA). T4 monoclonal antibody was obtained from the University of Iowa Developmental Studies Hybridoma Bank (Iowa City, IA, USA). β-Actin antibody was purchased from Abcam, Inc. (Cambridge, MA, USA). Secondary antibodies were from Zymed Laboratories (South San Francisco, CA, USA).
Statistical Analyses
All values are presented as mean ± s.e.m. with n values corresponding to the number of separate experiments. For each K influx experiment, all conditions were tested in at least quadruplicate. For Western blot analyses, samples were all tested in triplicate. Data shown were analyzed for significance using either analysis of variance for repeated measures with a Scheffe post hoc test or Student's t-test unless otherwise indicated. P-values less than 0.05 were considered to indicate statistical significance. SAS Stat-view software (Cary, NC, USA) was used for all data analyses.
Results
Effects of Chronic and Acute Estradiol Pretreatments on Cerebral Edema Formation in Permanent Middle Cerebral Artery Occlusion
To evaluate the possibility that estradiol offers neuroprotection in stroke at least in part by reducing ischemia-stimulated BBB Na–K–Cl cotransporter activity and consequent edema formation, we first evaluated the effects of estradiol treatments on brain swelling and ischemic lesion volume in rats subjected to MCAO. In these experiments, we also evaluated estradiol effects on MCAO-induced changes in DWI ADC values. For this, OVX rats were pretreated with either 17 β-estradiol or vehicle, then subjected to permanent MCAO (i.e., ischemia without reperfusion) or to sham surgery, as described in Materials and methods. Diffusion weighted imaging analysis of ADC values was then performed over a time course of 210 mins, followed by killing the rats and immediately evaluating brains for swelling by morphometric analysis and for ischemic injury volume by TTC staining as described in Materials and methods. In these studies, we first tested the effects of a 7-day estradiol pretreatment. Ovariectomized rats were implanted subcutaneously with a capsule containing estradiol or vehicle as described in Materials and methods. Plasma estradiol levels after 7 days treatment, as determined by radioimmunoassay (RIA), were 56.7 ± 8.4 pg/mL in estradiol-treated OVX rats compared with 9.3 ± 1.9 pg/mL for vehicle-treated OVX rats (mean ± s.e.m. for 8 and 4 rats respectively). Morphometric analysis of brain slices for swelling after 210 mins of permanent MCAO revealed mean cerebral swelling of 18.8% ± 2.0% versus 6.4% ± 2.2% for rats treated 7 days with vehicle and estradiol, respectively (mean values ± s.e.m., 5 rats each, P < 0.05 by Student's t-test). In addition, TTC evaluation of the brain slices for ischemic injury revealed that 7-day pretreatment with estradiol also reduced the volume of ischemic lesion detected after 210 mins of permanent MCAO as shown in Figure 1. Representative TTC-stained brain slices obtained after MCAO in rats treated with vehicle or estradiol are depicted in Figure 1A. The volume of TTC-defined lesion quantitated for each brain slice (2 mm to 16 mm from the frontal pole) was found to be significantly reduced by 7-day estradiol treatment compared with vehicle as shown in Figure 1B.
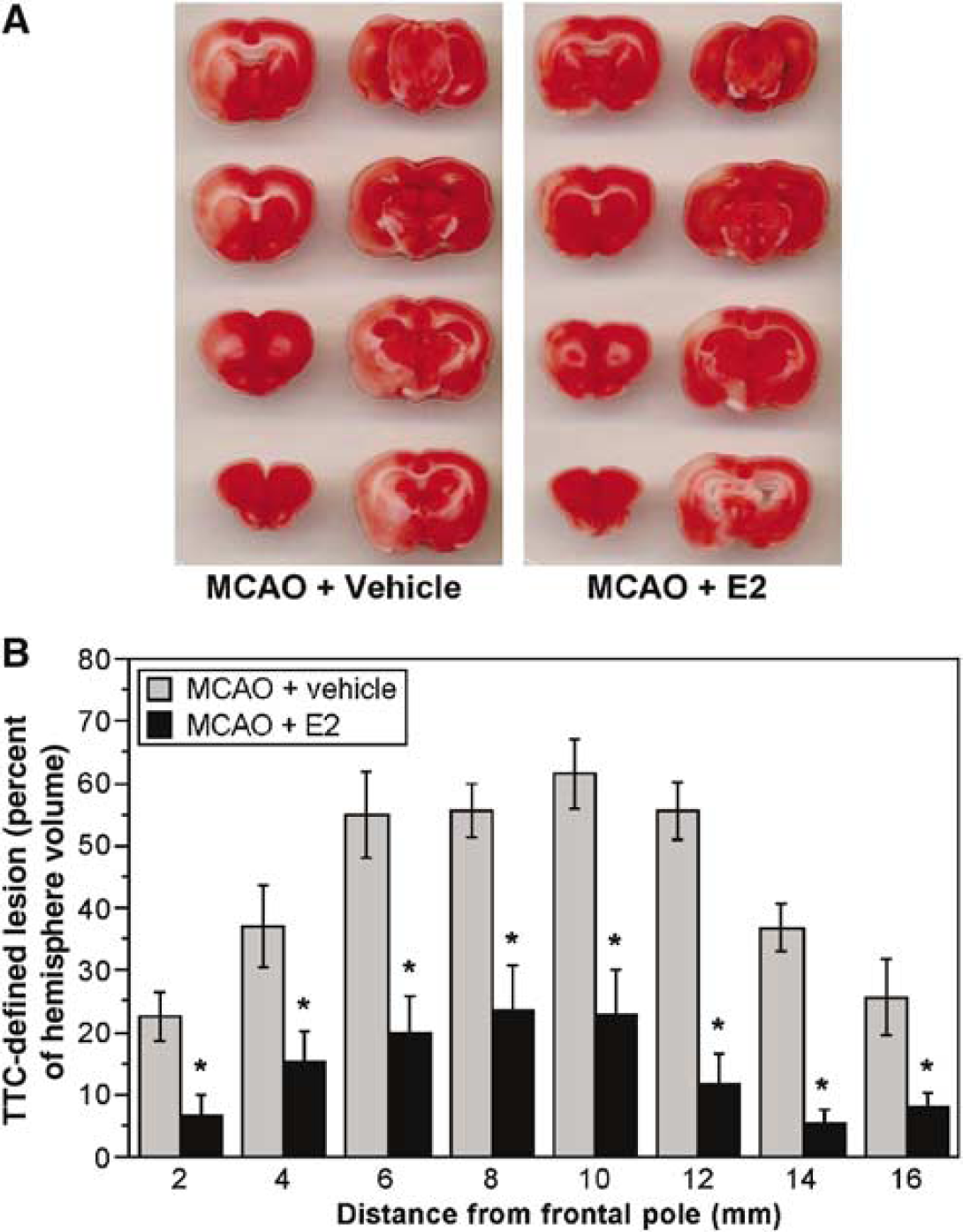
Estradiol reduction of TTC-defined lesion volume in rats subjected to MCAO.
For DWI studies of rats subjected to permanent MCAO or to sham surgery after 7-day estradiol pretreatment, ADC values were determined for cortical and striatal regions of interest over a time course of 210 mins. In these experiments, we determined both ipsilateral (left, occluded) and corresponding contralateral (right, control non-occluded) ADC values for the four regions of interest depicted in the representative diffusion weighted image shown in Figure 2A. We then calculated left to right ADC ratios for each region. Left/right ADC ratios for lateral cortex (L2/R2) and striatum (L4/R4) are shown in Figures 2B and 2C, respectively, for rats subjected either to MCAO or to sham surgery. In both the cortex and striatum, ADC ratios for sham-operated OVX rats (treated with estradiol for 7 days) were not significantly different from 1.0 throughout the MCAO period, as predicted for nonoccluded tissue. However, in OVX rats treated with vehicle and then subjected to MCAO, ADC ratios fell to approximately 0.55 to 0.6, indicating ischemia-induced changes in brain water distribution and mobility. Treating OVX rats for 7 days with estradiol resulted in a significantly attenuated decrease in ADC ratios on induction of MCAO. This reduction in the decrease of ADC ratio was observed at the earliest evaluation time (30 min) and maintained throughout 210 mins of permanent MCAO. Estradiol similarly attenuated the decrease in ADC values in regions of interest 1 and 3 (data not shown). Depending on the time after start of MCAO, reduction of the decrease in ADC ratios varied from 63% to 78% for region 1 and 55% to 65% for region 3, compared with 62% to 70% for region 2 and 55% to 77% for region 4. Thus, a 7-day estradiol pretreatment of OVX rats substantially attenuated ischemia-induced changes in brain water distribution in both cortex and striatum throughout the more than 3 h of permanent MCAO monitored in this study.
