
Abstract
The authors present two studies that investigate the biochemical and histologic effects of the nonimmunosuppressive neuroimmunophilin (NIMM) ligand V-10,367 in a mouse model of traumatic brain injury (TBI). In study 1, the authors examined the effect of V-10,367 (50 mg/kg x 2 per day, by mouth) on neurofilament M (NFM) protein levels and on α-spectrin breakdown products (SBDPs) when dosed for 2 days, starting 24 hours after TBI and killed on day 3. In study 2, V-10,367 was given for 10 days, starting 24 hours after TBI and the mice killed 6 weeks after TBI, to measure the extent of neurodegeneration (amino CuAg stain). The results in study 1 revealed that V-10,367-treatment significantly increased NFM protein levels in both sham and TBI mice. In addition, V-10,367 attenuated SBDP 150 levels in the cortex, striatum, and hippocampus. The results of study 2 indicated that TBI mice treated with V-10,367 demonstrated significantly less neurodegeneration compared to injured, vehicle-treated mice. In summary, these results suggest that NIMMs may be neuroprotective indirectly through inhibition of calpain-mediated cytoskeletal damage and perhaps via maintenance of neuronal plasticity. In the context of this mouse model of TBI, the therapeutic window for V-10,367's positive effects is at least 24 hours after injury, which, in the case of TBI models, is largely unprecedented for a neuroprotective compound.
During the past 15 years, there has been intense activity in the pharmaceutical industry and academia to discover and develop pharmacological agents for acute treatment of traumatic brain injury (TBI). The mechanistic targets of this effort have primarily been glutamate-mediated excitotoxicity, intracellular calcium overload, and oxygen radical-mediated cellular injury (Hall, 1995a). Several glutamate receptor antagonists and release inhibitors, calcium channel blockers, and free radical scavengers/antioxidants have been identified and were shown to be effective neuroprotectants in a variety of preclinical TBI models. As a result, at least five separate phase II and III clinical development programs have been carried out over the past 15 years to evaluate the potential benefits of the calcium channel blocker nimodipine, the glutamate antagonists selfotel (CGS19755) and aptiganel (CNS 1102), the free radical scavenger polyethylene glycol-conjugated superoxide dismutase, and the lipid peroxidation inhibitor tirilazad (U-74006F) (Langham et al., 2000; Marshall et al., 1998; Narayan and Michel, 2002). Although there has been some evidence of therapeutic efficacy in the case of patients with traumatic subarachnoid hemorrhage treated with nimodipine (Harders et al., 1996; Langham et al., 2000) and tirilazad (Marshall et al., 1998) these trials, as a whole, have been therapeutic failures (Doppenberg and Bullock, 1997; Narayan and Michel, 2002). However, a careful historical analysis of the pursuit of marketable neuroprotective agents has revealed that, in the case of most compounds destined for clinical trials, the preclinical data obtained prior to trial design have been grossly inadequate (Narayan and Michel, 2002). Even with compounds with more complete preclinical data packages, there has been a major disconnection between the preclinical data and the design of subsequent clinical efficacy trials. Perhaps the most significant of these disconnects is the common discrepancy between the therapeutic window demonstrated in animal models (e.g., 1 hour) and that employed in trials (e.g., 4–12 hours). Although preclinical definitions of the effective therapeutic windows for glutamate antagonists, calcium blockers, and antioxidants is sketchy at best, the available evidence suggests that these approaches only have neuroprotective efficacy in TBI models when given during the first few hours after injury. Thus, there is an obvious need for the discovery of novel neuroprotective strategies, which possess a longer, more clinically practical therapeutic window.
Focusing on mechanistic targets that encompass both neuroprotective and neuroregenerative properties (i.e., promoting neuronal plasticity) would be one approach to coming up with an extended therapeutic window for TBI and other acute central nervous system (CNS) insults. In that regard, a unique group of compounds with a combination of neuroprotective and neuroregenerative effects has been discovered, which interact with a family of proteins that are widely expressed in the CNS and are known as neuroimmunophilins (NIMMs). The discovery of the NIMMs and their ligands began with studies of the immunosuppressive compound FK506. FK506 suppresses T-cell-mediated immune responses by an action on the immunophilin FKBP 12 (12-kd FK506 binding protein) and possesses neuroprotective and neurotrophic effects in in vitro neuronal culture (Ankarcrona et al., 1996; Dawson et al., 1993; Lyons et al., 1994) and in vivo models (Bavetta et al., 1999; Butcher et al., 1997; Costantini et al., 1998; Gold et al., 1995; Guo et al., 2001; Kitamura et al., 1994; Sharkey and Butcher, 1994; Snyder and Sabatini, 1995; Toung et al., 1999). Immunophilins are highly expressed in CNS tissue, as revealed by binding of 3H-FK506 (Dawson et al., 1994) and are believed to be the locus of FK506's neuroprotective/neuroregenerative properties. Although the mechanisms of the neuroprotective effects of FK506 are unclear, some work implies that the interaction of FK506 with FKBP12 leads to an inhibition of calcineurin and, consequently, to an inhibition of the dephosphorylation of the apoptotic cell death regulatory protein BAD (Wang et al., 1999). This action serves to inhibit the proapoptotic effects of BAD (Guo et al., 2001). Mechanistically, the neuroregenerative effects of FK506 are unknown, but do not appear to involve an inhibition of calcineurin (Steiner et al., 1997a).
Several pharmaceutical companies have launched drug discovery programs aimed at the development of nonimmunosuppressive NIMM ligands with neuroprotective and neuroregenerative properties. These efforts have led to the successful discovery of NIMM ligands, such as GPI-1046 (Campbell et al., 1999; Harper et al., 1999; Steiner et al., 1997a,b) and V-10,367 (Armistead et al., 1995; Gold et al., 1997). In cultured and transplanted dopaminergic neurons, V-10,367 has been shown to stimulate neurite outgrowth (Costantini and Isacson, 2000) and to protect nigrostriatal dopaminergic neurons against MPTP toxicity (Costantini et al., 2001), suggesting its use in Parkinson's disease. In addition, V-10,367 has been documented to increase the regeneration of crushed motor nerve fibers (Gold et al., 1997), supporting its possible use in peripheral nerve injury. However, no studies have examined the potential utility of any of the NIMM ligands in TBI or other acute CNS injuries.
The purpose of the presently reported study was to investigate the neuroprotective efficacy of V-10,367 in a mouse weight-drop model of diffuse head injury (Hall, 1995b; Kupina et al., 2001). Recent studies in our laboratory with this model have documented the time course of posttraumatic cytoskeletal damage in terms of a loss of medium size neurofilament (NFM) and degradation of α-spectrin, showing that the peak of NFM loss and increased levels of spectrin breakdown products (SBDPs) occurs at 3 days after injury (Kupina et al., 2002). In those studies, the magnitude and time course of cytoskeletal damage is paralleled by the magnitude and timing of neurodegeneration, quantitatively assessed by silver staining (amino cupric silver staining; de Olmos method) (Switzer, 2000). The effects of V-10,367 on 3-day postinjury cytoskeletal damage in cortex, striatum, and hippocampus, following delayed posttraumatic administration (24 hours after injury, by mouth), was investigated and followed by an examination of the ability of the NIMM ligand to attenuate late (42-day) neurodegeneration.
MATERIALS AND METHODS
All procedures were carried out in strict compliance with the Institutional Animal and Use Committee of Pfizer Global Research and Development.
