
Abstract
Administration of the selective calpain inhibitor AK295 has been shown to attenuate motor and cognitive dysfunction after brain trauma in rats. To explore mechanisms underlying the behavioral efficacy of posttraumatic calpain inhibition, we investigated histologic consequences of AK295 administration. Anesthetized Sprague-Dawley rats received lateral fluid percussion brain injury of moderate severity (2.2 to 2.4 atm) or served as uninjured controls. At 15 minutes after injury, animals were randomly assigned to receive a 48-hour infusion of either 2 mmol/L AK295 (120 to 140 mg/kg) or saline via the carotid artery. At 48 hours and 1 week after injury, spectrin fragments generated specifically by calpain were detected by Western blotting and immunohistochemistry, respectively, in saline-treated, brain-injured animals. Interestingly, equivalent spectrin breakdown was observed in AK295-treated animals when cortical and hippocampal regions were evaluated. Similarly, administration of the calpain inhibitor did not attenuate cortical lesion size or the numbers of apoptotic cells in the cortex, subcortical white matter, or hippocampus, as verified by terminal deoxynucleotidyl transferase-mediated biotinylated deoxyuridine triphosphate nick-end labeling and morphology, at 48 hours after injury. These data suggest that an overt reduction in spectrin proteolysis, cortical lesion, or apoptotic cell death at 48 hours or 1 week is not required for behavioral improvements associated with calpain inhibition by AK295 after experimental brain injury in rats.
There is a growing body of literature that suggests that the calpains, a family of calcium-activated neutral proteases, may act as integral mediators in the pathophysiology of neurodegenerative diseases and injury to the central nervous system (CNS). Activation of calpain has been implicated in neuropathologic sequelae of traumatic brain injury (TBI), spinal cord injury, cerebral ischemia, CNS lesions and excitotoxic injury, and degenerative diseases such as Alzheimer's disease (for reviews, see Wang and Yuen, 1995; Bartus, 1997; Kampfl et al., 1997). Activation of these normally quiescent nonlysosomal proteases results from sufficient elevations of intracellular free calcium and can lead to limited proteolysis of a host of intracellular proteins (Saido et al., 1994) and perhaps to cell death (Bartus, 1997). Among the cellular calpain substrates, perhaps the most well-characterized are the cytoskeletal proteins (Shoeman and Traub, 1990). Proteolysis of the cytoskeletal protein spectrin yields stable, lower molecular weight fragments and is a well established consequence of calpain activation, having been demonstrated by a number of laboratories under in vitro and in vivo conditions (Bartus, 1997; Kampfl et al., 1997; Sorimachi et al., 1997).
Calpain activation with concomitant spectrin proteolysis occurs very early after experimental TBI and persists for several hours to days (Kampfl et al., 1996; Saatman et al., 1996a). Brain regions that exhibit calpain-mediated spectrin proteolysis also display progressive cell loss after experimental brain trauma (Saatman et al., 1996a; Newcomb et al., 1997). A large number of cells within these injured brain regions exhibit characteristics of necrotic cell death; however, a distinct population of cells display apoptotic morphology indicative of programmed cell death (Rink et al., 1995; Yakovlev et al., 1997; Conti et al., 1998). Although the involvement of calpains in mediating trauma-induced programmed cell death is currently unknown, evidence shows that calpain may play a role in apoptosis of neurons or neuronal-like cells under certain in vitro conditions (Behrens et al., 1995; Nath et al., 1996; Jordán et al., 1997).
We have recently shown that posttraumatic administration of AK295, a highly specific inhibitor of calpains that is effective in reducing neuronal pathology in a model of focal cerebral ischemia (Bartus et al., 1994, 1995), significantly reduces both cognitive and motor dysfunction induced by lateral fluid percussion (FP) brain injury in the rat (Saatman et al., 1996b). Therefore, in the current study, we evaluated the effects of an identical administration of AK295 on calpain-mediated spectrin proteolysis, overt cortical damage, and regional apoptotic cell death in the injured rat brain.
MATERIALS AND METHODS
Surgery, brain injury, and administration of inhibitor
Adult male Sprague-Dawley rats (370 to 420 g, n = 35) were anesthetized with 60 mg/kg sodium pentobarbital intraperitoneally, placed in a stereotactic frame, and surgically prepared for lateral FP brain injury as previously described (McIntosh et al., 1989). Anesthetized animals were then surgically prepared for intraarterial administration by ligating the left external carotid artery and retrogradely inserting a cannula to within 1 mm of the carotid bifurcation (Saatman et al., 1996b). The cannula was secured in place and externalized, allowing for continuous drug administration into the internal carotid artery.
A subgroup of animals were subjected to lateral FP brain injury of moderate severity (2.2 to 2.4 atm, n = 26). The remaining animals (n = 9), having received all surgical procedures without brain injury, served as uninjured controls (shams). All animals recovered on heating pads for 15 minutes, at which time they were placed in harnesses, tethered, and connected to continuous-drive syringe pumps. At 15 minutes after injury, animals were randomly assigned to receive either 2 mmol/L AK295 (n = 3 control, n = 13 injured) or saline vehicle (n = 6 control, n = 13 injured) delivered at a rate of 9 mL/h for 10 minutes. The flow rate was then reduced to 1.4 mL/h until 24 hours after injury, after which the rate was lowered to 0.7 mL/h until removing the animal from the syringe pump at 48 hours after injury. In this manner, drug-treated animals received 52.4 mg of AK295 (120 to 140 mg/kg) over 48 hours. This administration paradigm was identical to that used previously (Saatman et al., 1996b) and was designed to maximize the amount of inhibitor received in the acute posttraumatic phase (minutes to hours) while maintaining administration into the subacute phase (days). During the 48-hour drug administration period, animals were freely moving and had access to food and water.