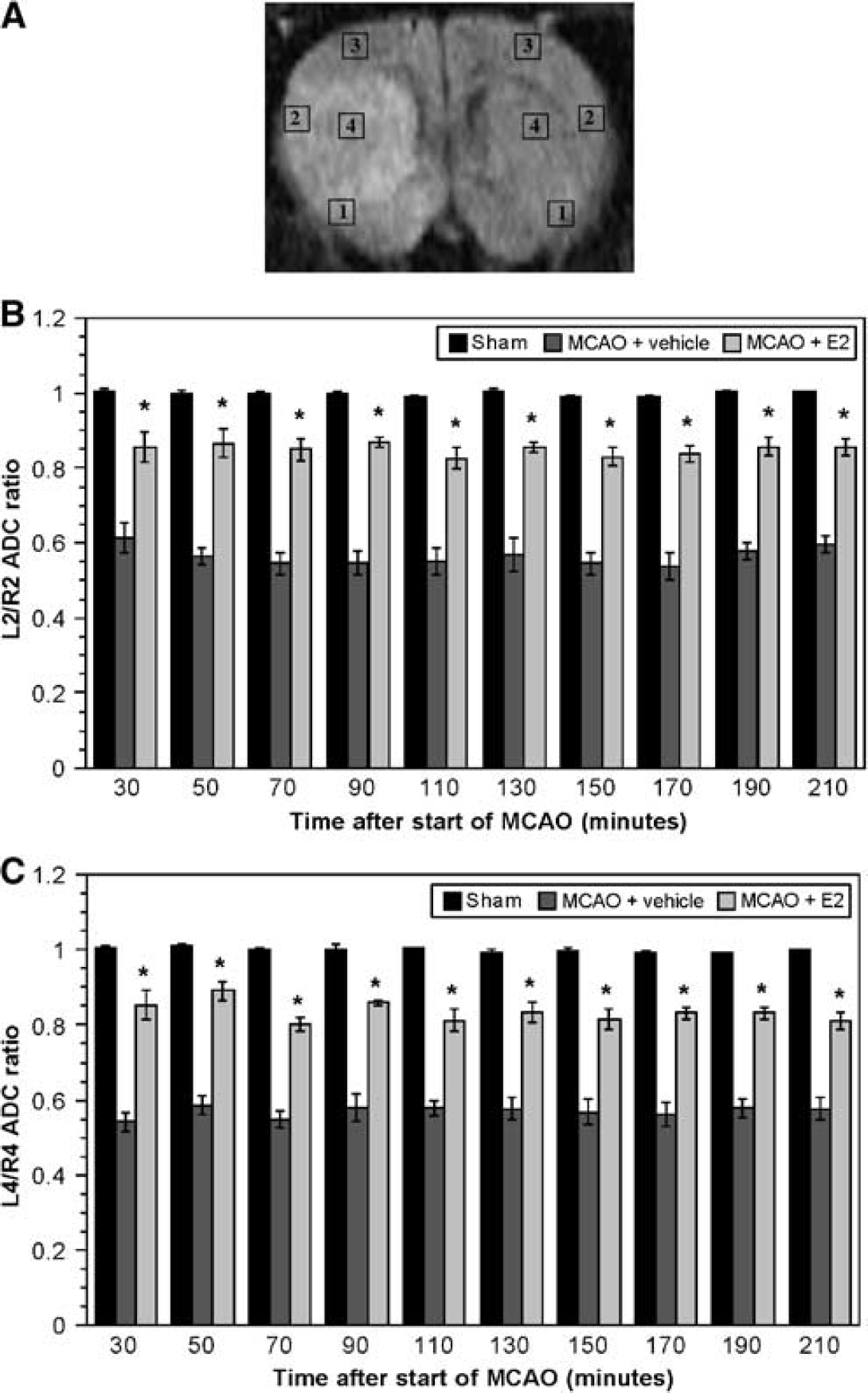
Seven-day estradiol pretreatment attenuates ischemia-induced decreases in ADC values in rats subjected to MCAO.
In the present study, we also evaluated the effects of acute estradiol pretreatment on MCAO-induced brain swelling, TTC-defined ischemic injury volume and decrease in DWI ADC values. For these experiments, OVX rats were administered either estradiol or vehicle (as described in Materials and methods) 30 mins before induction of permanent MCAO. Plasma estradiol levels of acutely treated rats, analyzed by RIA at the end of 210 mins MCAO, were found to be 144.9 ± 116.2 and 6.5 ± 0.8 pg/mL for estradiol-treated and vehicle-treated rats, respectively (mean ± s.e.m. for 3 and 4 rats, respectively). Morphometric analysis of brain slices after 210 mins of permanent MCAO revealed mean cerebral swelling of 28.7% ± 4.8% versus 10.8% ± 1.6% for rats treated 30 mins with vehicle and estradiol, respectively (mean ± s.e.m. of 3 and 4 rats, respectively, P < 0.05 by Student's t-test). Acute estradiol pretreatment also significantly reduced ischemic injury detected after 210 mins of permanent MCAO by TTC staining. Table 1 shows total TTC-defined lesion volumes (determined by summing lesion volumes of individual 2-mm slices taken 2 to 16 mm from the frontal pole) for both 30-min and 7-day estradiol- and vehicle-treated OVX rats. In both cases, estradiol significantly reduced total lesion volume. It should be noted that the relatively large lesion volumes observed after MCAO in our studies are consistent with a previous report that ischemic lesions in Sprague-Dawley rats are larger and occur more rapidly than those in Wistar Kyoto rats (Bardutzky et al, 2005).
Effect of 7-day or 30-mins estradiol pretreatments on total lesion volumes following 210 mins of permanent MCAO

Lesion volumes of TTC-stained brain slices (2-16 mm from the frontal pole) were quantitated as described in Materials and methods and summed to determine total lesion volumes. Data are expressed as percent of the total ipsilateral hemispheric volume. Rats were treated with estradiol or vehicle for 7 days or 30 mins and TTC staining of slices conducted immediately following 210 mins of permanent MCAO. Values are mean ± s.e.m. for the number of experiments shown in the parentheses. For both 7-day and 30-min pretreatments, total lesion volumes of estradiol-treated rats were found to be significantly different than those of vehicle-treated rats by Student's t-test.
The results of DWI experiments after acute estradiol pretreatment are shown in Figure 3. It can be seen that OVX rats treated with vehicle for 30 mins and then subjected to MCAO exhibited a decrease in ADC ratios for regions 2 and 4 (cortex and striatum, Figures 3A and 3B, respectively) as predicted. In OVX rats treated with estradiol for 30 mins, the decrease in ADC ratios was significantly attenuated in a manner similar to that observed for rats subjected to 7-day estradiol pretreatment (with the exception that the 30 mins value did not reach statistical significance for region 2). Apparent diffusion coefficient ratios for sham-operated rats are not shown in this figure. Estradiol showed similar effects on ADC ratios in regions of interest 1 and 3 (data not shown). For these experiments, attenuation of the decrease in ADC ratios (depending on the time after the start of MCAO) varied from 60% to 81% for region 1 and 47% to 74% for region 3, compared with 37% to 73% for region 2 and 47% to 73% for region 4.