Experimental design
Two studies were conducted to investigate the biochemical and histologic effects of the NIMM ligand V-10,367 in a mouse model of traumatic brain injury. The selected dosage of V-10,367 was based upon evidence for neuroprotective efficacy in the MPTP model of Parkinson's disease (Costantini et al., 2001). Study 1 was designed to evaluate whether V-10,367 could effect a change on posttraumatic degradation of cytoskeletal proteins. Twenty-four hours following head injury, mice were dosed with V-10,367 (50 mg/kg x 2 each day, by mouth) for 2 days and then killed on day 3. Western blot analysis of the levels of NFM and SBDPs was determined in areas of the cortex, striatum, and hippocampus. Study 2 was designed to determine whether V-10,367 could protect against posttraumatic neurodegeneration. Twenty-four hours following head injury, mice were dosed with V-10,367 (50 mg/kg x 2 each day, by mouth) for 10 days, killed on day 42, upon which the brains were silver stained for evidence of neurodegeneration. The choice of the 24-hour treatment delay was based upon the previous demonstration that NFM and spectrin breakdown in the TBI model used does not peak until 3 days after injury, subsiding thereafter (Kupina et al., 2002). The choice of the 10-day treatment duration, used in Study 2, was similarly based upon the prior demonstration that most of the posttraumatic cytoskeletal damage appears to be completed between 7 and 14 days after TBI.
Mouse model of diffuse head injury
The mouse impact-acceleration head-injury method was first introduced by Hall (Hall, 1995b) and was used as described with minor modification. CF-1 6- to 8-week-old male mice (Charles River, Portage, MI, U.S.A.), weighing 29 to 32 g, were fed ad libitum before injury. Mice were briefly anesthetized in a chamber containing 2.5% isoflurane, balanced with air and oxygen. Each mouse was grasped by the dorsal skin of the neck and its head was placed upon the metal base of the injury device. A round, flat, 6-mm-diameter Teflon impounder was positioned firmly against the top of the head, centered between the ears and eyes (encompassing the area over the frontal and parietal bones) and a 100-g stainless steel weight was released at a height of 10.5 cm, producing a velocity calculated at 143.53 cm/s. The biomechanics of this injury model involve both contact and inertial effects (see review by McIntosh et al., 1996). Contact effects, beginning from the point of impact, cause stress waves to spread throughout the brain. Inertial effects create pressure changes in the brain. The resulting injury creates a pattern of diffuse degeneration, which is diversely projected into the brain, with variable intensity, and can include contusions in the area of the cerebral cortex at angles of 90° or 180° from the point of impact. Although infrequent, animals incurring a skull fracture were removed from the study (approximately 1 in 60 or fewer). To prevent immediate posttraumatic hypothermia, injured mice were placed in a Hova-Bator incubator (model 1583, Randall Burkey Co., Boerne, TX, U.S.A.) at 37°C until consciousness was regained (20 to 30 minutes). In a previous study, rectal temperatures taken 2 hours following injury indicated TBI male mice to be approximately 2°C cooler than sham-injured mice (35.4 ± 0.3°C vs. 37.7 ± 0.1°C, mean ± SE). By 24 and 48 hours after injury, TBI mice remain about 1°C cooler compared to sham-injured mice (36.9 ± 0.2°C vs. 38.0 ± 0.2°C, mean ± SE).
Drug administration
V-10,367 was suspended in 0.5% carboxymethylcellulose (CMC) and dosed, by mouth, twice a day. In studies 1 and 2, each dose was 50 mg/kg, at a concentration of 5 mg/mL and a volume of 10 mL/kg. In study 1, beginning 24 hours after TBI, 40 mice were randomly assigned to 1 of 4 groups consisting of sham + drug/vehicle and TBI + drug/vehicle. Each group (N = 10) was dosed for 2 days and then killed on day 3. In study 2, beginning 24 hours after TBI, 18 mice were randomly assigned to 1 of 3 groups consisting of sham + vehicle and TBI + drug or vehicle. Each group (N = 6) was dosed for 10 days and then killed on day 42.
Measurement of cytoskeletal protein degradation
In Study 1, cytoskeletal protein degradation was quantified by Western immunoblot as previously described (Kupina et al., 2001). α-Spectrin degradation was expressed by measuring the density of the 150- or 145-kd α-spectrin fragment, while NFM was expressed by measuring the density of the 160-kd protein band. Four groups of mice [sham + vehicle/drug and TBI + vehicle/drug (N = 10 per group)] were deeply anesthetized with pentobarbital (0.1 mL; 65 mg/mL), and tissues from each brain (cortex, striatum and hippocampus) were dissected on a chilled stage and immediately frozen in isopentane over dry ice (−60°C) [previously described, (Kupina et al., 2002)]. Stored brain tissues (−80°C) were crushed into a powder using a pre-cooled mortar pestle over dry ice. Approximately 50 mg of each sample was suspended in Triton lysis buffer [1% Triton, 20 mmol/L Tris HCl, 150 mmol/L NaCl, 5 mmol/L EDTA, 1 mmol/L dithiothreitol and protease inhibitor cocktail (Roche Molecular Biochem, Santa Cruz, CA, U.S.A.)] and kept on ice for 3 hours with intermittent agitation. Following centrifugation at 15,000 g for 15 minutes at 4°C, protein concentrations were determined in the cleared lysates by a modified Lowry assay (Bio-Rad, Hercules, CA, U.S.A.). Each sample was normalized to contain 20 μg of protein, run on sodium dodecyl sulfate polyacrylamide gel electrophoresis [4–20% (wt/vol) acryl-amide; Invitrogen, Carlsbad, CA, U.S.A.] with a Tris/glycine running buffer system and then transferred to polyvinyl difluoride membranes using a semidry electrotransferring unit (Bio-Rad) at 20 mA for 2 hours. The blots were probed with an anti-α-spectrin antibody (monoclonal, Affinity, U.K.), an anti-SBDP 150 antibody (Senju Pharmaceuticals, Kobe, Japan), or an anti-neurofilament M antibody (monoclonal, Zymed, South San Francisco, CA, U.S.A.) and developed in a linear range using nitro blue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate (BCIP/NBT, Kirkegarrd & Perry). Densitometric analysis of Western blots was done using a color scanner (Epson 1600) and the NIH program, Image 1.5.
Western blot data are expressed as mean ± SD. Levels of SBDPs were statistically analyzed using a Kruskal-Wallis nonparametric analysis of variance (ANOVA) followed by a Dunn's multiple comparisons test for selected pairs. Statistical analysis of NFM protein levels was done using a one-way ANOVA followed by a Bonferroni multiple comparisons test for planned comparisons. A P value of 0.05 was considered significant.
Quantification of neuronal degenerative debris by AmCuAg staining
In Study 2, three groups of mice [sham + vehicle and TBI + vehicle/drug (N = 6 per group)] were deeply anesthetized with pentobarbital (0.1 mL; 65 mg/ml) and transcardially perfused with 0.9% sodium chloride (until clear of blood), followed by a fixative solution containing 4% paraformaldehyde, 4% sucrose, and 0.01 mol/L sodium cacodylate. The mice were decapitated, and the heads were stored in fixative for 24 hours, after which the brains were removed, placed in fresh fixative, and shipped (4°C) for histologic processing (NeuroScience Associates, Knoxville, TN, U.S.A.). The brains were embedded into a single block of gelatin, frozen, and coronal sections were cut at a thickness of 35 μm. Sections were stained for neuronal degeneration and debris using the amino cupric silver (silver) method of de Olmos (de Olmos et al., 1994) and counterstained with Neutral Red to reveal normal cell bodies.