All procedures described herein were approved by the University of Pennsylvania Institutional Animal Care and Use Committee and are in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (1996).
Immunoblotting for spectrin breakdown products at 48 hours after injury
For evaluation of spectrin breakdown at 48 hours after injury by immunoblot analysis, one group of animals (n = 3 injured vehicle-treated, n = 3 injured AK295-treated) was overanesthetized with sodium pentobarbital (200 mg/kg, intraperitoneally) immediately after termination of the drug infusion and decapitated. Each brain was quickly removed from the cranium, and tissue pieces of approximately 100 mg were dissected on ice from the ipsilateral hemisphere from the parietal/temporal cortex (“injured cortex,” region A, in Fig. 1), the occipital/ parietal cortex (“adjacent cortex,” region B), and the hippocampus (region C). The tissue samples were then snap-frozen in slurry of 95% EtOH and dry ice and stored at −80°C.
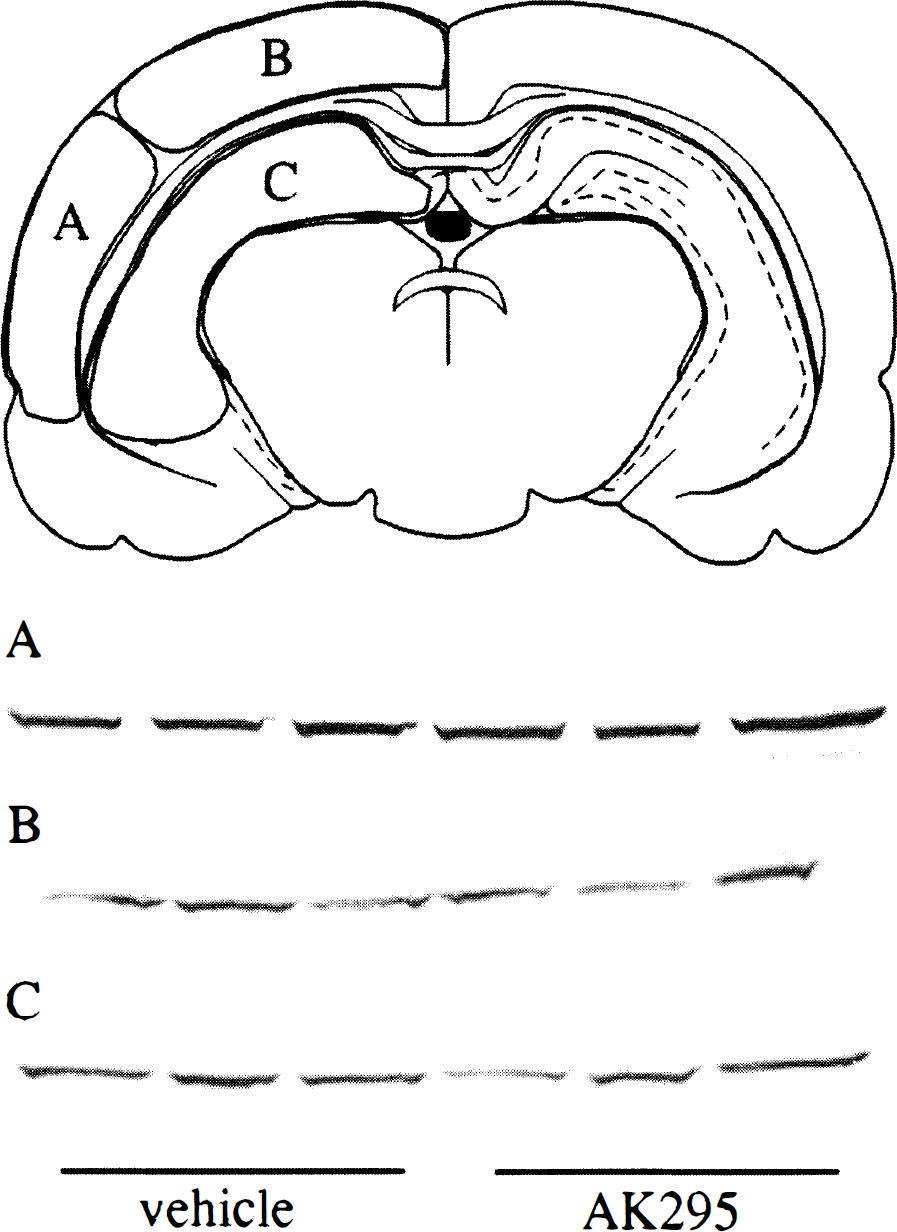
Immunoblots for spectrin proteolytic fragments generated by calpain at 48 hours after brain injury. A schematic drawing of a coronal brain slice illustrates the approximate regions of tissue dissected for immunoblot analyses. Calpain-mediated spectrin breakdown was observed in the injured cortex
Western blotting was performed essentially as described previously (Saatman et al., 1996a). Briefly, tissue samples were homogenized in buffer containing 20 mmol/L Tris-HCl (pH 8.0), 1% NP-40, 0.137 mol/L NaCl, 13 mmol/L EDTA and the recommended working concentration of protease inhibitor cocktail (Sigma Chemical Co., St. Louis, MO, U.S.A.; contains leupeptin, aprotinin, and serine protease inhibitor). After centrifugation, protein concentration of the supernatant was determined using a bicinchoninic acid micro protein assay (Bio-Rad Laboratories, Hercules, CA, U.S.A.). Three hundred micrograms of each sample were loaded into a 6% acrylamide gel and subjected to 2 hours of electrophoresis at 150 V, followed by transfer to nitrocellulose at 60 V for 2 hours. Blots were incubated 1 hour in Ab41, a polyclonal antibody that recognizes the NH2-terminus of the COOH-terminal fragment of α-spectrin generated by calpain (1:500). After incubation in alkaline phosphatase-conjugated goat anti-rabbit IgG (1:2000, Jackson ImmunoResearch, West Grove, PA, U.S.A.), Fast red (Sigma) was used as the chromogen for visualization.