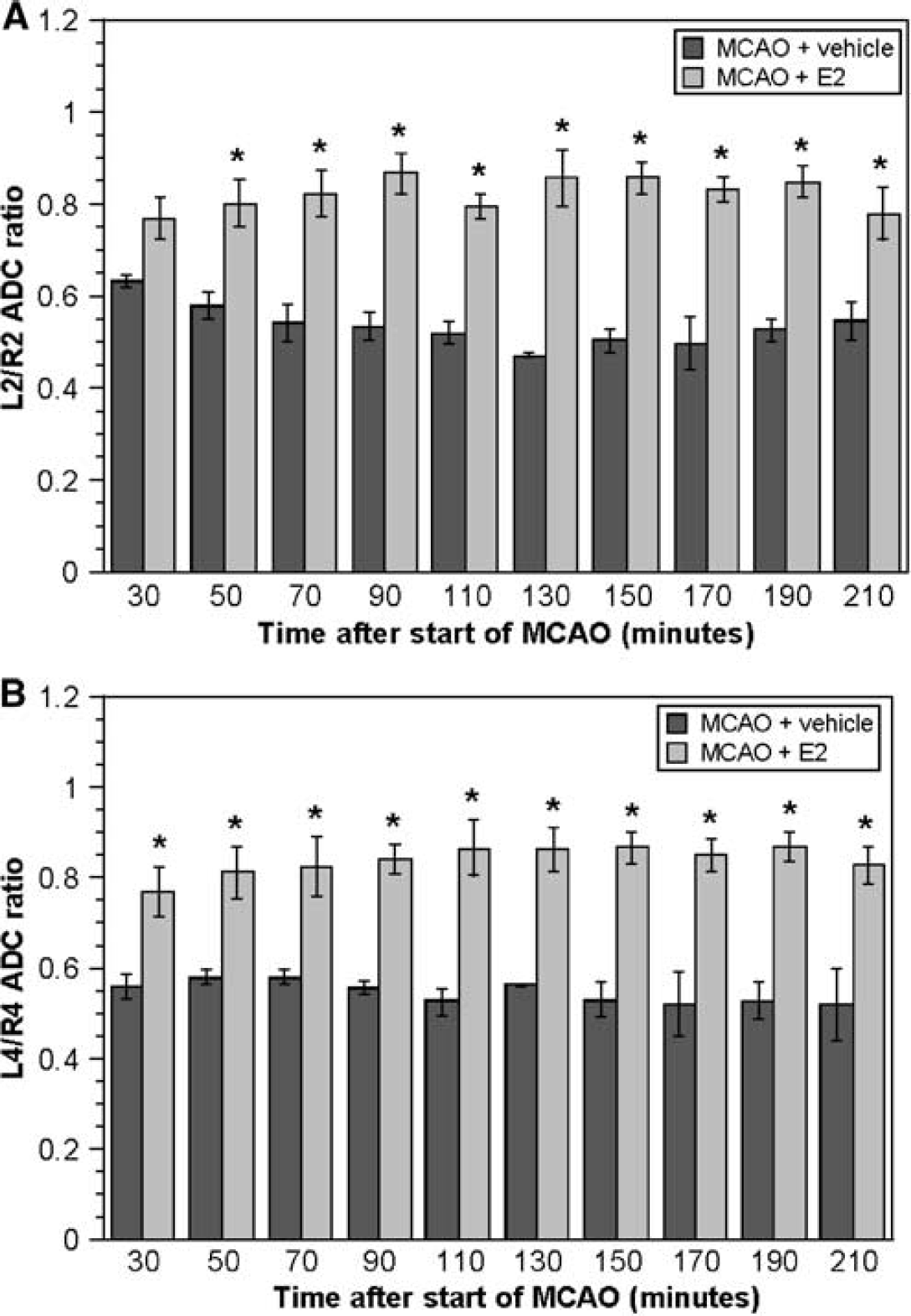
Thirty minutes estradiol pretreatment attenuates ischemia-induced decreases in ADC values in rats subjected to MCAO. Rats were administered an intravenous injection of 17 β-estradiol or vehicle 30 mins before induction of permanent MCAO and ADC values were determined by DWI as described in Materials and methods. Ipsilateral to contralateral (left/right) ADC ratios are shown for cerebral regions 2 (lateral cortex) and 4 (striatum) in (
Estradiol treatment, whether 7-day or 30-min, did not alter CBF in our experiments. Specifically, for rats subjected to a 7-day estradiol treatment, pre-occlusion CBF values were 282.0 ± 31.0 and 277.0 ± 11.4 laser Doppler units for estradiol and vehicle, respectively (mean ± s.e.m. for 5 rats each) and postocclusion CBF values were 74.0 ± 15.2 and 84.0 720.6 laser Doppler units for estradiol and vehicle, respectively (mean ± s.e.m. for 5 rats each). Similarly, for rats subjected to a 30-min estradiol treatment, preocclusion CBF values were 290.0 ± 13.5 and 270.0 ± 10.0 laser Doppler units for estradiol and vehicle, respectively (mean ± s.e.m. for 4 and 3 rats) and postocclusion CBF values were 85.0 ± 22.6 and 83.3 ± 6.7 laser Doppler units for estradiol and vehicle, respectively (mean ± s.e.m. for 4 and 3 rats). In the present study, we also evaluated a number of physiologic parameters in the OVX rats treated with estradiol or vehicle. Table 2 shows that MAP values were not significantly different among OVX rats treated with either estradiol or vehicle for 7 days and subjected to either MCAO or sham surgery. We also found that treating rats with estradiol for 7 days did not alter plasma levels of Na+, K+, Cl-, or HCO−3, nor did the treatments alter pH, pCO2, BUN, glucose, or hemoglobin compared with vehicle-treated rats. These physiologic parameters also did not vary significantly in OVX rats subjected to MCAO after 30 mins treatment with estradiol or vehicle (data not shown).
Physiologic parameters of rats subjected to MCAO with or without 7-day estradiol pretreatment
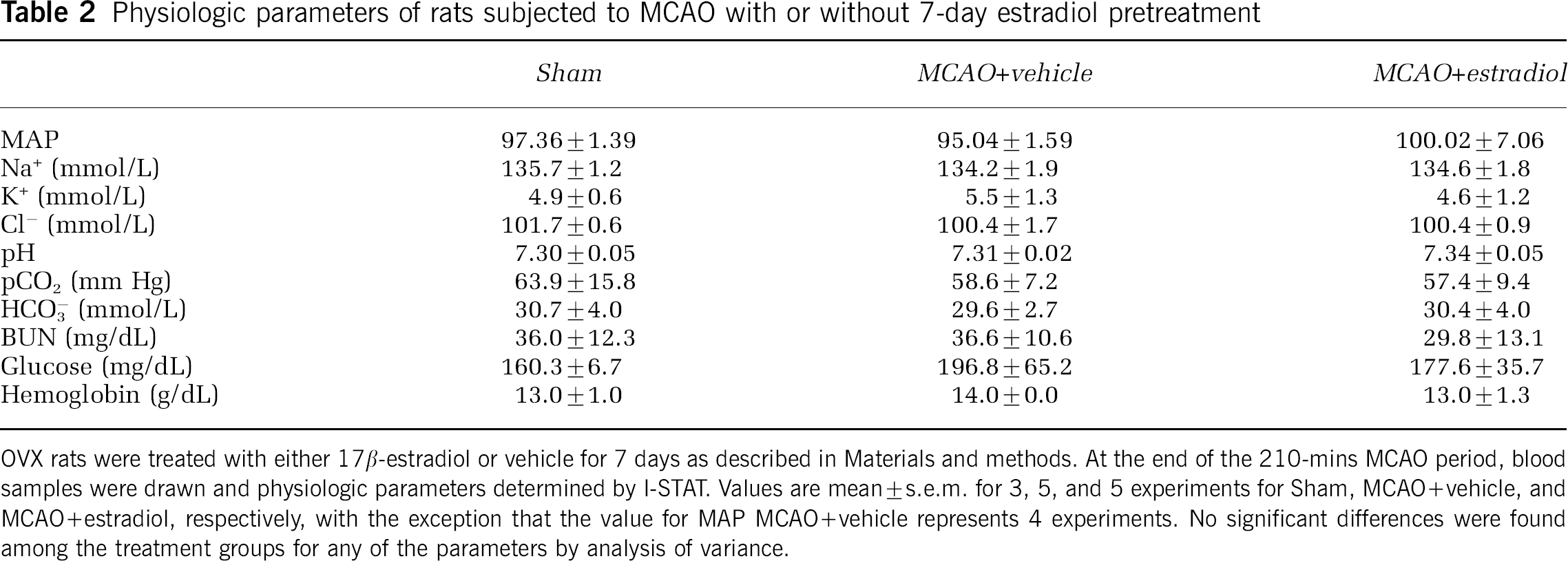
OVX rats were treated with either 17 β-estradiol or vehicle for 7 days as described in Materials and methods. At the end of the 210-mins MCAO period, blood samples were drawn and physiologic parameters determined by 1-STAT Values are mean ± s.e.m. for 3, 5 experiments for Sham, MCAO + vehicle, and MCAO + estradiol, respectively, with the exception that the value for MAP MCAO + vehicle represents 4 experiments. No significant differences were found among the treatment groups for any of the parameters by analysis of variance.