Thirteen equidistant sections were chosen for analysis of silver staining. Brain sections were photographed under a 1x objective lens and imported, calibrated (using a slide micrometer), and saved into the computer software program SigmaScan Pro 5. Utilizing a preprogrammed software method of contrast enhancement (all sections enhanced identically) and densitometric thresholding, the percent area of silver staining in each brain section was calculated by dividing the area of silver staining in each section by the area of the total brain section and multiplying by 100. The densitometric thresholding of each tissue section was completed by a blinded observer who visually compared each captured computer image with the matching microscope slide. Densitometric thresholding, beyond background noise, was not included. In an instance of unavoidable inclusion of background noise, care was taken to included the same elements in the thresholding of the total brain section, thereby discounting the noise in the final calculation of the section's total area of silver staining. Percent measurements were chosen primarily to take into account any posttraumatic tissue shrinkage that can occur over time. Percent volumetric silver staining was estimated by the equation % V = t* ∑ % a (s), where % V is percent silver stain volume, t is the distance between sections analyzed (420 μm), and ∑ % a (s) is the sum of percent area of silver staining in all sections examined (13 for each brain).
Percent silver stained areas and volumes are expressed as a mean ± SD. Percent area of silver staining was statistically analyzed by two-way repeated measures ANOVA, followed by an all pairwise Tukey multiple comparisons test. Statistical analysis of the percent volume of silver staining was done with a Kruskal-Wallis nonparametric ANOVA, followed by a Dunn's multiple comparisons test for selected pairs. A P value of 0.05 was considered significant.
RESULTS
Effect of V-10,367 on posttraumatic α-spectrin degradation
The cytoskeletal protein α-spectrin (280 kd) undergoes proteolytic degradation to form 150- and 145-kd protein bands (SBDP 150, SBDP 145), as demonstrated in striatal tissue in Fig. 1A (study 1). Figures 1B–D summarize the levels of SBDP 150 and SBDP 145 from Western immunoblots representing brain regions of the cortex, striatum, and hippocampus at 3 days after injury (mean ± SD, N = 10). The density of each protein band was densitometrically measured and normalized to a standard included on all comparable gels. In vehicle-treated TBI mice, significant increases in SBDP 150 and SBDP 145 were measured in the cortex, striatum, and hippocampus, compared to sham vehicle-treated mice. In contrast, within these same brain regions, there were no significant increases detected in SBDP 150 in drug-treated TBI mice when compared to vehicle-treated sham mice. However, treatment with V-10,367 did not significantly lower the increased levels of SBDP 145 [Kruskal-Wallis nonparametric ANOVA, P < 0.0001 cortex, striatum and P < 0.001 hippocampus; Dunn's multiple comparisons test for selected pairs, *P < 0.05, **P < 0.01, ***P < 0.001(compared to sham + vehicle)]. Degradation of α-spectrin can be mediated by both calpain and caspase-3 activation (with SBDP 150 resulting from both calpain and caspase-3 activation and SBDP 145 resulting from only calpain activation); therefore, we utilized a calpain-specific SBDP 150 antibody (Senju Pharmaceuticals) to further elucidate whether V-10,367 could protect against calpain-mediated α-spectrin degradation. Figure 2A shows an example of two Western immunoblots from the striatum (study 1), representing the portion of the SBDP 150 fragment, which specifically recognizes calpain-mediated α-spectrin degradation. Like the calpain/caspase-3 mixed-mediated SBDP 150 fragment, there was a significant increase in the calpain-specific SBDP 150 fragment in TBI vehicle-treated mice compared to sham vehicle-treated mice (Fig. 2B, shown in striatum; mean ± SD, N = 10). Again, there was a similarly significant decrease in calpain-specific SBDP 150 levels in drug-treated TBI mice compared to vehicle-treated TBI mice (Kruskal-Wallis nonparametric ANOVA, P = 0.0004; Dunn's multiple comparisons test for selected pairs; **P < 0.01 vs. sham + vehicle, #P < 0.05 vs. TBI + vehicle).
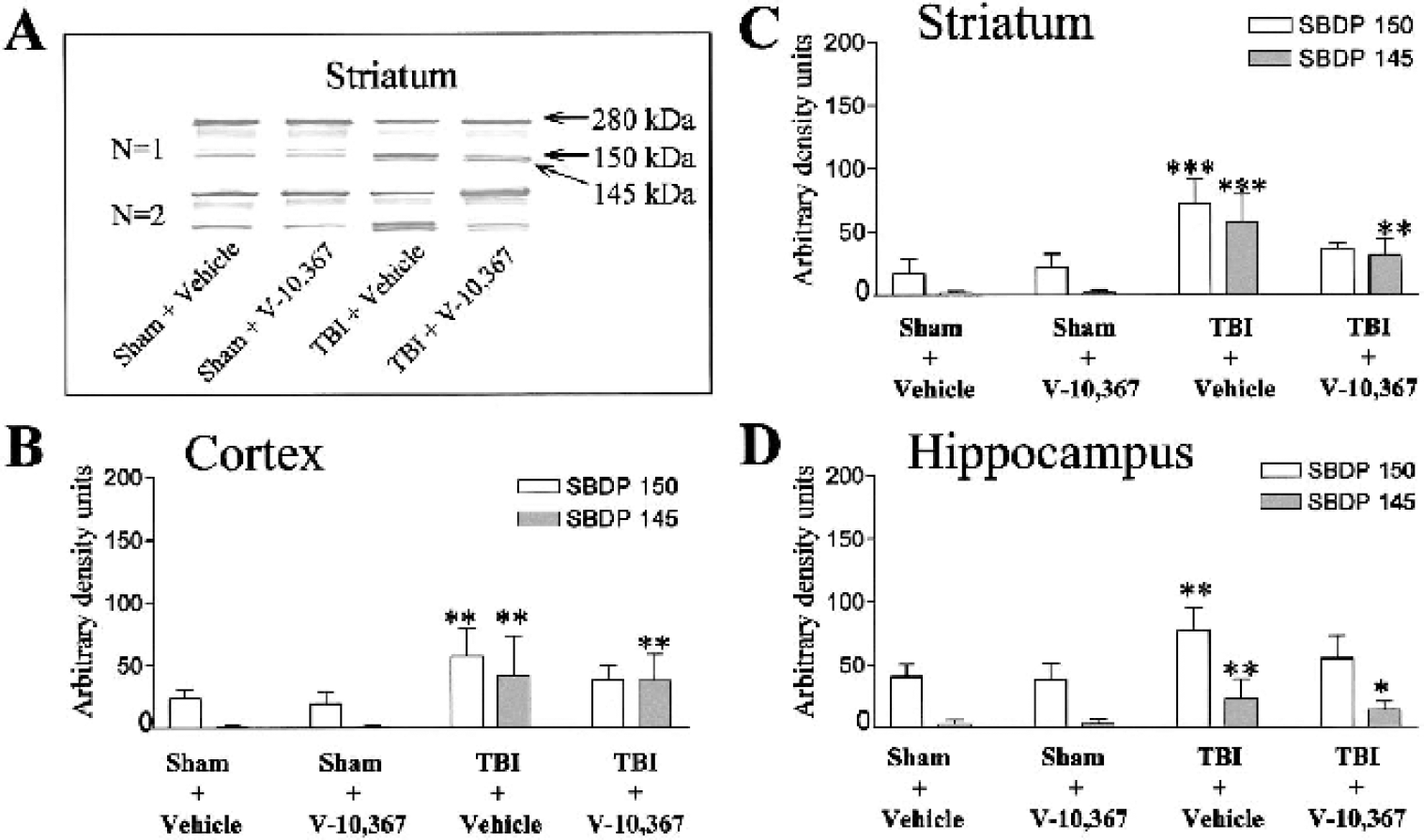
(A) Two sample Western immunoblots, from the striatum in study 1 (N = 10), demonstrating the degree of proteolysis of α-spectrin into the breakdown products SBDP 150 and SBDP 145 in sham-or traumatic brain-injured (TBI)-mice treated with and without V-10,367.
