
Abstract
17β-Estradiol (E2) was shown to exert neuroprotective effects both in
Introduction
17β-Estradiol (E2) has been shown to have protective effects against brain injury and neurodegeneration in several
Material and methods
Animals
All mouse strains were crossed to a C57BL/6 background. C57BL/6 mice were obtained from Charles River (Sulzfeld, Germany). Crossing the mice to a C57BL/6 background for four generations guarantees a nearly complete C57BL/6 background.
Myeloid-specific ERα mutant mice were generated by breeding mice expressing the Cre recombinase under control of the lysozyme M promoter (Clausen et al, 1999) with mice harboring loxP site-modified ERα alleles (ERfl/fl) (Wintermantel et al, 2006). To generate mice lacking ERα in the neurons of the forebrain, mice expressing the Cre recombinase under control of the CaMKII promoter were bred with ERfl/fl mice (Wintermantel et al, 2006).
Microglial Cell Culture
Microglial cells were isolated from newborn mice (P1) as described previously (Burudi et al, 1999). Flasks (one 75-cm2 flask for three brain samples) were coated with poly-L-lysine (0.01%). The brains of 1-day-old mice were isolated and collected in a cell culture dish containing Hank's balanced salt solution. The meninges were removed. The brains were then homogenized and lysates were incubated for 20 mins at room temperature after adding 1 mL of 1% trypsin. After 20 mins, Falcon tubes containing the cell lysate were filled to a volume of 50 mL with complete medium (complete Dulbecco's modified Eagle's medium (cDMEM): DMEM, 10% fetal bovine serum, 100 Units/mL penicillin, 100 μg/mL streptomycin, and 2 mmol/L glutamine) and centrifuged for 10 mins at 180
Estrogen Receptor-α Immunocytochemistry
Coverslips with attached microglial cells were washed with phosphate-buffered saline (PBS)/MgCl2 and fixed with 4% paraformaldehyde. Thereafter, the cells were treated for 10 mins with 50 mmol/L NH4Cl/PBS and permeabilized for 15 mins with 0.1% Triton-X-100/PBS. The cells were washed with PBS/MgCl2 and incubated for 20 mins with 5% NSS (normal sheep serum). Incubation with anti-ERα antibody (polyclonal anti-ERα MC-20, Santa Cruz (Heidelberg, Germany) sc-542, diluted 1:500 in 5% NSS) was performed overnight at 4°C. The cells were then washed thrice for 5 mins and incubated for 30 mins with the secondary antibody (anti-rabbit AlexaFluor 594, Invitrogen (Karlsruhe, Germany), diluted 1:500 in 5% NSS). After washing the cells thrice for 5 mins with PBS/MgCl2, they were incubated overnight with isolectin GS-IB4 AlexaFluor 488 from
For detection of ERα in sections, the brains were fixed in 4% paraformaldehyde for 48 h. After blocking of endogenous peroxidase activity, coronal vibratome sections (40-μm-thick) were incubated overnight with anti-ERα. The primary antibody was detected with biotinylated anti-rabbit antibody, the ABC-peroxidase system (Vector, Burlingame, CA, USA), and 3,3′-diaminobenzidine (Sigma, Munich, Germany) as substrate.
Real-Time Reverse Transcription-PCR
Cortices were dissected and homogenized using an Ultra-Turrax T8 homogenizer (IKA Werke, Staufen, Germany). For RNA isolation, we used the RNeasy Mini Kit (Qiagen, Hilden, Germany). We removed genomic DNA from the lysate by using RNase-free DNase (Qiagen). RNA was transcribed using the SuperScript II Reverse Transcriptase Kit (Invitrogen). For real-time PCR, the Absolute QPCR-mix (Thermo Scientific, Karlsruhe, Germany) and the following primers and probes were used: ERα, Mm 00433149_m1; hypoxanthine guanine phosphoribosyl transferase (HPRT), Mm 00446968_m1 (Applied Biosystems, Darmstadt, Germany). Results were normalized to HPRT expression.