Estradiol Reduces Na–K–Cl Cotransporter Activity of Cerebral Microvascular Endothelial Cells
Our next goal was to determine whether estradiol reduces activity of the BBB Na–K–Cl cotransporter and, in particular, whether estradiol prevents stimulation of the cotransporter under conditions of cerebral ischemia. For these studies, we evaluated the effects of estradiol on Na–K–Cl cotransporter activity of cultured bovine CMECs as described in Materials and methods. In previous studies, we have shown that two prominent components of cerebral ischemia, i.e., hypoxia and AVP, are both potent stimulators of CMEC cotransporter activity (Foroutan et al, 2005; O'Donnell et al, 2005). Thus, in the present study we were particularly interested in evaluating the effects of estradiol on CMEC cotransporter activity stimulated by hypoxia and AVP. First, we found that when we exposed CMECs to estradiol (80 nmol/L in these initial studies) for either 5 mins or 3h, Na–K–Cl cotransporter activity was significantly reduced, as shown in Figure 4A. In testing the effects of exposing CMECs for 5 mins to a range of estradiol doses, we found that concentrations as low as 1 nmol/L significantly reduced activity of the Na–K–Cl cotransporter (Figure 4B). While estradiol decreased cotransporter activity within minutes, this effect was sustained through hours (Figure 4A) and even days. Specifically, a 5-day treatment with estradiol (80 nmol/L) reduced CMEC cotransporter activity to 77.2%± 3.7% of control (vehicle only) (mean ± s.e.m., n = 3 experiments, P < 0.05). To address the question of whether estradiol may attenuate hypoxia-stimulated CMEC cotransporter activity, in the present study we exposed cells to oligomycin in the presence or absence of 80 nmol/L estradiol. As a mitochondrial ATP synthase inhibitor that reduces cell ATP levels, oligomycin is frequently used to induce ‘chemical hypoxia’ and mimic true hypoxia (Dubinsky and Rothman, 1991; Kagawa and Racker, 1966; Nicholls and Budd, 2000). Others and we have shown previously that oligomycin stimulates CMEC cotransporter activity, as does true hypoxia (Foroutan et al, 2005; Kawai et al, 1996). Figure 4A shows that oligomycin (1 mg/mL) stimulated Na–K–Cl co-transporter activity of the CMECs and that either 5-min or 3-h treatments with 80 nmol/L estradiol significantly reduced the oligomycin stimulation and, in fact, even decreased cotransporter activity below the baseline value (in absence of oligomycin or estradiol). In these studies, we also tested the ability of estradiol to reduce CMEC cotransporter activity stimulated by AVP. For this, CMECs were treated for 5 mins with AVP (100 nmol/L) alone or in the presence of a range of estradiol doses from 0.01 to 10 nmol/L. As shown in Figure 5, cells treated for 5 mins with AVP alone exhibited significantly elevated cotransporter activity over control (no AVP or estradiol). However, when cells were treated for 5 mins with AVP plus estradiol at concentrations of 0.10 to 10 nmol/L, cotransporter activity was decreased to levels not significantly different from control. At 0.01 nmol/L, estradiol appeared to partially reduce AVP stimulation of the cotransporter but the effect did not reach statistical significance. The Na–K–Cl cotransporter activities shown in the present study are expressed relative to control cotransporter activity to facilitate comparing the magnitudes of estradiol, oligomycin and AVP effects. The absolute cotransporter activity value for control conditions in Figure 4B was 25.14 ± 2.60 (mean ± s.e.m., n = 8).
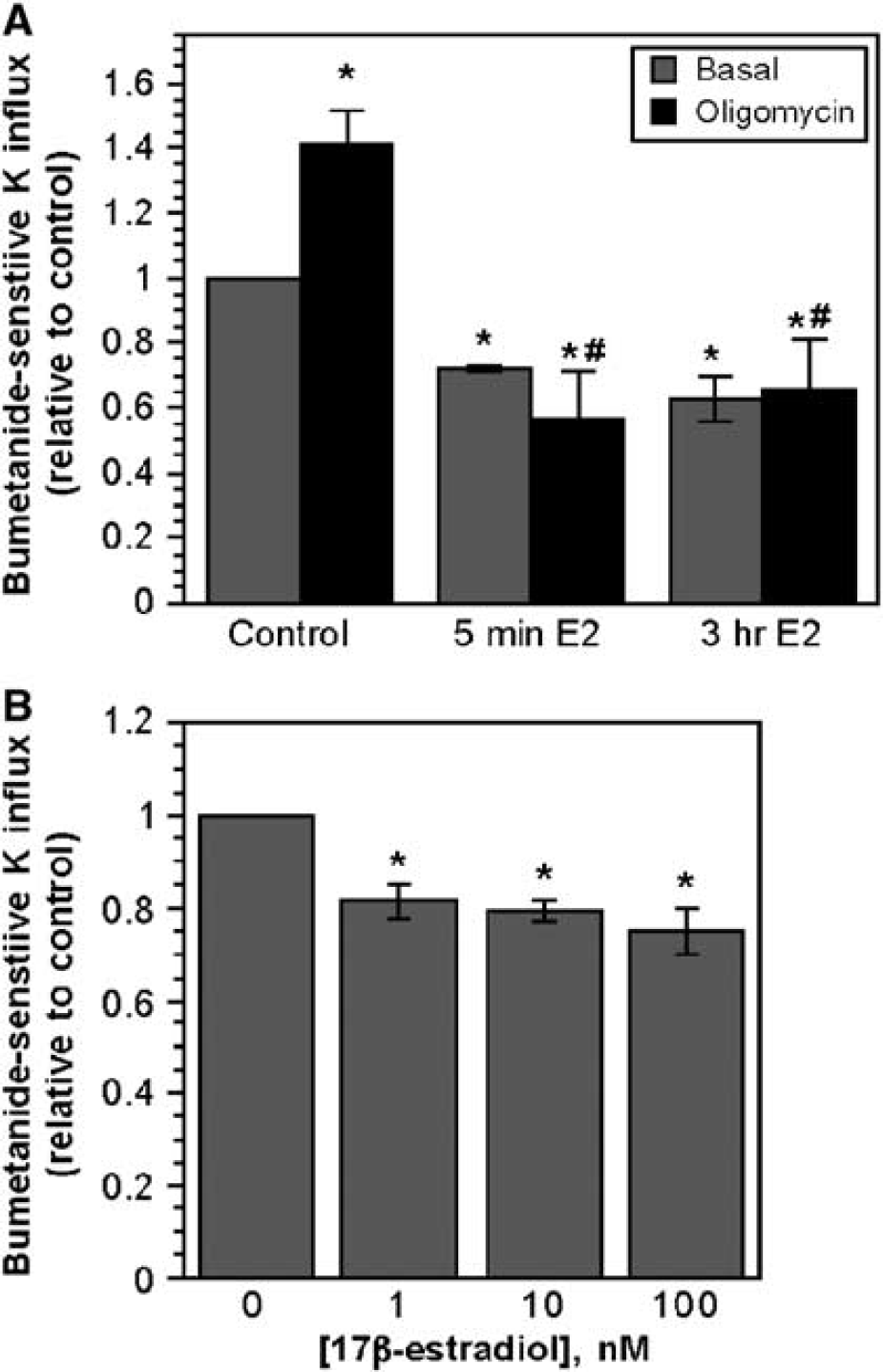
Estradiol inhibition of both basal and oligomycin-stimulated CMEC Na–K–Cl cotransporter activity after short-term exposure to 17 β-estradiol.