Immunohistochemistry for calpain-mediated spectrin proteolysis at 7 days after injury
To those animals designated to survive for 1 week (n = 5 injured vehicle-treated; n = 5 injured AK295-treated; n = 3 control vehicle-treated; n = 3 control AK295-treated), 2% iso-fluorane anesthesia was administered at the termination of the 48-hour infusion, allowing the externalized cannula to be tied off and embedded beneath the skin. At 1 week after injury, animals were overanesthetized with sodium pentobarbital (200 mg/kg, intraperitoneally) and transcardially perfused with heparinized saline followed by fixative (4% paraformaldehyde in 0.1 mol/L phosphate buffer, pH 7.4). The brain was then removed from the cranium, immersed in fixative for 4 hours at 4°C, infiltrated with 20% sucrose at 4°C, and then frozen. Selected coronal tissue sections (40 μm) were treated with 0.9% H2O2 in 50% methanol, blocked in 5% normal goat serum, and then incubated overnight at 4°C in Ab37, a polyclonal antibody that recognizes the COOH-terminus of the NH2-terminal fragment of α-spectrin generated by calpain (1:10,000, rabbit IgG) or in Tris-buffered saline with 0.1% Triton-X as a negative control. The tissue was then incubated in biotinylated goat antirabbit IgG (1:200, Vector Laboratories, Burlingame, CA, U.S.A.) followed by horseradish peroxidase-conjugated streptavidin (1:1000, Jackson ImmunoResearch). Nickel sulfate-enhanced diaminobenzidine tetrahydrochloride solution was used to visualize the reaction product.
Under light microscopy (Nikon, Optical Apparatus, Ardmore, PA, U.S.A.), the area of cortical Ab37 immunoreactivity was quantified by an evaluator blinded to treatment using image analysis software (MCID, Imaging Research, Ontario, Canada). Two separate but related measurements of cortical injury were taken. The first measurement yielded the area of cortical Ab37 immunoreactivity on a single coronal section by automatically detecting the region stained with an intensity above a preset threshold (that of uninjured cortical staining). The second cortical measurement, total cortical lesion area, included the area of cortical Ab37 immunoreactivity plus that of the cortical cavity (tissue loss). The total lesion measurement accounted for two possibilities: (1) without accounting for tissue loss, animals with less calpain activation or a slower progression of cell loss might paradoxically appear to have a greater extent of spectrin breakdown because of the presence of a greater number of cells in the damaged area, and (2) necrotic tissue at the center of the lesion might have washed away during the free-floating tissue staining procedure leading to an underestimation of spectrin breakdown. In addition, the spatial extent of spectrin breakdown products (BDPs) in the hippocampal CA3 region was scored on a scale of 0 to 4, where 0 = no spectrin BDPs; 1 = BDPs in the stratum oriens; 2 = BDPs in the stratum oriens (as in 1) and pyramidal cells; 3 = BDPs as in 2 and in the stratum radiatum; and 4 = BDPs as in 3 and in other hippocampal areas.
Cortical lesion and in situ nick-end labeling at 48 hours after injury
Those animals to be evaluated histochemically at 48 hours after injury (n = 5 injured vehicle-treated; n = 5 injured AK295-treated; n = 3 control vehicle-treated) were anesthetized with an overdose of sodium pentobarbital (200 mg/kg, intraperitoneally) immediately after termination of the drug infusion and perfused as described above. After postfixation, the brains were processed into paraffin blocks from which 6-μm coronal sections were cut.
For analysis of cortical injury, sections were collected from each brain at 2.3, 3.3, 4.3, 5.3, and 6.3 mm posterior to Bregma. Sections were deparaffinized, rehydrated, and stained with hematoxylin and eosin (H&E). The area of cortical damage was circumscribed on each section to include the region of decreased H&E staining as well as adjacent regions exhibiting eosinophilic, shrunken, or dystrophic neurons. Lesion volume was calculated using the areas obtained from each coronal section and the known distance separating these sections. Quantification was performed using image analysis software (MCID) under blinded conditions.
Terminal deoxynucleotidyl transferase-mediated biotinylated deoxyuridine triphosphate nick-end labeling (TUNEL) was used to detect damaged DNA. TUNEL histochemistry was performed with a procedure used previously in our laboratory (Rink et al., 1995; Conti et al., 1998). Three sections spaced at least 36 μm apart were selected from each brain at −3.3 mm Bregma. Briefly, sections were heated to 60°C before deparaffinization and rehydration. After proteinase K digestion, tissue was incubated in cacodylate buffer and labeled with biotinylated-16-dUTP (20 μmol/L, Boehringer Ingelheim, Indianapolis, IN, U.S.A.), TdT (0.3 U/μL, Boehringer Ingelheim) and 1.5 mmol/L cobalt chloride in cacodylate buffer. Labeling was terminated by rinsing with a solution of sodium chloride and sodium citrate. The tissue was blocked in 10% normal goat serum and incubated in streptavidin alkaline-phosphatase (1:40, Biogenex, San Ramon, CA, U.S.A.) which was reacted using Fast red (Sigma). Counterstained sections were analyzed under light microscopy by an evaluator blinded to the injury and treatment status of each animal. The number of nonapoptotic and apoptotic TUNEL-positive cells was manually counted in six regions: the cortex, subcortical white matter tract, and hippocampus of both the ipsilateral and contralateral hemispheres. Only those cells which stained with TUNEL and exhibited distinct and verifiable apoptotic morphology, as described in Rink et al. (1995), were classified as apoptotic. Reported counts represent the total number of cells for the three nonadjacent tissue sections evaluated for each animal.