Middle Cerebral Artery Occlusion
Eight-week-old female mice were anesthetized with intraperitoneal injection of 150 μL of 2.5% tribromoethanol per 10 g body weight and ovariectomized. Both female and male mice received a pellet containing 0.025 mg E2 with a release time of 21 days (Innovative Research of America, Sarasota, FL, USA). Control animals received a placebo pellet. Mice were randomly assigned to treatment groups. Ten days after ovariectomy and implantation of pellets, E2 plasma levels were determined by a chemiluminescent immunoassay and the ADVIA centaur (Siemens, Nürnberg, Germany). 17β-Estradiol-treated ovariectomized mice had E2 levels in the physiologic range (ovariectomized, E2-treated, 68.4 ± 13.3 pg/mL,
Coronal serial frozen sections of the forebrain were prepared. Silver staining was performed to determine the infarct volume as described previously (Schneider et al, 1999). Stained sections were scanned at 300 d.p.i., and the infarct area was measured using the Scion ImageJ software (Scion, Frederick, MD, USA). To correct for edema formation, the difference between areas of the left and right hemispheres was subtracted from the measured silver-negative area (Swanson et al, 1990). For calculating the whole brain infarct volume, the infarct areas were added and multiplied by the distance between the sections (0.4 mm). Surgery was performed, and ischemic damage was measured by an investigator who had no knowledge of the treatment group or the genotype. Animal experiments were approved by the Regierungspräsidium Karlsruhe.
Measurement of Physiologic Parameters
Measurement of arterial blood pressure, pulse, and blood gases was carried out both before and after ischemia in a separate cohort of mice. Mice were kept under tribromoethanol anesthesia on a heating pad at 37°C, and the body temperature was measured using a rectal thermometer. For measuring blood pressure and pulse, a cannula was inserted into the right femoral artery. A blood sample of 150 μL per mouse was collected in a heparin-coated glass capillary for analysis of arterial blood gas, hemoglobin, and glucose levels. The catheter was washed with 200 μL NaCl solution mixed with 50 IE (international units) of heparin before measuring blood pressure. For laser Doppler measurements, the electrode (P415-205; Perimed, Jarfalla, Sweden) was placed 3 mm lateral and 6 mm posterior to the bregma. Relative perfusion units were determined (Periflux 4001; Perimed).
Statistical Analysis
Student's
Results
17β-Estradiol treatment is neuroprotective in various rodent models of stroke (Gibson et al, 2006). To verify the efficacy of E2 treatment in a model of permanent distal MCAO using C57BL/6 mice, we implanted ovariectomized female mice with E2 or placebo pellets. Ten days later, mice were subjected to MCAO and killed after 48 h to determine the infarct volume. 17β-Estradiol treatment significantly reduced the infarct volume (Figure 1).
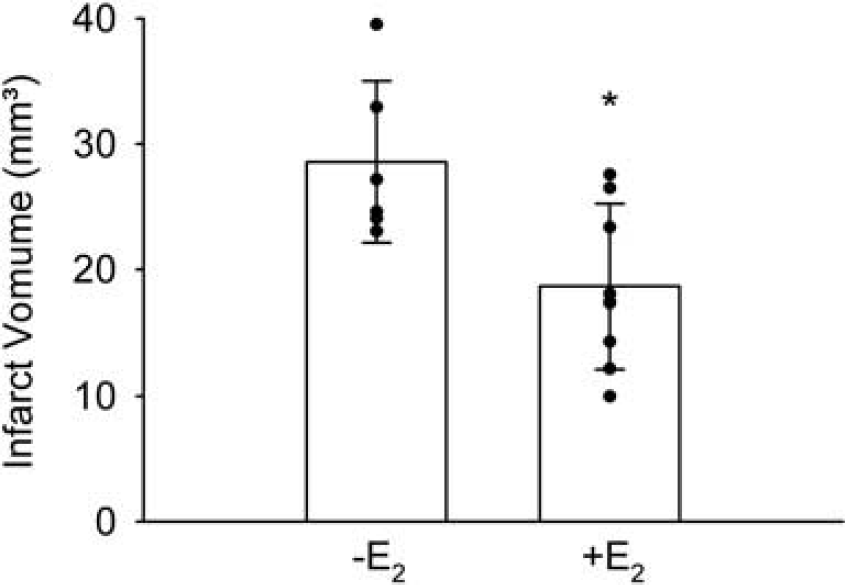
17β-Estradiol (E2) reduced the infarct volume in ovariectomized mice that were subjected to permanent distal MCAO. The infarct volume was determined 48 h after MCAO. Means ± s.d. and individual values are depicted (
To investigate the role of ERα in myeloid cells including microglia, we generated myeloid lineage-specific ERα mutant mice by breeding mice expressing the Cre recombinase (Cre) under control of the lysozyme M promoter (Clausen et al, 1999) with mice harboring a conditional ERα allele (LysCre/ERfl/fl) (Wintermantel et al, 2006). To investigate the recombination efficiency, we detected nuclear ERα expression in isolated microglial cells by immunocytochemistry. All the microglia obtained from control mice were stained (Figures 2A and 2C), whereas 92% of the microglial cells isolated from LysCre/ERfl/fl mice showed no immunoreactivity for ERα (Figures 2B and 2C). 17β-Estradiol treatment reduced the infarct volume of control mice (ERfl/fl). After E2 treatments, the infarct size of LysCre/ERfl/fl mice and control mice did not differ (Figure 3), indicating that the absence of microglial ERα does not affect ischemic brain damage in E2-treated mice.