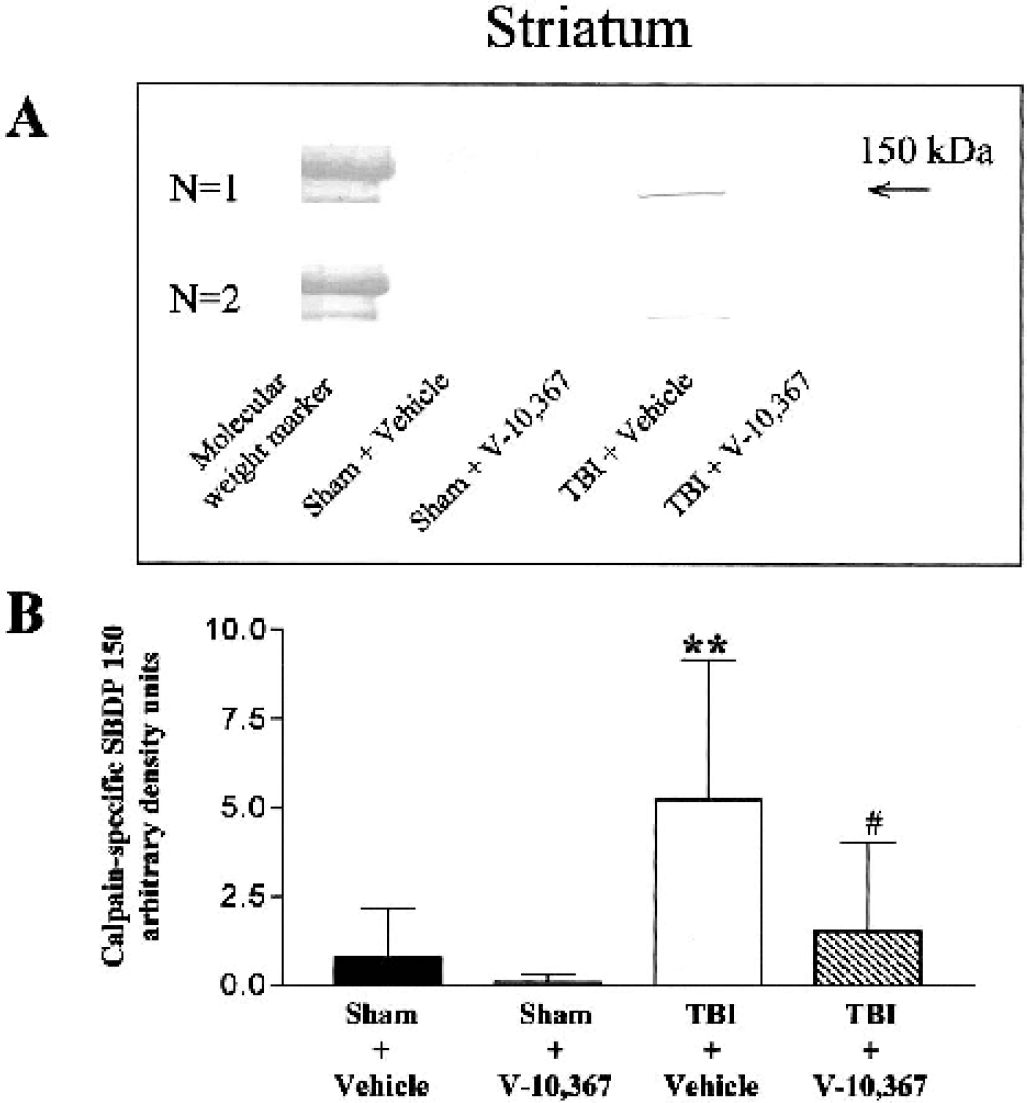
(A) Two sample Western immunoblots, from the striatum in study 1 (N = 10), demonstrating the degree of calpain-mediated (not caspase-3-mediated) α-spectrin proteolysis.
Effect of V-10,367 on posttraumatic neurofilament M degradation
The intermediate NF protein, NFM, is represented by the 160-kd band and is demonstrated in the striatum (study 1) in Fig. 3A. Figures 3B–D summarize the NFM protein levels from Western immunoblots representing brain regions of the cortex, striatum, and hippocampus (mean ± SD, N = 10). The density of each protein band was measured and normalized to a standard included on all directly compared gels. TBI vehicle-treated mice had a significantly lower level of NFM protein in the cortex and striatum compared to sham vehicle-treated mice. However, drug-treated TBI mice resulted in significantly greater levels of NFM protein than in vehicle-treated TBI mice. Interestingly, a significant increase in NFM protein levels, over that of vehicle-treated sham mice, was also evident in V-10,367-treated sham mice (one-way ANOVA, P < 0.001 for cortex, striatum, and hippocampus; Bonferroni multiple comparisons test for selected pairs; **P < 0.01, ***P < 0.001 [vs. sham + vehicle], ###P < 0.001 [vs. TBI + vehicle]).
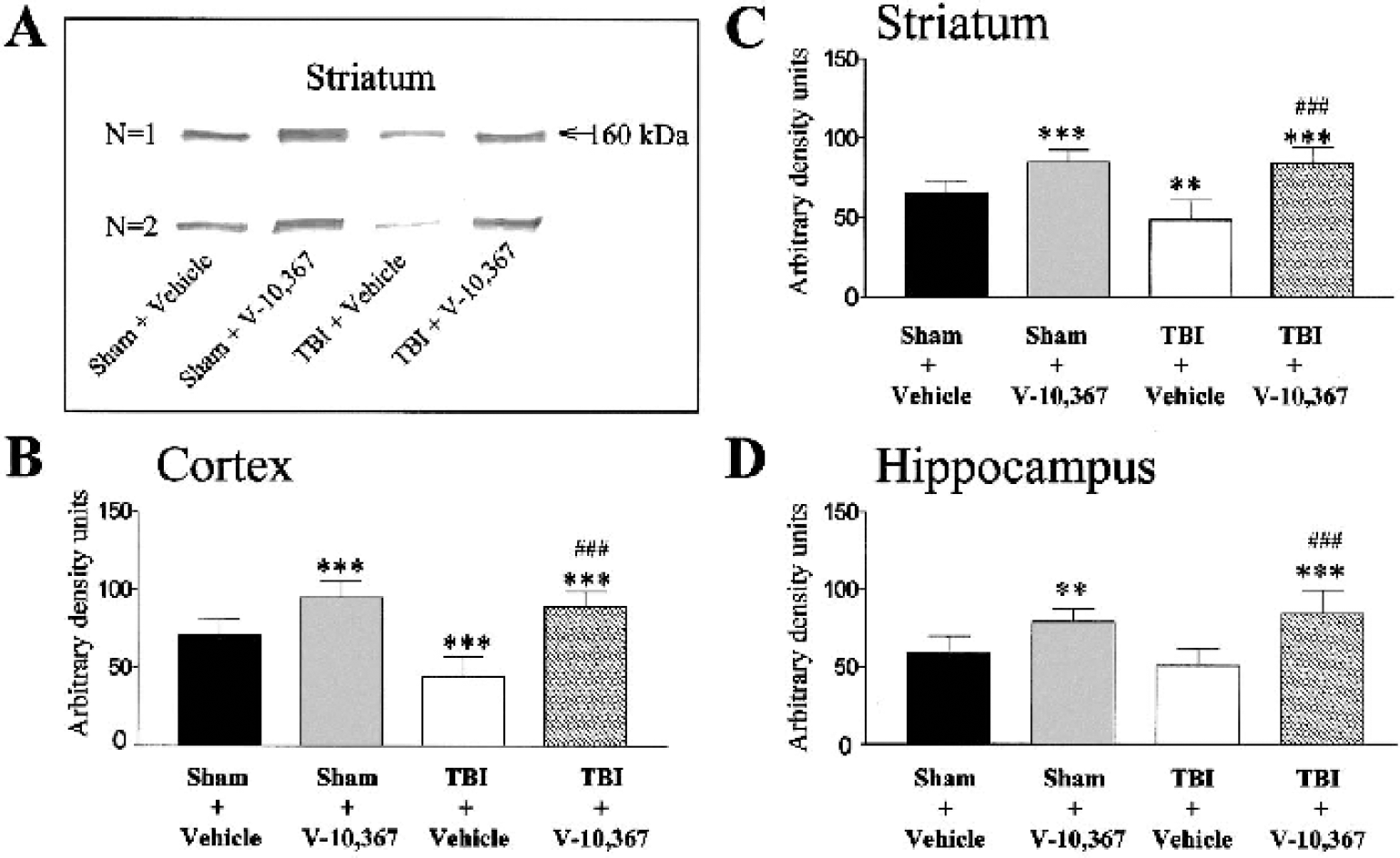
(A) Two sample Western immunoblots, from the striatum in study 1 (N = 10), demonstrating medium chain neurofilament (NFM) protein levels in sham or traumatic brain-injured (TBI) mice treated with and without V-10,367.