Statistical analyses
Lesion areas and cell counts were considered parametric data and were therefore expressed as means with standard deviations. Comparisons of cell counts, cortical injury, or cortical lesions were made using Student's t tests. A P value of less than 0.05 was considered statistically significant.
RESULTS
Calpain-mediated spectrin breakdown
Immunoblot analysis of tissue samples taken from the injured cortex (region A) using an antibody specific for calpain-mediated spectrin BDPs revealed spectrin breakdown in all brain-injured animals at 48 hours after injury. The intensity of the single band at approximately 150 kD was equivalent for all animals regardless of treatment, indicating that the animals receiving AK295 had comparable levels of spectrin breakdown as those receiving vehicle (Fig. 1A). Similarly, no reduction in the level of spectrin breakdown in animals treated with AK295 was observed in tissue samples from either region B of the cortex (Fig. 1B) or the hippocampus (Fig. 1C).
At 7 days after injury, no staining for spectrin BDPs was observed in any region in control (sham) animals. In contrast, all brain-injured animals showed evidence of spectrin proteolysis surrounding a necrotic cavity in the ipsilateral cortex at 1 week after injury (Fig. 2). The area exhibiting spectrin breakdown began, on average, at −0.8 mm Bregma and continued beyond −7.0 mm Bregma. The region exhibiting immunoreactivity for spectrin fragments was diffusely stained but interspersed with small puncta and contained no labeled cells other than an occasional neuron. There were no apparent differences in the pattern or intensity of cortical calpain-mediated spectrin proteolysis observed in vehicle-treated animals (Fig. 2, top row) when compared to those given AK295 (Fig. 2, bottom row). Quantification of the area of spectrin BDP immunoreactivity (Fig. 3A) and of the total cortical lesion area (which includes the area of spectrin BDP immunoreactivity plus that of the cortical cavity) (Fig. 3B) revealed no significant differences between brain-injured animals receiving vehicle and those receiving AK295 at any of the three rostrocaudal levels that were examined.
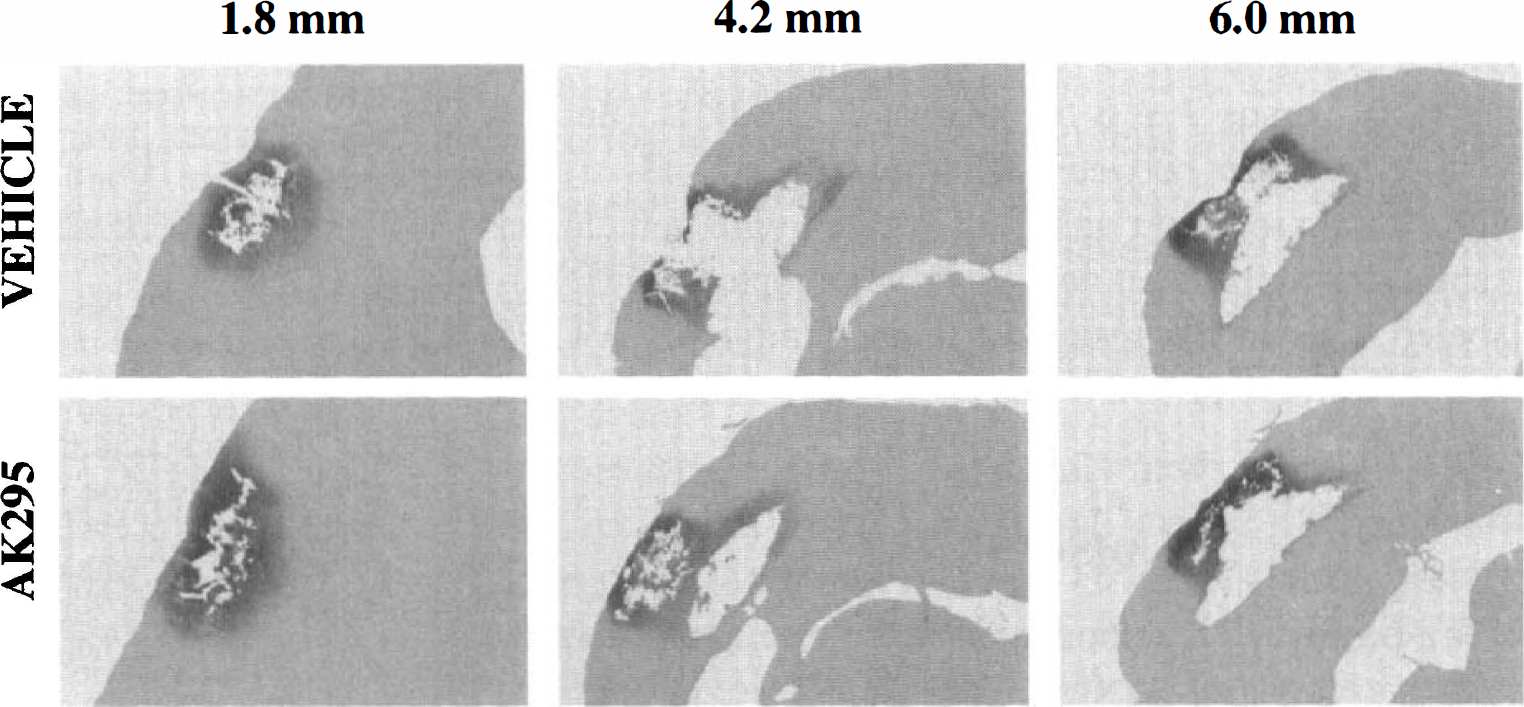
Immunohistochemistry for spectrin breakdown products at 1 week after injury. Coronal sections of rat brain are shown at 1.8, 4.2, and 6.0 mm posterior to Bregma. The extent of calpain-mediated spectrin proteolysis was comparable after vehicle (top row) and AK295 (bottom row) treatment.