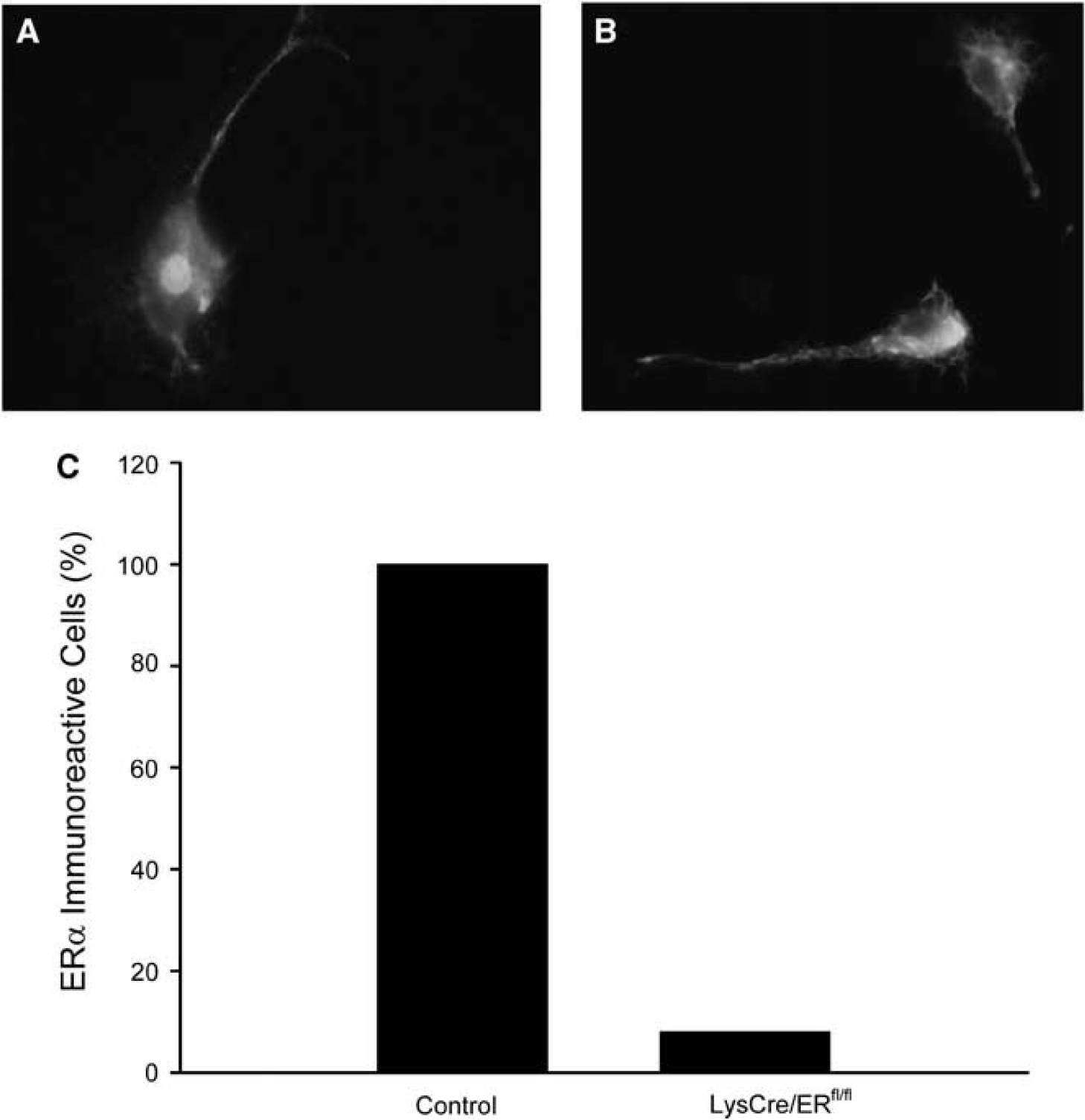
Deletion of ERα in microglial cells of LysCre/ERfl/fl mice. (
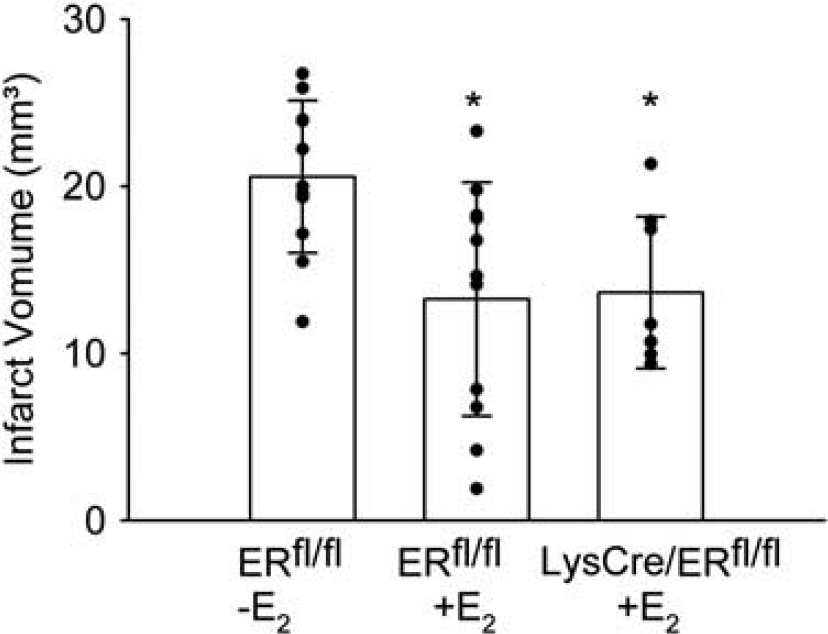
Deletion of ERα in microglial cells did not interfere with the neuroprotective effect of 17β-estradiol (E2). The infarct volume of E2-treated female LysCre/ERfl/fl (
To analyze the contribution of neuronal ERα, we deleted ERα in the neurons of the forebrain by breeding mice expressing the Cre recombinase under control of the CaMKII promoter with ERfl/fl mice (Wintermantel et al, 2006). As described previously for ERα expression in hypothalamic neurons, we found a marked reduction of ERα immunoreactivity in the cortices of CaMKIICre/ERfl/fl mice (Figure 4A). Quantification of ERα mRNA by real-time reverse transcription PCR 24 h after MCAO confirmed this finding. Estrogen receptor-α mRNA was upregulated in the ischemic ipsilateral cortex compared with the contralateral side (Figure 4B) in accordance with a report that cerebral ischemia induces ERα expression (Dubal et al, 2006). However, in CaMKIICre/ERfl/fl mice, ERα expression was markedly reduced both in the ischemic and the contralateral sides (Figure 4B).