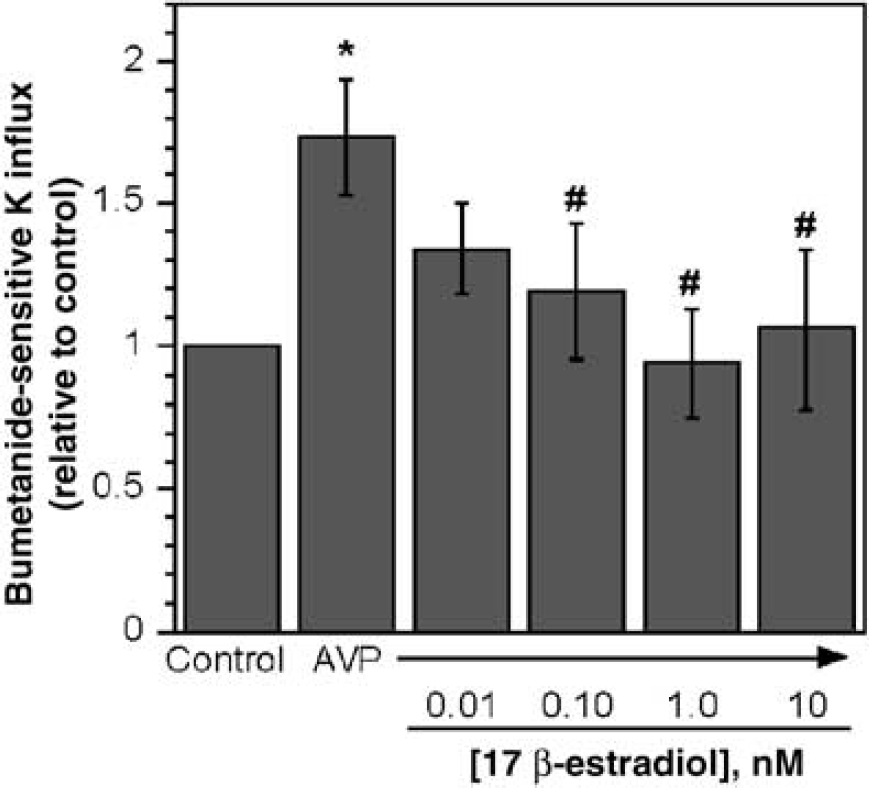
Inhibition of AVP-stimulated Na–K–Cl cotransport activity in CMECs by 17 β-estradiol. Cell monolayers were exposed to 100 nmol/L AVP and varying concentrations of estradiol as indicated for 5 mins then assayed for Na–K–Cl cotransporter activity for 5 mins (in the presence of AVP and estradiol or vehicle) as described in Materials and methods. Data are mean ± s.e.m. of eight experiments. *Value is significantly different from control without AVP or estradiol. *Values are significantly different from AVP without estradiol. Comparisons were made by analysis of variance with a Fisher's PLSD pos thoc test.
As a steroid hormone, estradiol freely permeates the BBB and thus has access to cells in the brain parenchyma. Previous studies have provided evidence that the Na–K–Cl cotransporter of cerebral astrocytes contributes to ischemia-induced swelling of the astrocytes, (Su et al, 2002; Walz, 1992; Yan et al, 2001) and, similar to BBB endothelial cells, the astrocyte cotransporter is stimulated by hypoxia and AVP (Katay et al, 1998; Lenart et al, 2004). In studies conducted as an initial investigation of possible estradiol effects on astrocytes, we found that estradiol also reduces activity of the Na–K–Cl cotransporter in astrocytes, as it does in CMECs. Specifically, cotransporter activity of cultured primary astrocytes was 14.95 ± 1.91 and 11.02 ± 1.75 μmol K/g prot min for cells treated 5 mins with vehicle and 10 nmol/L estradiol, respectively (mean ± s.e.m., 6 experiments each, P < 0.01 by Student's paired t-test). Thus, it appears that estradiol influences activity of the Na–K–Cl cotransporter in both BBB endothelial cells and astrocytes. This will be considered further in the Discussion. Because our previous studies have provided evidence that the BBB cotransporter contributes significantly to cerebral edema formation during the early hours of cerebral ischemia when the BBB is still intact, a primary goal of the present study was to investigate the effects of estradiol on the BBB cotransporter. For this reason, the remainder of the experiments in the present study focused on the CMEC cotransporter.
Estradiol Modulates Na–K–Cl Cotransporter Protein Abundance in Cerebral Microvascular Endothelial Cells
The finding that estradiol can reduce CMEC cotransporter activity after just a 5-min incubation indicates that this effect of estradiol very likely occurs through a mechanism independent of de novo protein synthesis. However, the observation that reduced cotransporter activity is maintained in the continued presence of estradiol for hours or even days raises the possibility that estradiol could also influence cotransporter activity through slower acting mechanisms, for example, transcription and/or translation events leading to change in the amount of cotransporter protein expressed in the cells. As an initial investigation of this possibility, we evaluated the effects of estradiol on abundance of the CMEC Na–K–Cl cotransporter protein. For this, CMECs were exposed to varying doses of estradiol over varying times, then cotransporter abundance determined by Western blot analysis as described in Materials and methods. Figure 6A is a representative Western blot of cotransporter abundance in CMECs exposed to 1 nmol/L estradiol (equivalent to 272 pg/mL) for 1 to 3h. This blot shows that at 1 nmol/L, estradiol caused a marked reduction in cotransporter protein abundance relative to control even after just 1 h of exposure. Figure 6B is a summary of quantitated Western blot results for 1 to 3 h treatments with 1, 10, and 100 nmol/L estradiol. Both 1 and 10 nmol/L estradiol caused significant reductions in cotransporter protein abundance after 1, 2, or 3-h exposures. At 100 nmol/L, cotransporter protein significantly decreased only after 3 h of exposure. It should be noted that in these experiments, the greatest decrease in cotransporter abundance was observed with 1 nmol/L estradiol. Thus, the values for 1- and 3-h exposures to 1 nmol/ L estradiol are significantly different than values for 1-h exposure to 10 nmol/L estradiol and 3-h exposure to 100 nmol/L estradiol, respectively (P < 0.01, paired t-tests). We also tested the effects of exposing CMECs to estradiol for 1 to 7 days. Figure 6C is a representative Western blot of cotransporter protein abundance in CMECs after exposure to 1 nmol/L estradiol for 1, 2, or 3 days. It can be seen here that cotransporter abundance was not reduced by 1 to 3 days exposure to estradiol. Summarized Western blot quantitated results for 1- and 7-day exposures to 1, 10, and 100 nmol/L estradiol are shown in Figure 6D. We found that a 1-day exposure to 1 or 100 nmol/L estradiol caused a slight but significant increase in cotransporter protein (significance was not reached for the 10 nmol/L 1-day treatment). Estradiol exposures of 2 and 3 days (1 to 100 nmol/L) gave similar results (data not shown). However, after 7 days of exposure to 1, 10, or 100 nmol/L estradiol, the cotransporter protein abundance was again reduced to levels below control. Thus, although estradiol exerts a sustained inhibitory effect on cotransporter activity, it appears to cause a marked early decrease in protein abundance (within hours) followed by recovery to levels at or slightly above control (1 to 3 days) and then a subsequent return to levels below control after 7 days. This issue will be considered further in the Discussion.