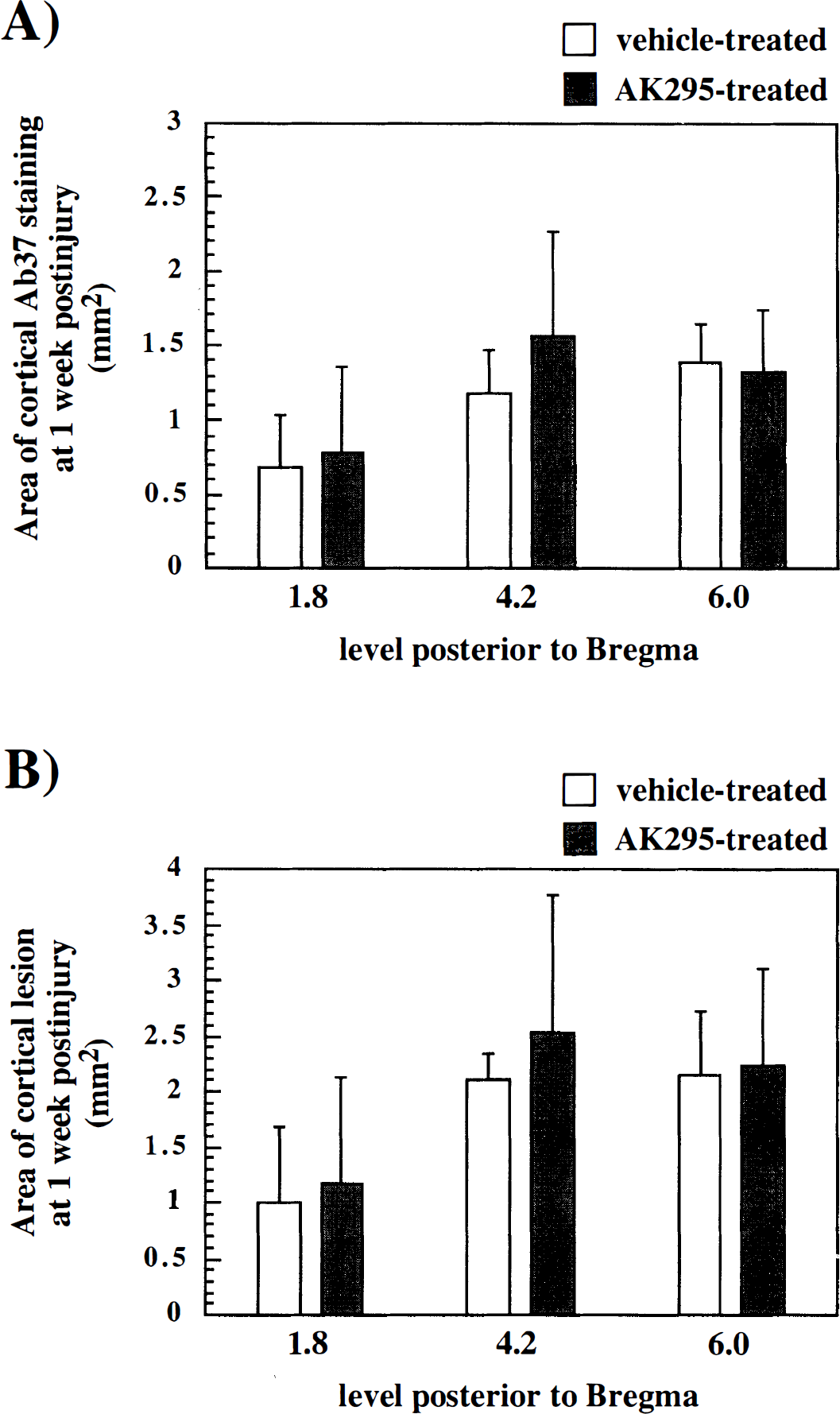
The extent of cortical injury at 1 week after injury was not significantly influenced by treatment with calpain inhibitor.
In 8 of the 10 brain-injured animals, calpain-mediated breakdown was also observed in the CA3 region of the ipsilateral hippocampus. No intact cells were immunolabeled. Rather, punctate spectrin BDP immunoreactivity was detected, most frequently in the stratum oriens, followed by the pyramidal cell layer and then the stratum radiatum. When animals were rated on a 0 to 4 scale according to the spatial extent of spectrin breakdown in the CA3 region, no significant effect of treatment was observed (1.8 ± 1.5 for vehicle treatment and 1.4 ± 1.1 for AK295 treatment, mean ± SD; P > 0.05).
Cortical lesion
At 48 hours after FP brain injury, all animals exhibited loss of H&E-stained neurons in the ipsilateral cortex. Loss of H&E staining was observed at all rostrocaudal levels examined, from 2.3 to 6.3 mm posterior to Bregma. Quantification of the area of cortical damage, which included regions containing eosinophilic and pyknotic neurons, revealed no significant differences because of treatment with vehicle versus calpain inhibitor at any of the rostrocaudal levels (Fig. 4). The cortical lesion volume, calculated from 2.3 to 6.3 mm posterior to Bregma, was 15.6 ± 3.8 mm3 for vehicle-treated animals and 18.6 ± 5.8 mm3 for AK295-treated animals (P > 0.05).