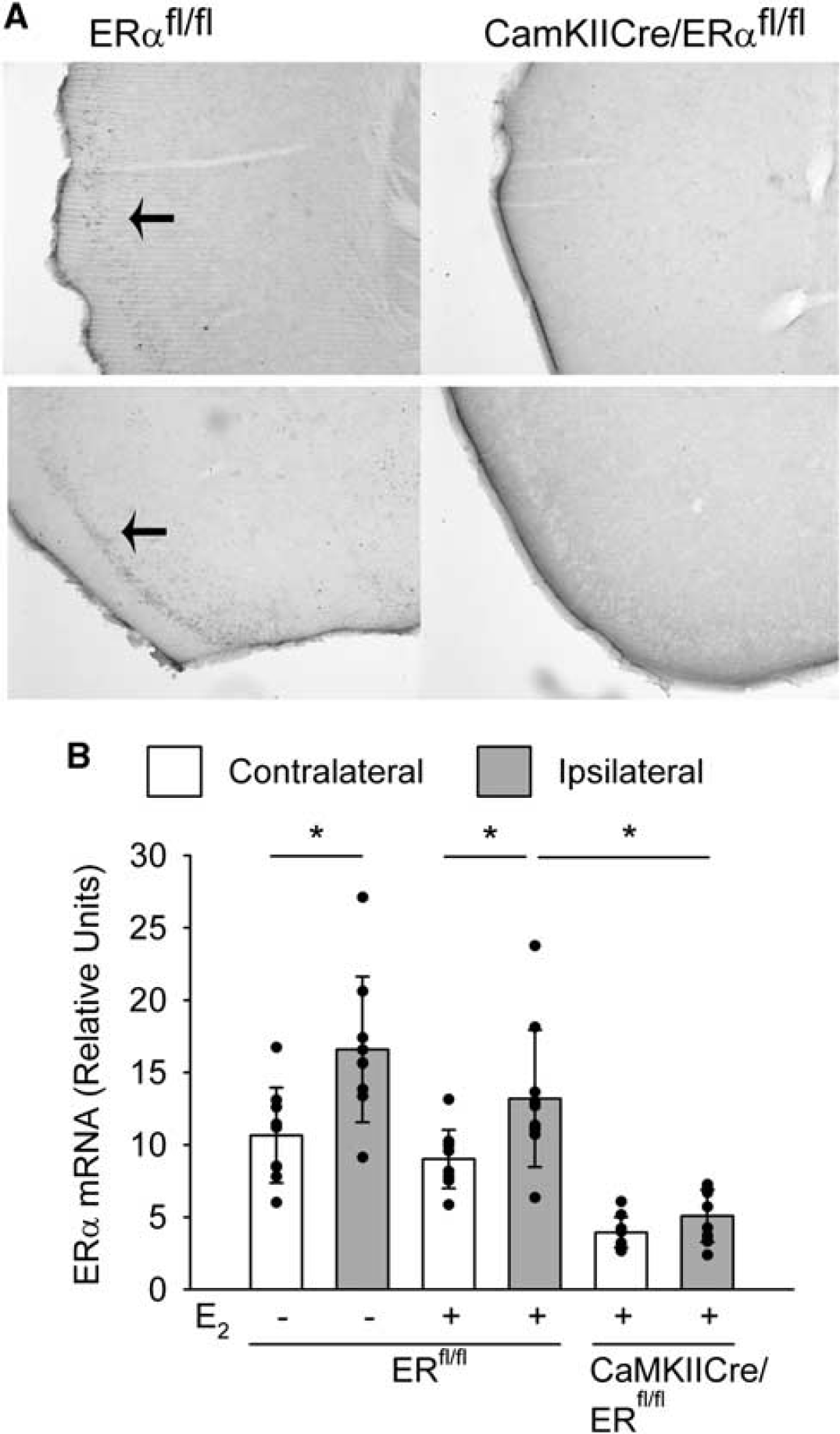
Reduced ERα expression in CaMKIICre/ERfl/fl mice. (
17β-Estradiol-treated female CaMKIICre/ERfl/fl mice showed an increased infarct volume compared with E2-treated control mice 48 h after MCAO (Figures 5A and 5B). Infarct volumes of E2-treated CaMKIICre/ERfl/fl mice were similar to those of the placebo-treated control animals. This finding was reproduced by an independent experiment (infarct volume of ovariectomized ERfl/fl mice without E2 treatment, 21.0 ± 3.6 mm3,