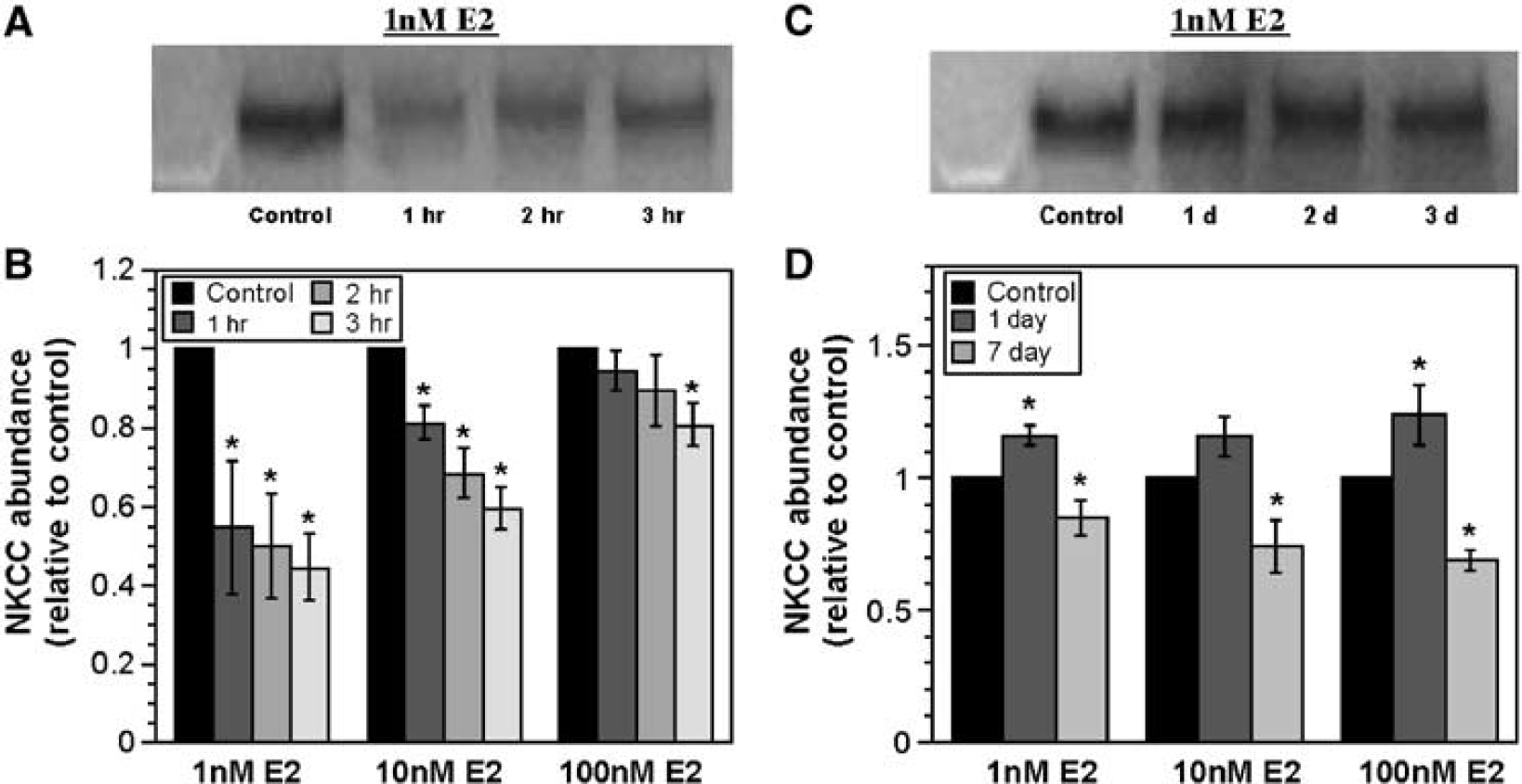
17 β-Estradiol effects on CMEC Na–K–Cl cotransporter protein abundance. Cell monolayers were exposed to growth media containing 1 to 100 nmol/L 17 β-estradiol or vehicle for 1 to 3 h or 1 to 7 days. At the end of the exposure period, cells were lysed and prepared for gel electrophoresis and Western blotting as described in Materials and methods. Blots were probed for Na–K–Cl cotransporter (NKCC) protein with T4 antibody and bands detected by ECL. (
Investigation of the signaling pathways responsible for estradiol modulation of CMEC cotransporter activity and protein abundance is clearly beyond the scope of the present study. However, we have conducted a limited number of experiments to address the question of whether estradiol effects on the cotransporter are estradiol receptor-mediated. For this, we tested the effect of ICI 182,780, a highly selective estrogen receptor antagonist (Howell et al, 2001), on CMEC cotransporter protein abundance in the presence and absence of estradiol. As shown in Figure 7, ICI 182,780 (10 mmol/L) significantly attenuated the decrease in cotransporter protein induced by 3-h exposure to 1 or 10 nmol/L estradiol. This suggests that estradiol effects on the CMEC cotransporter, at least with respect to cotransporter protein abundance, occur via an ICI 182,780-sensitive estradiol receptor.
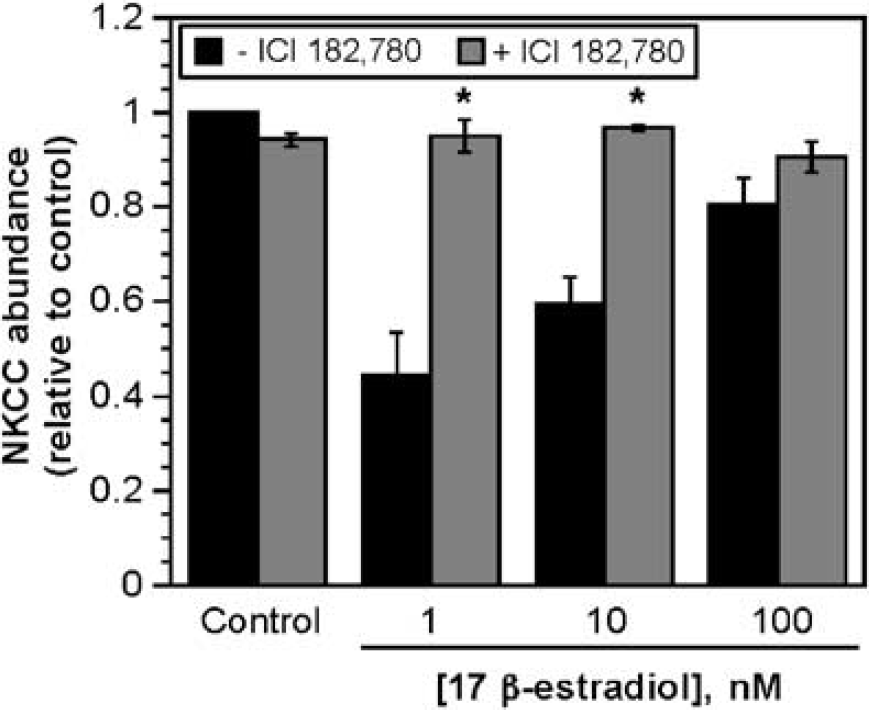
Effects of acute estradiol exposure on CMEC Na–K–Cl cotransporter protein: inhibition by ICI 182,780. Cell monolayers were exposed for 3 h to 1, l0, or 100 nmol/L estradiol in the presence or absence of 10 μmol/L 182,780, a selective estrogen receptor antagonist. Western blot analysis of cell lysates was then performed as described in Materials and methods. Data are mean ± s.e.m. of 4 experiments for all conditions in the presence of ICI 182,780. Data are mean ± s.e.m. of 22, 4, 15, and 4 for control, 1, 10, and 100 nmol/L estradiol in the absence of ICI 182,780. *Values for estradiol and ICI 182,780 are significantly different from values for estradiol without ICI 182,780.