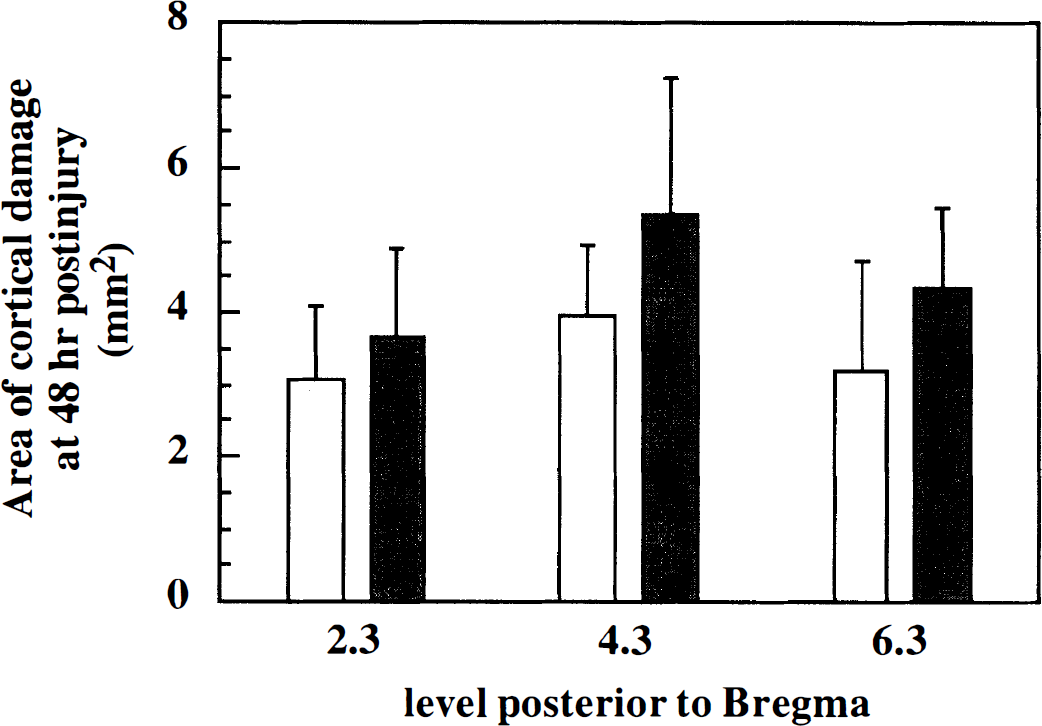
Measurement of cortical injury at 48 hours after injury in treated and untreated animals. At each of three levels relative to Bregma, the area of cortical damage, which included areas exhibiting loss of H&E staining as well as eosinophilic and dysmorphic neurons, was quantified. No significant treatment effect was observed. Bars represent means of vehicle-treated (open) and AK295-treated (filled) animals. Error bars represent standard deviations.
Apoptosis
TUNEL histochemistry labeled cells with fragmented DNA, and a subpopulation of TUNEL-positive cells displayed distinct morphologic features of apoptosis. Apoptotic cells were round and shrunken, with round, condensed, intensely stained nuclei (Fig. 5A, left). Cells which lacked these features and exhibited predominantly cytoplasmic TUNEL staining (Fig. 5A, right) were considered nonapoptotic. At 48 hours, very few, if any, TUNEL-labeled cells with apoptotic morphology were present in uninjured (sham) animals (Figs. 5B and 5C). In contrast, brain-injured animals treated with vehicle exhibited a significantly greater number of apoptotic cells in the ipsilateral cortex (P < 0.05), hippocampus (P < 0.05), and white matter (P < 0.005) than did sham animals (Fig. 5B). Interestingly, there was also a slight but significant increase in the number of apoptotic cells attributable to brain injury in the contralateral cortex (P < 0.05). Brain-injured animals who received AK295 displayed a significant increase in the number of apoptotic cells only in the ipsilateral subcortical white matter when compared to sham animals (P < 0.05) but exhibited no differences in any region from vehicle-treated, brain-injured animals.
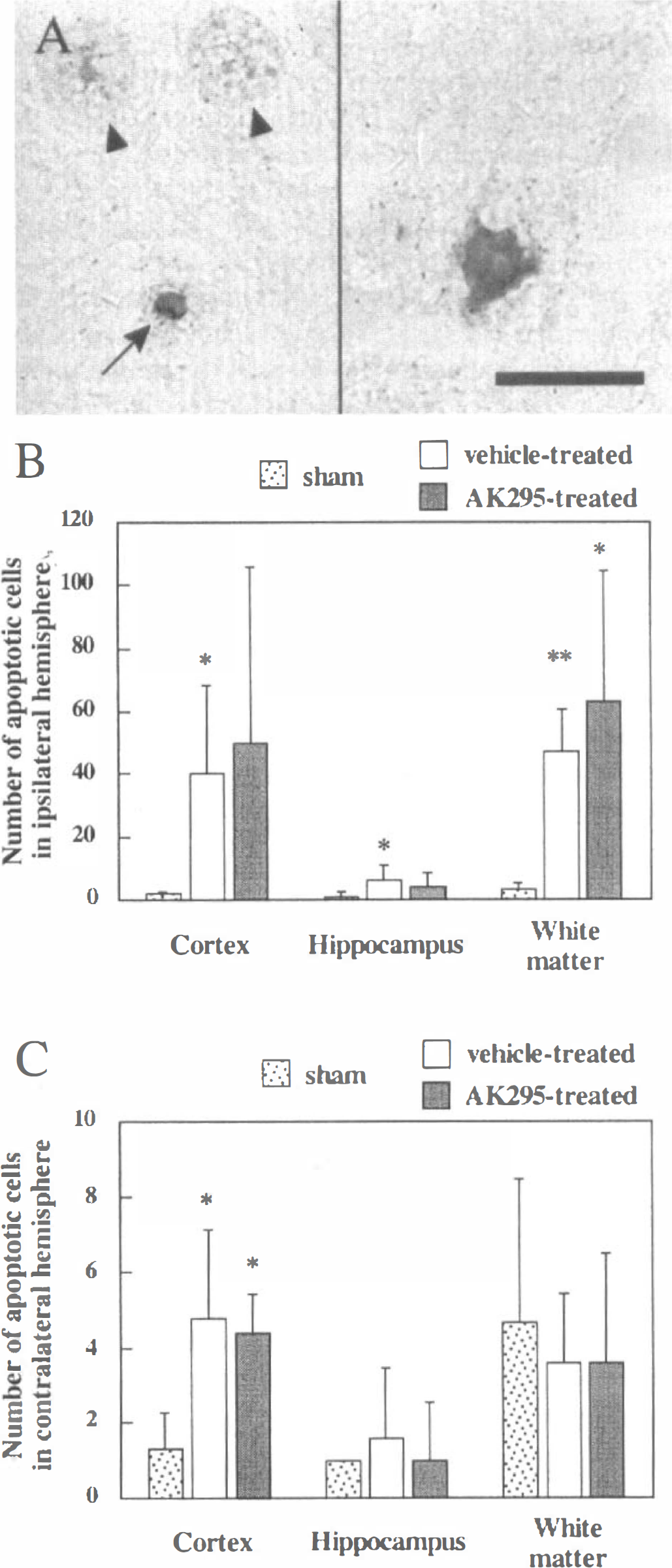
Evaluation of effect of calpain inhibitor treatment on regional apoptotic cell death at 48 hours after injury. Apoptotic cells were identified by terminal deoxynucleotidyl transferase-mediated biotinylated deoxyuridine triphosphate nick end labeling (TUNEL) positivity and by morphologic criteria such as shrinkage and chromatin condensation.
Similarly, vehicle-treated, brain-injured animals exhibited a significant increase in the number of nonapoptotic, TUNEL-positive cells in the ipsilateral cortex (110 ± 100 versus 4 ± 1 in shams; P < 0.05), hippocampus (6 ± 3 versus 1 ± 1 for sham; P < 0.05), and white matter (84 ± 34 versus 7 ± 6 for sham; P < 0.005). AK295-treated animals showed a significant increase in TUNEL-stained nonapoptotic cells in the ipsilateral white matter (89 ± 48 versus 7 ± 6 for shams; P < 0.05). The only difference noted between brain-injured animals treated with calpain inhibitor and those treated with vehicle was a small but significant decline in nonapoptotic, TUNEL-labeled cells in the ipsilateral hippocampus of AK295-treated animals (1 ± 2 versus 6 ± 3 for vehicle-treated; P < 0.05).
DISCUSSION