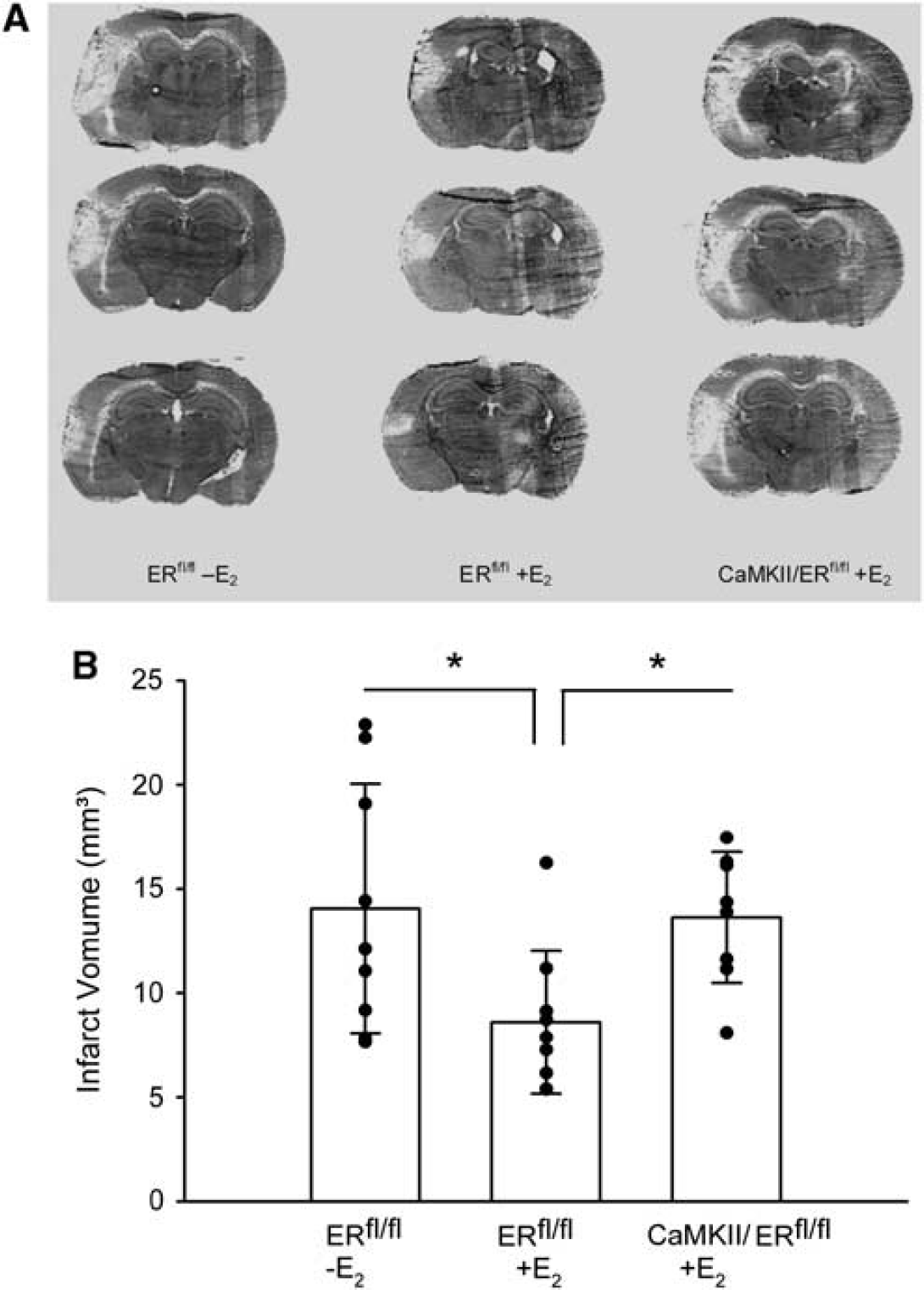
Deletion of ERα in neurons abolished the neuroprotective effect of 17β-estradiol (E2) in female mice. (
To investigate whether this mechanism is sex-specific, E2-treated male CaMKIICre/ERfl/fl and control mice were also investigated in the MCAO model. Similar to female mice, E2-treated male CaMKIICre/ERfl/fl mice showed an increased infarct volume compared with E2-treated control mice (Figure 6), suggesting that neuroprotection by E2 depends on neuronal ERα.