Effect of V-10,367 on the area and volume of neurodegeneration
In study 2, 13 equidistant (420 μm) coronal brain sections were selected for densitometric analysis of silver staining, beginning at approximately 2.22 mm bregma and 6.02 mm interaural (section 10) and ending at approximately −2.92 mm bregma and 0.88 mm interaural (section 34) (Paxinos and Franklin, 2001). Three representative silver-stained coronal brain sections from each dosing group are shown in Fig. 4. Quantitative analysis of the distribution of silver staining, by treatment and across all tissue sections analyzed, is summarized in Table 1 and graphically depicted in Fig. 5A (mean ± SD, N = 6 brains x 13 sections). Statistically (two-way repeated measures ANOVA) there was a significant treatment difference (P < 0.001), as well as an interaction between treatment and day after injury (P < 0.001). In vehicle-treated TBI mice, 11 of the 13 section levels evaluated had significantly greater percent area silver staining than in vehicle-treated sham mice (*Tukey multiple comparisons test). However, in drug-treated TBI mice, only 7 of the 13 section levels had significantly greater percent area silver staining than in vehicle-treated sham mice. Also, in 5 of 13 sections levels evaluated, TBI mice treated with V-10,367 had significantly less percent area silver staining than did TBI mice treated with vehicle alone (P < 0.05 # = Fig. 5A, † = Table 1).
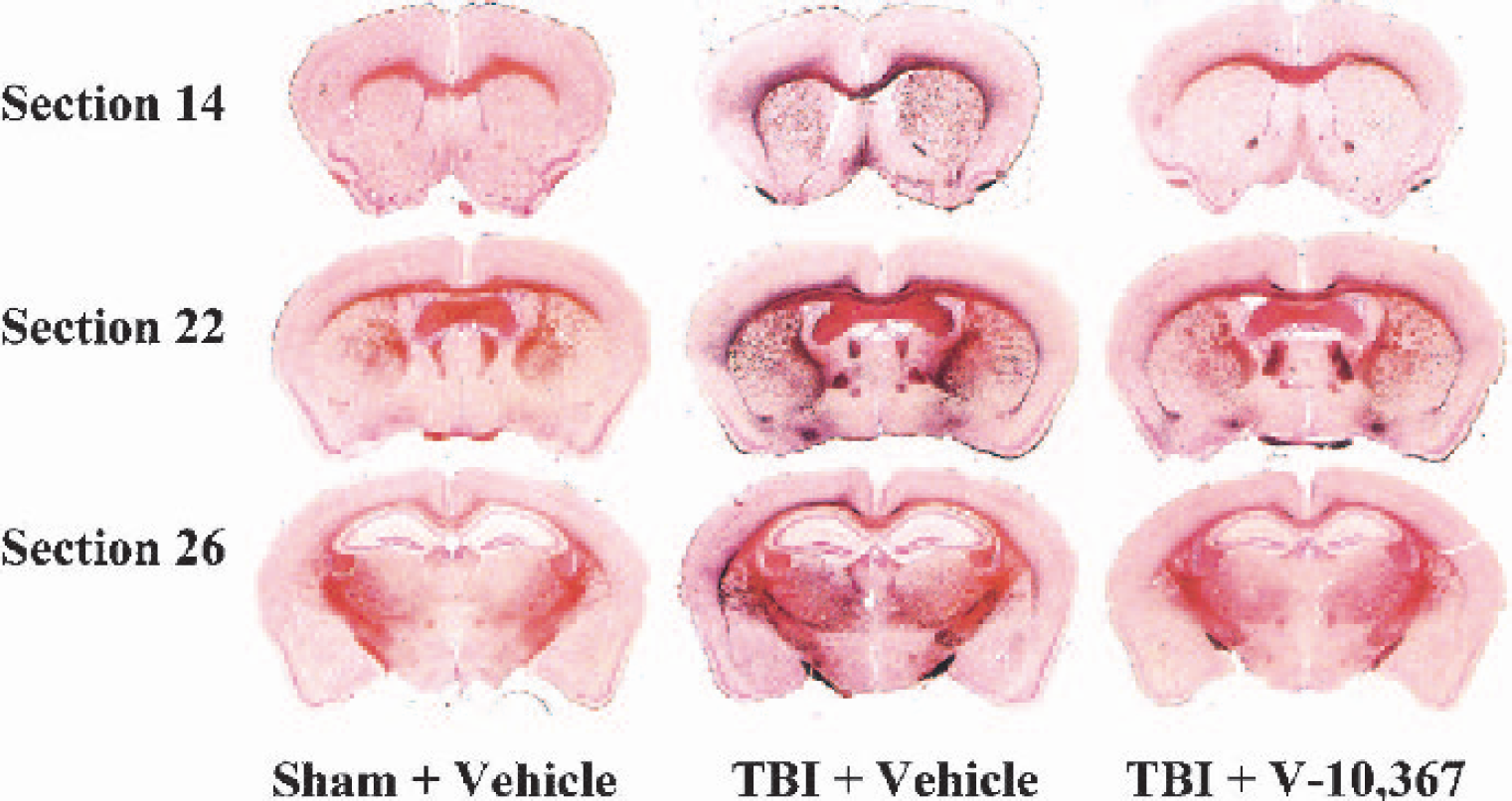
Three representative coronal brain sections from each dosing group (study 2, N = 6; 13 sections per brain). Brains were embedded in a gelatin matrix, frozen, and sectioned at a thickness of 35 μm, impregnated with amino cupric silver stain, and counterstained with Neutral Red. V-10,367–treated traumatic brain-injured (TBI) mice had significantly less silver staining (percent area in select sections and percent total volume) when compared to vehicle-treated TBI-mice (see Results, Fig. 5, and Table 1).