Discussion
In the present study, we used morphometric analysis and TTC staining of brain slices to evaluate estradiol effects of brain swelling and ischemic lesion size after permanent MCAO. In addition, we used magnetic resonance DWI methods to monitor the effects of 17 β-estradiol on the distribution and mobility of water in the ischemic lesion that occurs during the early hours of permanent MCAO. Our results show that both chronic (7-day) and acute (30-min) estradiol treatments of OVX rats significantly attenuate brain swelling and TTC-defined ischemic lesion volume associated with permanent MCAO. Further, these estradiol treatments attenuate MCAO-induced reduction of ADC values in a manner that correlates well with estradiol reductions in brain swelling and lesion volume. In this study, we also used cultured CMECs to assess the effects of 17 β-estradiol on activity and expression of the BBB Na–K–Cl cotransporter. Our studies reveal that estradiol inhibits both hypoxia- and AVP-induced stimulation of the cotransporter and also modulates expression of the BBB Na–K–Cl cotransporter protein. Together, these findings support the hypothesis that estradiol reduces ischemia-induced stimulation of the BBB Na–K–Cl cotransporter, resulting in attenuation of cerebral edema formation.
Our present DWI studies were designed to assess the effects of estradiol on changes in the distribution and mobility of water in the ischemic lesion occurring during the first 3 to 4 h of ischemia in the permanent MCAO model and to compare those changes with brain swelling and TTC-defined lesion volumes obtained in each experiment. The overall goal of our studies has been to better understand the early events occurring during cerebral ischemia that promote brain edema formation before BBB breakdown (4 to 6 h after the onset of ischemia). In contrast, a majority of studies evaluating edema formation in stroke have focused on ischemia/reperfusion, a situation that promotes relatively rapid BBB breakdown. Further, although a variety of studies have shown that estradiol reduces lesion volume in both the permanent MCAO model of ischemia (Dubal et al, 1988) and the reversible MCAO model of ischemia/reperfusion (Rusa et al, 1999; Shi et al, 2001; Toung et al, 1998; Yang et al, 2000; Zhang et al, 1998), little is known about estradiol effects on edema formation occurring during ischemia. To our knowledge, the results of the present study provide the first evidence that estradiol causes a significant reduction of cerebral edema throughout the early hours of permanent MCAO. In addition, our studies show that this is the case whether estradiol is administered to OVX rats via subcutaneous capsule 7 days before MCAO or as an intravenous injection only 30 mins before MCAO (giving mean plasma estradiol levels in our study of ∼57 and ∼ 145 pg/mL, respectively). Similar methods were used in a previous study to investigate estradiol effects on stroke-induced changes in cerebral water distribution during reversible MCAO (Shi et al, 2001). In those experiments, estradiol was administered as an intravenous injection to OVX rats (100 mg/kg) 2h before MCAO and was found to reduce DWI-defined lesion size occurring 30 mins into the occlusion and also several hours after reperfusion, interpreted as reduction of ‘cytotoxic edema’ and ‘vasogenic edema’, respectively, in that study. Our studies now show that the estradiolattenuated decrease in ADC values is maintained throughout at least 3.5 h of permanent MCAO (when the BBB is still intact, i.e., before ‘vasogenic’ edema formation) and further, that edema (assessed as brain swelling) and ischemic injury volume (assessed as TTC staining) are reduced in both cortex and striatum by either 7-day or 30-min estradiol treatment. Other studies have reported varied results with respect to whether estradiol reduces edema and/or ischemia-induced lesion in both cortex and subcortex. Shi and coworkers (Shi et al, 2001) found that injecting estradiol 2 h before MCAO significantly reduced DWI signal intensity in the cortex observed 30 mins after the start of MCAO, while in the subcortex the signal intensity fell without reaching statistical significance. It should be noted that in that study, DWI signal intensity (inversely related to ADC values) of the ipsilateral lesion was monitored rather than determining the ipsilateral/ contralateral ADC ratios for selected cerebral regions as in the present study. Dubal et al (1988) reported that a 7-day estradiol treatment reduced cortical but not striatal infarct volume (determined by TTC staining) after 24 h of permanent MCAO. In that study, estradiol given at the start of MCAO was without effect. Similar to our findings, estradiol has been reported to reduce lesion volume in both the cortex and striatum as determined by TTC staining (Yang et al, 2000), with lesion evaluated after 2 days of permanent MCAO in this case and estradiol administered up to 3 h after the onset of ischemia. It is important to note that these studies vary with respect to estradiol dose, treatment time, and/or occlusion time. Thus, clarifying the role of each of these factors in determining the ability of estradiol to reduce edema and/or ischemic lesion occurring in the cortex and striatum during ischemia will require further investigation.
It is noteworthy that in our studies, the greatest decrease in ADC ratios (80% to 85%) observed occurred within 30 mins of the onset of permanent MCAO. This is consistent with previous reports that within the first 15 mins of stroke onset ADC decreases to 75% of its control value (Sotak, 2004) and thereafter ADC values decrease more slowly. Thus, while the decrease in ADC provides a good index of early changes in brain water distribution and mobility, due at least in part to swelling of astrocytes and some neurons (while other neurons shrink and interstitial brain extracellular volume decreases) (Liu et al, 2001) (Nicholson and Syková, 1998), subsequent further increases in brain water (occurring as water along with Na and Cl continues to enter the brain across the BBB) are not accompanied by a proportional decrease in ADC values (Hasegawa et al, 1994; Knight et al, 1994; Liu et al, 2001; Moseley et al, 1990; Sotak, 2004). In our studies, we found that while cortical ADC values continued to decrease slightly at times beyond the first 30 mins of occlusion, the decrease was not statistically significant. Further, no decrease in ADC values was observed in striatum beyond the initial decrease at 30 mins after induction of MCAO. This is in contrast to a previous report that ADC values can continue to decrease throughout 210 mins of permanent MCAO in both Sprague-Dawley and Wistar Kyoto rats (Bardutzky et al, 2005). However, in that study CBF was reduced to ∼ 36% of preocclusion flow whereas in our experiments MCAO reduced CBF to 26% to 30% of preocclusion flow, suggesting the presence of a more pronounced ischemia in our studies. Thus, it is possible that in our studies the lower CBF produced a more rapid onset of edema and thus a more pronounced early decrease in ADC values. Despite the fact that the MCAO-induced decrease in ADC is an early event, our experiments were designed to extend over a time course of 210 mins to determine whether the effects of estradiol on attenuating the decrease in ADC are sustained throughout the period of permanent MCAO. Our data show that the effects of estradiol are indeed sustained throughout 210 mins of MCAO. It is also noteworthy that the effects of estradiol on reduction of MCAO-induced brain swelling, TTC lesion volume, and decrease in ADC values are in reasonable agreement. For example, 7-day estradiol pretreatment was found to reduce brain swelling by 66% and TTC lesion by 43% after 210 mins of permanent MCAO while attenuating the decrease in ADC by 55% to 78%, depending on the brain region and time after onset of MCAO. Similarly, 30-min estradiol treatment reduced brain swelling by 62%, TTC lesion volume by 77% and attenuated the decrease in ADC by 37% to 81% depending on brain region and time after onset of MCAO. This suggests that for our permanent MCAO model of early cerebral ischemia, the changes in brain water distribution and mobility, observed as a decrease in ADC values, correlate well with both brain edema as assessed by brain swelling and brain lesion as assessed by TTC staining.