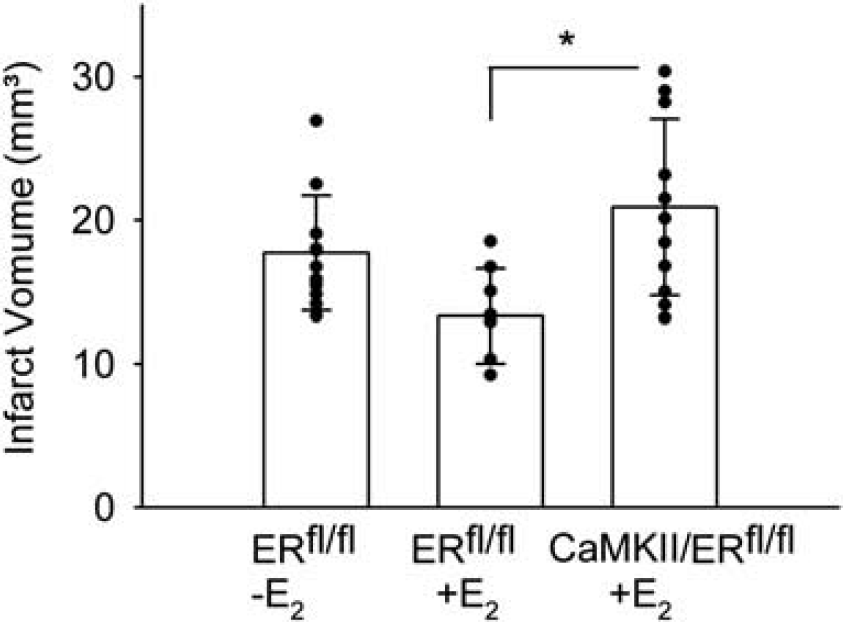
Deletion of ERα in neurons abolished the neuroprotective effect of 17β-estradiol (E2) in male mice. The infarct volumes of E2-treated male CaMKIICre/ERfl/fl mice (
To exclude differences in cardiovascular parameters between control and CaMKII/ERfl/fl mice responsible for the phenotype, we measured blood pressure, pulse, blood gases, and glucose levels before and after MCAO (Table 1). No significant differences were detectable between genotypes.
Preischemic and postischemic physiologic parameters of female ERαfl/fl and CaMKIICre/ERfl/fl mice, both treated with E2
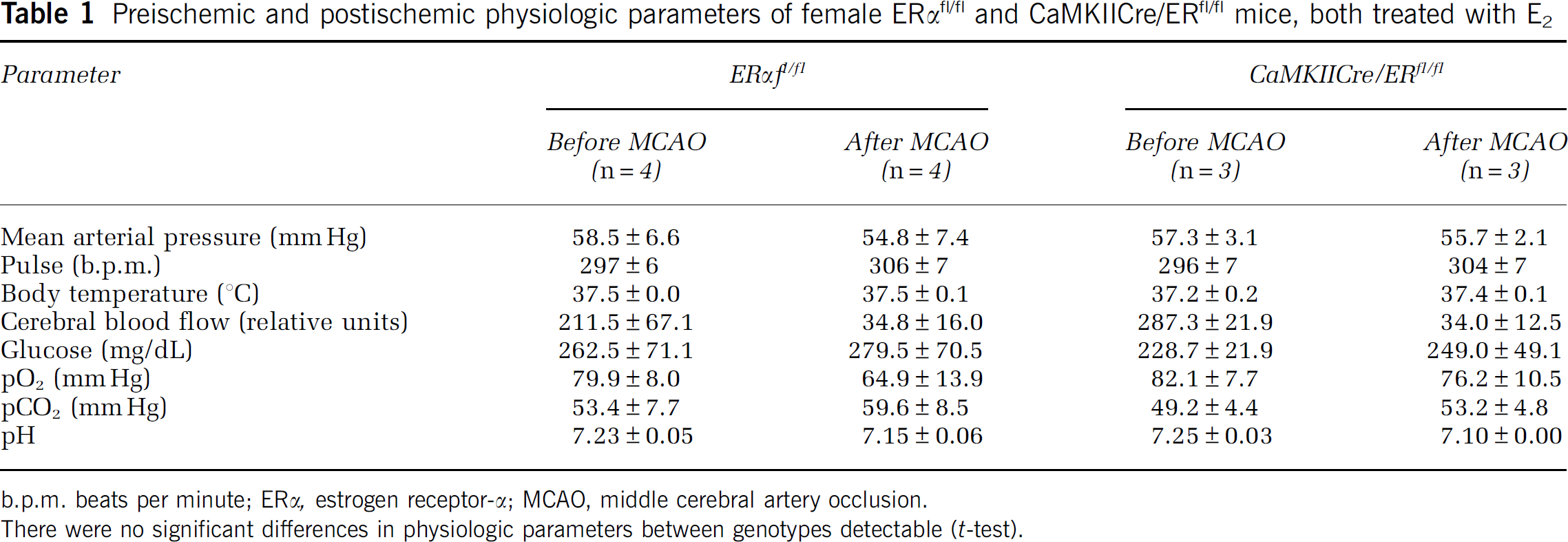
b.p.m. beats per minute; ERα, estrogen receptor-α; MCAO, middle cerebral artery occlusion.