We previously demonstrated that administration of the selective calpain inhibitor AK295 conferred significant protection against both neuromotor and cognitive deficits resulting from experimental brain injury (Saatman et al., 1996b). However, in the present study, we found no evidence that treatment of brain-injured animals with AK295 attenuated cortical lesion volume, regional apoptosis, or calpain-mediated spectrin proteolysis in the vulnerable regions of the cortex and hippocampus at 48 hours or 1 week after injury. These data suggest that overt histologic protection in the areas of maximal damage after brain trauma is not a necessary condition for behavioral improvements associated with posttraumatic calpain inhibition. Our findings with the compound AK295 reiterate the importance of using multiple, clinically relevant outcome measures when evaluating the therapeutic potential of novel compounds in trauma.
Although evidence for a role for calpains in the pathology of CNS injury has been increasing (Bartus, 1997; Kampfl et al., 1997), few studies have investigated the effects of calpain inhibitors in models of traumatic CNS injury. Administration of cysteine protease inhibitors has been shown to reduce axonal degeneration and neurofilament degradation after spinal cord injury in rats (Iwasaki et al., 1987). A more recent study demonstrated that treatment with calpain inhibitor II attenuated cortical neuropathology and cytoskeletal proteolysis 24 hours after cortical impact brain injury in rats (Posmantur et al., 1997). Our data demonstrating a lack of overt neuroprotection attributable to calpain inhibition with AK295 in brain-injured rats are in sharp contrast to these studies. In comparison to AK295, however, the inhibitors used in previous trauma studies were less specific for calpain (Bartus et al., 1995; Wang and Yuen, 1995; Sorimachi et al., 1997) and may have affected other proteases involved in the processes of cell damage and death. It is also possible that differences in pharmacokinetics and tissue penetration of the inhibitors and in the injury models contributed to the disparate findings.
Using an administration paradigm similar to that employed in the present study, Bartus et al. (1994) demonstrated that AK295 significantly reduced infarct size when given after focal cerebral ischemia in the rat. The related calpain inhibitor CX216 has been shown to protect rat CA1 neurons when administered after global cerebral ischemia (Bartus et al., 1995). Although treatment with these and other calpain inhibitors has been shown to confer neuroprotection in models of cerebral ischemia (Lee et al., 1991; Rami and Krieglstein, 1993; Hong et al., 1994; Markgraf et al., 1998), we found no reduction in spectrin proteolysis or overt cell death after treatment of brain-injured rats with AK295. These data suggest that the histologic effects of AK295 in the traumatically injured brain may be uniquely different from those in the ischemic brain, indicating the necessity for thorough evaluation of potentially therapeutic calpain inhibitors in models of TBI as well as cerebral ischemia.