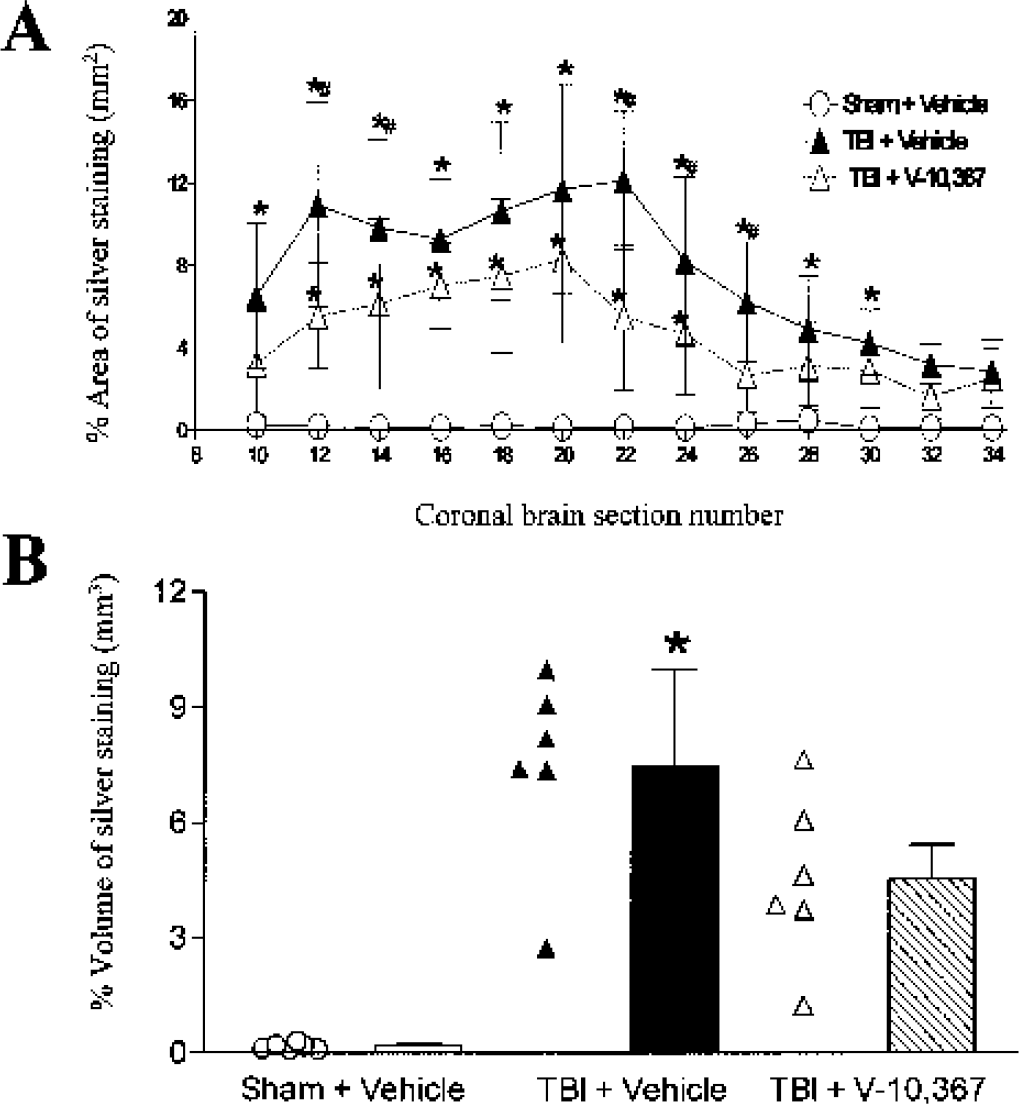
(A) Graphic summary of the percent area (area of silver stain density/total section area) of silver stain measured in injured mice treated with vehicle or V-10,367 (study 2, N = 6; 13 sections per brain; mean ± SD). A significant treatment difference (P < 0.001), as well as an interaction between treatment and day after injury (P < 0.001) was identified by two-way repeated measures analysis of variance. Tukey multiple comparisons tests revealed greater percent area silver staining (*P < 0.05) in vehicle-treated traumatic brain-injured (TBI) mice compared to vehicle-treated sham mice. However, drug-treated TBI mice had less percent area silver staining (#P < 0.05) than in vehicle-treated TBI mice.
Percentage area of posttraumatic silver staining in coronal brain sections
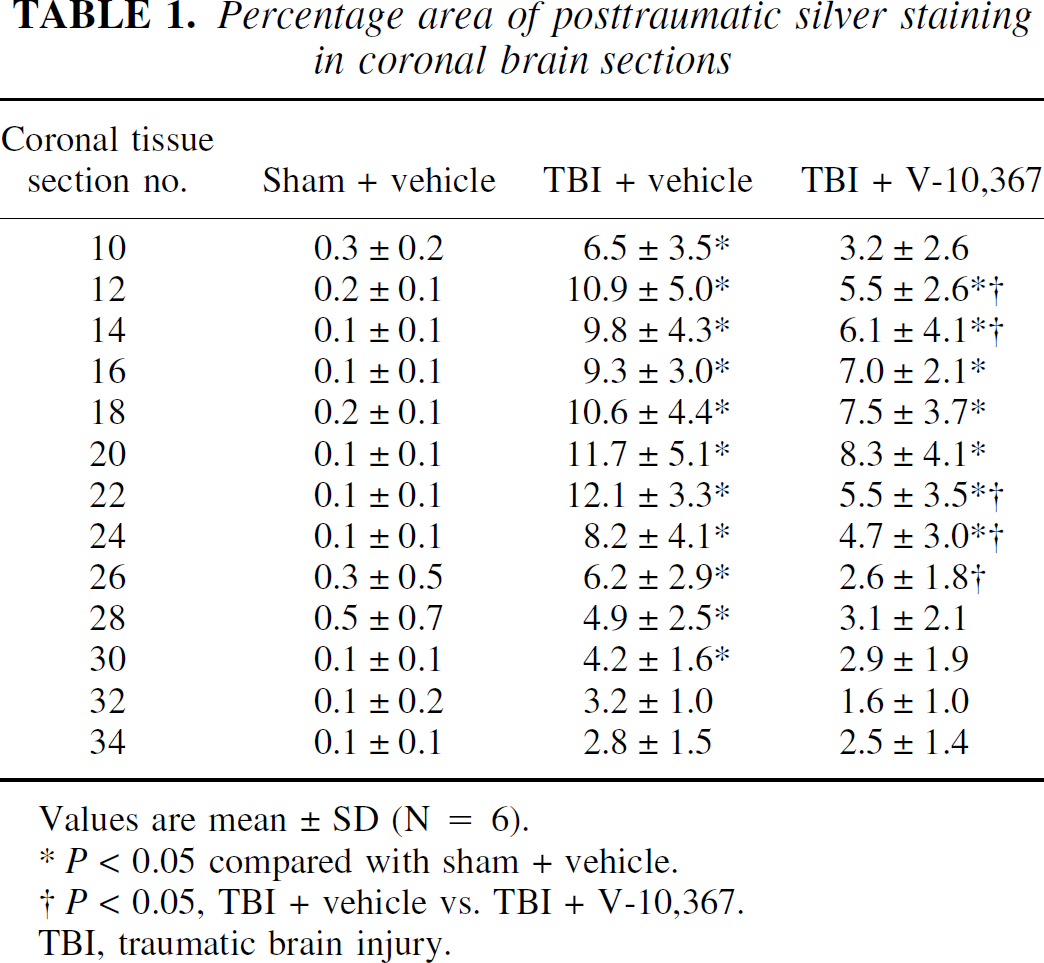
Values are mean ± SD (N = 6).
P < 0.05 compared with sham + vehicle.
P < 0.05, TBI + vehicle vs. TBI + V-10,367.
TBI, traumatic brain injury.