There were no significant differences in physiologic parameters between genotypes detectable (
Discussion
The neuroprotective effect of estrogens in physiologic doses is well established (Gibson et al, 2006; Strom et al, 2009), but the molecular and cellular mechanisms remain controversial. Although receptor-independent effects of E2 have been reported (Culmsee et al, 1999), there is ample evidence that suggests that ERs mediate most of the neuroprotective effect. In line with this concept, the ER antagonist ICI182780 increased the infarct volume after MCAO (Sawada et al, 2000). Selective ER agonists have been used to define which of the two ERs reduces ischemic brain damage. These studies suggested that in models of global ischemia, ERβ is involved in neuroprotection (Carswell et al, 2004; Miller et al, 2005), but in focal cerebral ischemia only an ERα agonist was neuroprotective (Farr et al, 2007). However, a neuroprotective function of ERα has been challenged by the finding that after transient MCAO, the infarct size did not differ in control and ERα knockout mice (Sampei et al, 2000). In this study, mice were not ovariectomized. As ERα knockout mice have highly elevated serum levels of E2 (Couse et al, 1995), the effect of ERα may have been compensated by an ERα-independent effect of high E2 levels. Indeed, deficiency of ERα but not of ERβ abolished the protective effect of E2 in ovariectomized mice subjected to focal cerebral ischemia (Dubal et al, 2001). Our data in CaMKIICre/ERfl/fl mice support the neuroprotective role of ERα.
Estrogen receptor-α is expressed in many cells of the brain, including neurons, astrocytes, microglia, and endothelial cells. Therefore, the localization of ERα involved in the protection against ischemic damage is not self-evident. Numerous
The results show that ERα in the microglia and possibly in other cells of myeloid lineage does not mediate the effect of E2 on infarct volume. We investigated the infarct volume 48 h after MCAO, when the microglia was activated in the MCAO model we used (Muhammad et al, 2008). However, we cannot exclude the fact that at other time points ERα deletion in myeloid cells may affect the infarct size.
In contrast to LysCre/ERfl/fl mice, neuronal ERα knockout animals had larger infarcts than did controls after E2 treatment. In fact, the infarct size of E2-treated CaMKIICre/ERfl/fl mice was identical to that of untreated controls. We cannot exclude that ERα exerts an E2-independent effect on the infarct size. It is also conceivable that androgen levels were altered in male CaMKIICre/ERfl/fl mice, as reported in ERα knockout mice (McDevitt et al, 2007), and that androgens may have affected ischemic brain damage, because male mice were not castrated in our experiments. However, the most likely explanation of our findings is that neuroprotection by E2 is mediated by neuronal ERα. The introduction of the CaMKIICre gene to delete ERα from neurons is a possible confounding factor of our results. As previous experiments found no effect of other Cre genes on ischemic brain damage (Inta et al, 2006; data not shown), it seems very unlikely that the CaMKIICre gene itself could lead to enlarged infarct volumes.
This study suggests neuronal ERα as one of the major targets of E2-mediated neuroprotection in a model of cerebral stroke. To our knowledge, this is the first time that the neuroprotective actions of ERα in stroke were investigated on a cellular level
In view of these
Footnotes
TMW is an employee of Bayer Schering Pharma AG.
Acknowledgements
We are grateful to R Geibig and M Westphal for technical support.