The lack of overt neuroprotection observed with AK295 administration may be attributable, in part, to a delay in the establishment of therapeutic levels of calpain inhibitor in areas of maximal damage. Calpain activation in the rat brain after TBI has been detected as early as 15 minutes and shown to peak before 1 day after injury, with maximal appearance of calpain-mediated spectrin breakdown products at approximately 1 day after injury (Kampfl et al., 1996; Saatman et al., 1996a). The paradigm for administration of the calpain inhibitor AK295, beginning at 15 minutes and continuing for 48 hours, was selected to target early posttraumatic calpain activation and maintain inhibition beyond the period of maximal activation. The ability of AK295 to inhibit the degradation of neurofilament protein in rat spinal cord has been shown to be dose-dependent, with near complete inhibition at concentrations of 25 to 50 μmol/L (James et al., 1998). Although extensive and prolonged compromise of the blood-brain barrier after experimental brain injury (Cortez et al., 1989; Tanno et al., 1992; Baldwin et al., 1996) should have facilitated penetration of the drug into damaged tissues, the time course and level of AK295 achieved in the traumatically injured brain in the current study are unknown. Therefore, a delay in reaching local, therapeutic calpain inhibitor levels may have allowed sufficient calpain activation to initiate cascades of destructive intracellular events that ultimately resulted in cell death.
Our data showing significantly more apoptotic cells in the ipsilateral cortex and white matter 48 hours after lateral FP brain injury was consistent with previous studies using this model (Rink et al., 1995; Conti et al., 1998). We also found small increases in the numbers of apoptotic cells in the ipsilateral hippocampus and contralateral cortex. However, calpain inhibitor treatment did not significantly attenuate apoptotic cell death in any of these regions. Although the role of calpain in apoptosis after TBI is currently unknown, calpain has been implicated in pathways of apoptotic cell death in vitro using a number of cell types including thymocytes (Squier et al., 1994), hepatocytes (Gressner et al., 1997), neutrophils (Brown et al., 1997), and neurons and neuroblastoma cell lines (Behrens et al., 1995; Nath et al., 1996; Jordán et al., 1997). The effectiveness of calpain inhibitors in preventing apoptosis may depend on the cell type, initiating stimulus, and characteristics of the inhibitor (Nath et al., 1996; Sorimachi et al., 1997), suggesting that results from in vitro studies may not adequately reflect the environment after trauma in which multiple cell types experiencing both physical and biochemical stimuli undergo apoptosis (Conti et al., 1998).
Although posttraumatic calpain inhibition did not attenuate damage in the vulnerable regions of the cortex and hippocampal CA3 pyramidal layer, it is possible that the behavioral improvements documented in our previous study were associated with subtle histologic benefits resulting from AK295 treatment that were not detected in the present study. Lateral FP brain injury has been shown to result in diffusely distributed axonal damage (Yaghmai and Povlishock, 1992; Pierce et al., 1996; Saatman et al., 1998) and progressive cell loss in regions other than the cortex and hippocampus (Dietrich et al., 1994; Hicks et al., 1996; Bramlett et al., 1997; Smith et al., 1997; Conti et al., 1998). Our histologic assessment techniques may not have been sensitive enough to detect treatment effects related to scattered axonal injury or delayed (secondary) degeneration. A small reduction in the number of nonapoptotic, TUNEL-positive cells in AK295-treated animals was detected in the ipsilateral hippocampus, which may reflect an effect of calpain inhibition on cells in early stages of cellular injury or with sublethal damage. Future studies should address these possibilities.
The robust behavioral improvements derived from posttraumatic calpain inhibition were not accompanied by an overt reduction in cortical cell loss or spectrin degradation. There is evidence to suggest that cell loss may not directly correlate with functional outcome after experimental brain injury (Lyeth et al., 1990; Sabel, 1997). For example, despite the ability of untreated, brain-injured animals to partially recover neuromotor function over days, weeks, and months after trauma (McIntosh et al., 1989; Pierce et al., 1998), histologic evidence shows that both necrotic and apoptotic cell death continue during this same time period (Dietrich et al., 1994; Hicks et al., 1996; Bramlett et al., 1997; Smith et al., 1997; Conti et al., 1998). Therefore, it is likely that inhibition of calpain may improve recovery of function in a manner independent of direct sparing of vulnerable cells. Pretreatment with calpain inhibitors has been shown to preserve neuronal ability to exhibit long-term potentiation (Lee et al., 1991) and facilitate recovery of synaptic transmission (Arai et al., 1990; Hiramatsu et al., 1993) after CNS injury. Furthermore, because calpain acts on a great number of intracellular substrates, its inhibition may have led to functional/behavioral efficacy in part by preventing the proteolysis of intracellular signaling or membrane-associated molecules not evaluated in the current study.
We have shown that the behavioral efficacy observed at 1 week after brain injury by rats treated for 48 hours with the calpain inhibitor AK295 (Saatman et al., 1996b) was not accompanied by an overt attenuation of calpain-mediated spectrin breakdown, cortical lesion size, or number of apoptotic cells. These data suggest that the behavioral effects produced by posttraumatic administration of AK295 may be mediated through subtle or diffuse histologic and biochemical alterations that may be uniquely different from those observed in experimental cerebral ischemia. Importantly, these findings suggest that the sparing of a large number of cells in the areas of maximal damage inflicted by trauma may not be a necessary condition for improving functional outcome by the inhibition of calpain after brain injury.
Footnotes
Acknowledgments
The authors thank Dr. Hisayuki Murai for lending his considerable surgical skills to this study and Drs. Ramesh Raghupathi and Michelle LaPlaca for their help with tissue preparation and Western blotting. Antibodies Ab37 and Ab41 were generously donated by Dr. Robert Siman. The authors also thank Kelli L. McDermott, Brian R. Perri, and Kristin L. Sanderson for their technical assistance and Jeanne Marks for assistance in preparation of the manuscript.