The second aspect of the present studies focused on evaluating the possibility that estradiol reduces the ability of the BBB Na–K–Cl cotransporter to contribute to edema formation during the early hours of cerebral ischemia. The results of our studies reveal that estradiol abolishes stimulation of the cotransporter by oligomycin (chemical hypoxia) and by AVP, and even reduces basal Na–K–Cl cotransport activity in cultured CMECs. The cotransporter activity-lowering effect of estradiol appears to be mediated via a rapidly acting mechanism, occurring with as little as 5 mins of exposure, but can also be sustained for hours and even days in the continued presence of estradiol. In addition, estradiol significantly reduces AVP-stimulated Na–K–Cl cotransporter activity at physiologic to supraphysiologic doses (e.g., 0.1 and 1 nmol/L, equivalent to 27.2 to 272 pg/mL estradiol), within the range of doses observed to reduce brain swelling, lesion volume and falling ADC values in our MCAO DWI studies. The lowest dose of estradiol that appears to effectively lower basal CMEC cotransporter activity is 1 nmol/L. This suggests that while ischemia-stimulated Na–K–Cl cotransporter activity can be reduced by physiologic levels of estradiol, reducing basal activity of the cotransporter occurring in normoxic conditions (i.e., in the absence of stimulatory ischemic factors such as AVP and hypoxia) requires higher doses of estradiol. It should also be noted that while estradiol causes a modest reduction of basal cotransporter activity, it abolishes stimulation of cotransporter activity by AVP or by chemical hypoxia. It is also noteworthy that estradiol decreases cotransporter activity to the basal level in AVP-stimulated CMECs but additionally decreases cotransporter activity below the basal level in oligomycin-stimulated CMECs. The reasons for this are as yet unclear but are likely because of differences in oligomycin- and AVP-induced signaling pathways that stimulate CMEC cotransporter activity. These studies examining the effects of estradiol on AVP- and oligomycin-stimulated cotransporter activity were conducted as an initial evaluation of whether estradiol might reduce ischemia-stimulated cotransporter activity. In recent studies, we have found that CMEC Na–K–Cl cotransporter activity is very sensitive to stimulation by true hypoxia, with significant stimulation occurring over a range of hypoxia levels from modest to severe and further, that aglycemia, whether alone or in combination with hypoxia, also stimulates the CMEC cotransporter (Foroutan et al, 2005). Thus, additional investigations will need to examine the effect of estradiol on cotransporter activity in the presence of true hypoxia. Nevertheless, these findings collectively support the hypothesis that during cerebral ischemia, estradiol significantly reduces or prevents stimulation of Na–K–Cl cotransporter activity by AVP and hypoxia, resulting in reduced cerebral edema formation. Further, the finding that estradiol inhibition of hypoxia- and AVP-stimulated cotransporter activity occurs within minutes of exposure is consistent with our observation that estradiol reduces brain swelling and the decrease in ADC values even when administered only 30 mins before induction of MCAO. In addition, our finding that estradiol reduction of hypoxia-stimulated cotransporter activity is sustained over 3 h is consistent with the observation that the estradiol-attenuated decrease in ADC values is sustained throughout 3 h of permanent MCAO.
Few other studies have examined estradiol effects on BBB transporters. However, a number of studies have provided evidence that the BBB is a target for estradiol protective effects in stroke. In this regard, it has been shown previously that estradiol, when administered to OVX rats (100 mg/kg) 2 h before MCAO, increases expression of glucose transporter-1 and that cultured CMECs treated with estradiol (2 nmol/L) show increased survival after ischemic conditions (in this case, anoxia and hypoglycemia) (Shi et al, 1997). Treating cultured CMECs with estradiol has also been found to reduce the rise in intracellular [Ca] ([Ca]i) and resulting cell damage caused by 3-nitropropionic acid-induced chemical hypoxia (Mogami et al, 2002). The findings of our present study suggest that the Na–K–Cl cotransporter is another important target for the beneficial effects of estradiol at the BBB during ischemia. Further, our observation that estradiol also reduces activity of the astrocyte Na–K–Cl cotransporter suggests that the neuroprotective effects of estradiol extend beyond the BBB to include astrocytes. This is important because BBB endothelial cells and astrocytes comprise two major cell types of the neurovascular unit, and appear to be functionally coupled in a number of respects, including metabolism and electrolyte homeostasis of the brain (Hawkins and Davis, 2005; Leybaert, 2005; Simard and Nedergaard, 2004). Thus, it is likely that reduction of cerebral edema formation in permanent MCAO includes estradiol effects on both BBB and astrocyte Na–K–Cl cotransporters. Additional studies are needed to further explore these issues.
While estradiol reduces Na–K–Cl cotransporter activity in a manner that is sustained over time, the effects of estradiol on cotransporter protein abundance vary both with time of exposure and dose of estradiol. We were somewhat surprised to observe that the CMEC cotransporter protein is relatively quickly (within 1 to 3 h of exposure) decreased on exposure of the cells to estradiol (1 to 100 nmol/L). Additional experiments will be needed to clarify the mechanisms responsible for this decrease. However, there are at least two possibilities. Estradiol could increase degradation of the cotransporter protein or alternatively, if there is a relatively high turnover rate of the protein, estradiol could simply reduce synthesis to bring about decreased cotransporter abundance. While the cells appear to recover and even slightly increase cotransporter protein by 1 to 3 days of estradiol exposure, abundance of the protein decreases again after 7 days of exposure. Sorting out the events that bring about these modulations in the amount of CMEC cotransporter protein will require further investigation. Future experiments will also need to address whether estradiol alters cotransporter protein abundance under ischemic conditions as well as normoxic conditions as evaluated in this study.
The signaling pathways whereby estradiol reduces CMEC Na–K–Cl cotransporter activity and cotransporter protein abundance are as yet unclear. While our studies reveal that the early effect of estradiol on decreasing cotransporter protein abundance is blocked by ICI 182,780, and therefore likely to be estradiol receptor-mediated, we do not yet know whether estradiol effects on cotransporter activity are blocked by ICI 182,780. Although an investigation of signaling processes that mediate the actions of estradiol on the CMEC Na–K–Cl cotransporter is beyond the scope of the present studies, previous studies provide some insight regarding the pathways that may be involved. First, we have shown previously that the CMEC cotransporter is stimulated by elevation of [Ca]i and also that AVP stimulates cotransporter activity through a mechanism dependent on elevation of [Ca]i (O'Donnell et al, 2005, 1995). In addition, it has been shown that transient increases in [Ca]i occur within 1 h of exposing CMECs to hypoxia (Brown et al, 2003) (1% O2 in those studies). We have also shown previously that both hypoxia- and AVP-induced CMEC cotransporter stimulation is associated with increased phosphorylation of the cotransporter protein (Foroutan et al, 2005). Further, we know that CMEC cotransporter activity is reduced by elevation of cGMP (O'Donnell et al, 1995) and estradiol has been shown to elevate endothelial nitric oxide levels (Haynes et al, 2000; Hisamoto et al, 2001), leading to increased CMEC cGMP (Palmon et al, 1998). Thus, it is possible that estradiol could reduce CMEC cotransporter activity through an elevation of cGMP. It is also possible that estradiol could inhibit AVP- and hypoxia-induced stimulation of the cotransporter by interfering with elevation of [Ca]i and/or the resulting phosphorylation of the cotransporter protein. However, estradiol is well known to activate a number of other signaling pathways, including Akt kinase and MAP kinase. For example, estradiol has been shown to activate Akt kinase in endothelial cells (Haynes et al, 2000; Hisamoto et al, 2001) and evidence has been provided that activation of MAP kinase is involved in estradiol-induced neuroprotection (Belcher and Zsarnovszky, 2001; Dhandapani and Brann, 2002; Linford et al, 2000). Clarifying whether any or all of these signaling pathways are involved in estradiol effects on the CMEC Na–K–Cl cotransporter awaits further investigation.
In summary, the key findings of the present study are: (1) that estradiol reduces brain swelling, ischemic injury volume, and the accompanying changes in brain water distribution and mobility that occur in the early hours of cerebral ischemia; and (2) that estradiol also inhibits AVP- and hypoxia-induced stimulation of the BBB Na–K–Cl cotransporter. Together with our previous demonstration that the BBB Na–K–Cl cotransporter is a major contributor to ischemia-induced cerebral edema formation, these findings support the hypothesis that estradiol neuroprotective effects in stroke include reduction of cerebral edema through inhibiting ischemia-induced stimulation of BBB Na–K–Cl cotransporter.