Percent volumetric reconstruction of the percent area of silver staining is illustrated in Fig. 5B (mean ± SD, N = 6 brains x 13 sections). Traumatic brain injured vehicle-treated mice had a significantly greater percent volume of silver staining compared to vehicle-treated sham mice. However, drug-treated TBI mice were not significantly different than vehicle-treated sham mice (Kruskal-Wallis nonparametric ANOVA, P = 0.0013; Dunn's multiple comparisons test, P < 0.001 vs. sham + vehicle).
Effect of V-10,367 on posttraumatic total body weight change
Total body weight was measured each morning in vehicle- and drug-treated sham and TBI mice over a posttraumatic time course of 28 days (mean ± SD, N = 12). Figure 6 illustrates the weight loss that occurs within 24 hours following TBI (all mice were of similar weight range prior to injury; see Materials and Methods). There was a statistically significant interaction between treatment and day after injury (two-way repeated measures ANOVA, P = 0.004). A significant improvement in weight gain in TBI drug-treated mice, compared to TBI vehicle-treated mice, occurred on days 3, 4, 5, and 6 following injury (Tukey multiple comparisons test, P < 0.05).
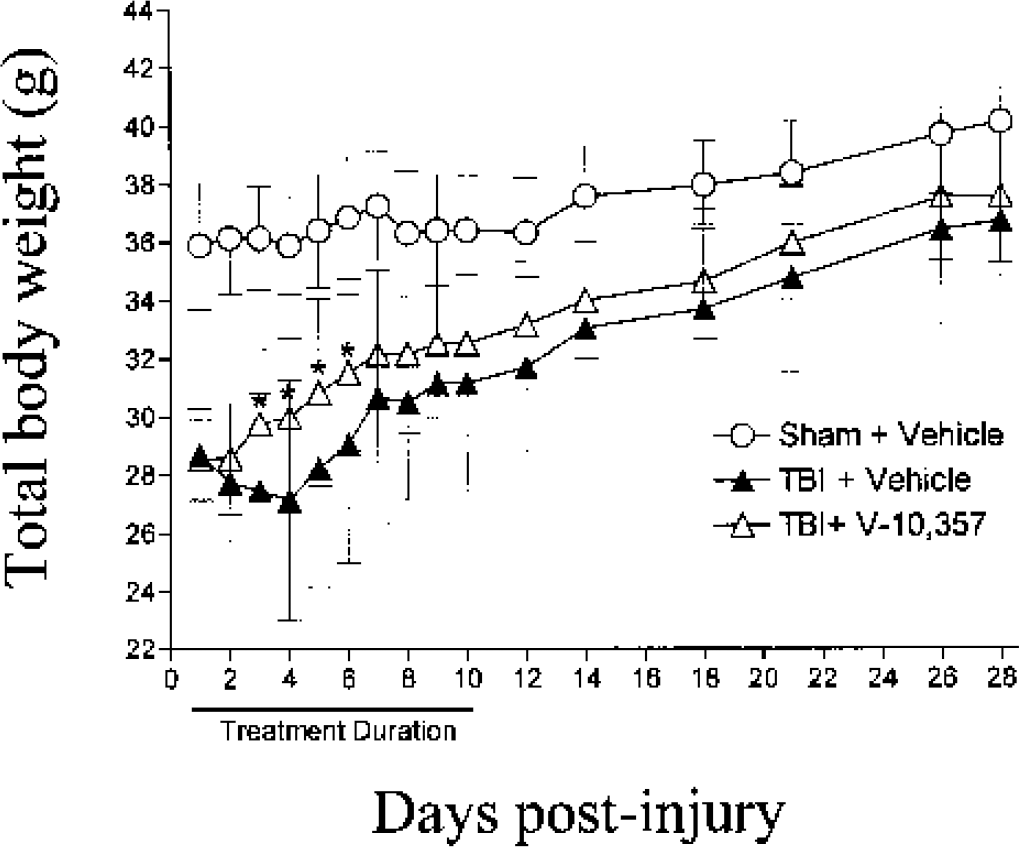
Graphic summary of the total body weight measured over a posttraumatic time course of 28 days (mean ± SD, N = 12) in sham + vehicle- and traumatic brain-injured (TBI) + vehicle- or TBI + drug-treated mice. A significant interaction between treatment and day after injury was measured by two-way repeated measures analysis of variance, P = 0.004. Note an improvement in weight gain in TBI drug-treated mice, compared to TBI vehicle-treated mice, which occurred on days 3, 4, 5, and 6 following injury (*Tukey multiple comparisons test, P < 0.05).
DISCUSSION
The data that we present show, for the first time, impressive evidence that the nonimmunosuppressive NIMM ligand V-10,367 can provide neuroprotection in a model of TBI, even when dosing is delayed until 24 hours after injury. Although a precise determination of the therapeutic window remains to be made, the 24-hour delay in treatment still resulted in a significant reduction in posttraumatic cytoskeletal degradation and neurodegeneration. The nature and extent of this therapeutic window is quite unprecedented compared to other neuroprotective agents tested in various TBI animal models and in clinical trials (Narayan and Michel, 2002). The only exception to this is the immunophilin cyclosporin A, which is also reported to possess a 24-hour therapeutic window in the rat controlled cortical impact injury model (Sullivan et al., 2000).
In the current study, a 24-hour posttraumatic dosing delay of V-10,367 (by mouth, 50 mg/kg x 2 per day for 2 days) was shown to protect against α-spectrin degradation in areas of the cortex, striatum, and hippocampus [measured 3 days after injury (study 1)]. Also, V-10,367 had a significant effect on the elevation of NFM protein levels, including those in noninjured, drug-treated mice. These results provide evidence of a short-term treatment benefit in TBI and suggest the likelihood that the neuroprotective effect of V-10,367 involves inhibition of calpain-mediated cytoskeletal damage. However, the fact that the short-term attenuation of cytoskeletal damage translates into lasting neuroprotection is evidenced (study 2) through histologic findings following a similar 24-hour delay in treatment (by mouth, 50 mg/kg x 2 per day for 10 days). On posttraumatic day 42 (31 days after cessation of drug treatment), vehicle- and drug-treated sham and TBI mice were killed and their brains were silver stained for quantitative densitometric analysis of neurodegeneration. The more current de Olmos method of amino cupric silver staining (de Olmos et al., 1994) selectively detects neuronal degeneration and associated debris by chemical reduction of accumulated silver ions, resulting in a black stain that can discern degeneration of synaptic terminals, cell bodies, dendrites, and axons (Switzer, 2000). The evidence for this type of selective staining (which also includes the Fink and Heimer method) has been shown, ultrastructurally, by several laboratories (Grafe and Leonard 1980a, 1980b; Heimer, 1970; Ikonomidou et al., 2000; Yamamoto et al., 1986). V-10,367-treated TBI mice had significantly less silver staining (percent area in select sections and percent total volume) when compared to vehicle-treated TBI mice. It should be noted that the lesser density of silver stain in the drug-treated mice at 42 days after injury could be due to either less residual neurodegenerative debris remaining as a result of attenuation of early degeneration or possibly due to inhibition of delayed (apoptotic) degeneration. The present experiments do not allow for a distinction between these two possibilities.
The rationale for a 24-hour posttraumatic delay in treatment with the NIMM ligand V-10,367 and the subsequent measurement of degraded cytoskeletal proteins at 3 days after injury was based upon previous knowledge of the time course of cytoskeletal degradation in the mouse TBI model. Recently, we provided evidence for the correlated timing of cytoskeletal degradation and neurodegeneration following experimental TBI (Kupina et al., 2002). In male head-injured mice, degradation of the cytoskeletal proteins α-spectrin (into 150- and 145-kd breakdown products) and NFM were shown to reach peak levels between 3 and 7 days after injury (Western blot analysis), which coincided with the peak magnitude of neurodegeneration as assessed by silver impregnation. These studies indicated a critical role for calpain- and, perhaps, caspase-3-mediated events in the pathophysiology of TBI, since both calpain and caspase-3 share similar proteolytic substrates (i.e., cytoskeletal proteins) (Wang, 2000). The fact that cytoskeletal damage and neurodegeneration did not peak until 3 days after injury suggested that, for some neuroprotective strategies, a therapeutic window of 24 to 48 hours might be feasible. The present results confirm the concept that agents that target cytoskeletal damage may indeed possess a 24-hour window of opportunity.
As already noted, degradation of cytoskeletal elements, including α-spectrin and NFM, can be carried out by either calpain or caspase-3 (Wang, 2000). The relative roles of the two enzymes in cytoskeletal damage can be partially dissected. For instance, portions of the SBDP 150 fragment can be formed by both calpain and caspase-3 degradation of α-spectrin. In the present experiments, we were able to employ an antibody specific for the calpain-mediated portion of the SBDP 150 fragment [Senju Pharmaceuticals; see (Pike et al., 2001) for previous antibody usage]. This approach allowed us to demonstrate that calpain activation had a significant role in the development of the SBDP 150 fragment that occurred 3 days after injury in untreated TBI mice. In contrast, treatment with V-10,367 completely prevented this increase. These results show that most, if not all, of V-10,367's cytoskeletal-protective effect is due to a mechanism or mechanisms that ultimately attenuate calpain activation. However, the present experiments do not rule out a contribution of caspase-3 activation in the degradation of the cytoskeleton, nor do they eliminate the possible inhibition of caspase-3 activation as being part of the protective effect of V-10,367. The statement of this caveat is particularly appropriate in view of recent studies showing caspase-3 activation in rodent models of TBI (Beer et al., 2000; Buki et al., 2000; Pike et al., 1998).
It should be noted that the caspase-3 generated 120-kd SBDP (Wang, 2000) was not examined in detail. Previous studies (Kupina et al., 2001, 2002) in our laboratory with the presently employed mouse TBI model have failed to demonstrate a consistently measurable level of SBDP 120 in any of the brain regions examined either before or after TBI. This is in contrast to the small, but measurable, increase in SBDP 120 seen in the rat controlled cortical impact injury model, particularly at the epicenter of the cortical injury (Pike et al., 1998). However, the severity of the focal rat TBI model is more intense at least at the injury epicenter than that of the diffuse mouse TBI paradigm.
In addition to the neuroprotective action of V-10,367, the present results also suggest that V-10,367 may stimulate an increase in neuronal plasticity. Previously, we reported evidence of an endogenous increase in NFM protein levels in male TBI mice between 2 and 3 weeks after injury (Kupina et al., 2002). Indeed, very recently, Zheng et al. (2001) have shown that injured (sciatic nerve crush) adult sensory neurons (DRG) are able to synthesize intraaxonal proteins, which act to facilitate regeneration of the axon. They also reported similar translational factors, ribosomal proteins, and rRNA in motor axons (ventral spinal roots) following a 7-day sciatic nerve crush, suggesting that these mechanisms are not limited to sensory neurons. In the present study, as early as 3 days after injury, TBI drug-treated mice (2 days, oral dosing) had significantly higher levels of NFM proteins in the cortex, striatum, and hippocampus than in vehicle-treated TBI mice. Furthermore, otherwise normal, uninjured mice treated with V-10,367 had significantly higher levels of NFM proteins compared to vehicle-treated uninjured mice. While additional work is required, such an action would not be unexpected, considering the ability of V-10,367 to increase axonal sprouting and neurite outgrowth in neuroblastoma cells (Gold et al., 1997).
Another significant effect of V-10,367 on mice subjected to TBI is a demonstrable improvement in recovery of body weight during the first postinjury week. Within the first 24 hours following TBI, the mice lost, on average, 7.5 g of body weight. This could be due to the well-documented generalized increase in protein degradation (i.e., negative nitrogen balance) that has been shown to occur in rodent TBI models (Krishnappa et al., 1999) and in head-injured humans (Dickerson et al., 1990; Young et al., 1991). Furthermore, the loss of weight could be attributed to a certain degree of dehydration (since it is not practical to maintain the mice on intravenous fluids), although the animals did not show any signs to indicate this as an obvious concern. Over a period of 27 days, vehicle-treated TBI mice gradually recovered their body weight, while V-10,367-treated mice showed significant improvement in weight gain as early as 3 to 6 days following injury. This effect, while modest, could be secondary to a reduction in posttraumatic brain damage and subsequent greater ability to eat and drink, and/or a systemic action to mitigate protein degradation.
Although a growing amount of research has been published to demonstrate neuroprotective and neuroregenerative effects of NIMM ligands, the mechanisms to account for these effects are not well understood. One mechanistic focus is directed to non-calcineurin-mediated activities, such as that related to the mitochondrial transition pore and mitogen-activated protein kinase pathways that have protective/neuroregenerative roles in TBI and stroke (Albensi et al., 2000; Gogarten et al., 1998; Pratt and Toft, 1997). A second area of focus is on FK506 binding proteins (FKBPs), particularly those that are highly expressed in the nervous system, such as FKBP 12, 38, 52, and 65. Despite the fact that the role of FKBP 12 has been widely considered as a site for immunophilin ligand activity, more recent studies (utilizing FKBP12-deficient mice) have provided evidence that this binding protein is not necessarily required for NIMM-mediated neurotrophic effects (Gold et al., 1999; Guo et al., 2001). Gold et al. (1999) have reported a potentially important role for FKBP 52/59, which is part of a steroid receptor complex. Through the use of a monoclonal antibody to FKBP-52, they were able to completely block the neurotrophic action of FK506 in human neuroblastoma SH-SY5Y cells. The current study was not designed to shed any new light on the present mechanistic quandary that underlies the neuroprotective/neuroregenerative properties of the NIMMs. However, the results strongly support the conclusion that the manifestation of these properties concerns the protection and possible growth of the neuronal cytoskeleton. In the case of the neuroprotective effects, a main consequence of NIMM action appears to be inhibition of calpain-mediated cytoskeletal degradation.
At present, it is uncertain whether we can extrapolate the time course of posttraumatic cytoskeletal degradation/neuronal degeneration, as well as the favorable therapeutic window for V-10,367, in our mouse model of TBI to the human TBI condition. However, there are some clinical data available to suggest that the mouse data may have some relevance to the time course of human cytoskeletal damage following TBI. For instance, posttraumatic secondary axonal injury in humans has been shown to become apparent after 12 hours (Blumbergs et al., 1995; Gentleman et al., 1993; Grady et al., 1993). More specifically, McCracken et al. (1999) have recently described SBDP increases and neurofilament degradation in the corpus callosum of patients who sustained blunt TBIs at time points greater than 24 hours following injury.
Future experiments are needed to further examine the neuroprotective and possible neuroregenerative effects of V-10,367 and other NIMMs in this and other models of TBI. Although the present results are consistent with other published effects of V-10,367 in related central and peripheral neurodegenerative models, it will be necessary to do dose-response studies, as well as evaluate different treatment durations, since 10 days of treatment may not be optimal and dosing earlier or later than 24 hours might prove more or less effective. Nevertheless, the current demonstration that a pharmacological compound can exert a significant protection of the neuronal cytoskeleton and attenuate neurodegeneration after a moderately severe TBI is heretofore unprecedented, with the possible exception of the immunosuppressive immunophilin cyclosporin A (Sullivan et al., 2000).