
Abstract
This study examined the effect of posttraumatic hypoxia on cerebral vascular responsivity and axonal damage, while also exploring hypothermia's potential to attenuate these responses. Rats were subjected to impact acceleration injury (IAI) and equipped with cranial windows to assess vascular reactivity to topical acetylcholine, with postmortem analyses using antibodies to amyloid precursor protein to assess axonal damage. Animals were subjected to hypoxia alone, IAI and hypoxia, IAI and hypoxia before induction of moderate hypothermia (33°C), IAI and hypoxia induced during hypothermic intervention, and IAI and hypoxia initiated after hypothermia. Hypoxia alone had no impact on vascular reactivity or axonal damage. Acceleration injury and posttraumatic hypoxia resulted in dramatic axonal damage and altered vascular reactivity. When IAI and hypoxia were followed by hypothermic intervention, no axonal or vascular protection ensued. However, when IAI was followed by hypoxia induced during hypothermia, axonal and vascular protection followed. When this same hypoxic insult followed the use of hypothermia, no benefit ensued. These studies show that early hypoxia and delayed hypoxia exert damaging axonal and vascular consequences. Although this damage is attenuated by hypothermia, this follows only when hypoxia occurs during hypothermia, with no benefit found if the hypoxic insult proceeds or follows hypothermia.
Introduction
Hypoxic insults typically occur early after traumatic brain injury (TBI) or during the subsequent intensive care management of the patient. Clinically, hypoxia can arise from posttraumatic airway obstruction, reduced ventilation, neurogenic pulmonary edema, or acute respiratory distress syndrome (Rockswold et al, 2006). As reported in epidemiologic investigations (McHugh et al, 2007), the prevalence of posttraumatic hypoxic insult varies from 5.9% to 34.5%. Further, the occurrence of a hypoxic episode after injury negatively impacts on patient outcome, with the odds for an adverse outcome now doubling (Murray et al, 2007). Although published evidence affirms that hypoxia is one of the most common and devastating secondary insults after TBI (Rosner 2003; Thomas et al, 2000), postinjury interventional strategies to attenuate its damaging consequences have not been critically evaluated in either the clinical or laboratory setting. Unfortunately, this failure has most likely negatively impacted the treatment of TBI, which is currently associated with significant morbidity and mortality (Geeraerts et al, 2008; Stahel et al, 2008).
Recently, there has been renewed interest in the use of posttraumatic mild hypothermia (Dietrich et al, 2009; Povlishock and Wei, 2009; Clifton et al, 2009; Jiang, 2009). Further, it has been suggested that mild (33°C to 35°C) prolonged hypothermia can provide significant improvement in the patient's Glasgow outcome scale (Jiang et al, 2006; Jiang and Yang, 2007). To date, the presumed targets of hypothermia are multiple, with the suggestion that some of hypothermia's protective effects reside in its ability to blunt the damaging consequences of posttraumatic secondary insults. Yamamoto et al (1999) in an animal model of TBI complicated by hypotension and hypoxia showed brain protection after the introduction of posttraumatic hypothermia. In contrast, Robertson et al (2000) showed no long-term benefit of posttraumatic hypothermia in a rodent animal model of controlled cortical injury and hypoxic insult in terms of behavioral and cognitive performance as well as lesion volume and hippocampal neuronal loss. In light of the potential benefits associated with post-TBI hypothermia as well as the controversy surrounding its potential attenuation of the damaging consequences of secondary insults, we have revisited the potential that posttraumatic hypothermia can reduce the adverse consequences of traumatically related secondary insults. To this end, we evaluated cerebral vascular functional activity and brain stem axonal damage, exploring whether the mild hypothermia can attenuate the adverse effects of hypoxia after impact acceleration injury (IAI).
Materials and methods
General Preparation
All experiments were carried out in adult male Sprague–Dawley rats (
Studies of the Cerebral Microcirculation
A middle sagittal incision was performed to expose the skull bone, which was cleaned and dried. A rectangular opening, 3 × 4 mm, was made in the skull over the left parietal cortex and removed, together with careful resection and removal of the underlying dura mater. Next, a cranial window was installed over the exposed brain surface, and fixed in place by bone wax and dental acrylic. As described previously (Ellis et al, 1983; Levasseur et al, 1975), the cranial window consisted of a stainless steel ring with three outlets and a circular glass plate inside a ring. Two of the outlets served as inflow and outflow paths for the perfusion and clearance of selected vasoactive agents, whereas the free end of the other outlet was set at a predetermined height to achieve an intracranial pressure of 5 mm Hg. The space under the cranial window and the three outlets were then filled with sterile artificial cerebrospinal fluid, pH of which was adjusted to 7.35 by equilibration with a gas mixture containing 6% O2 and 6% CO2 balanced with N2. The underlying pial microcirculation was visualized with a microscope and pial arteriolar diameters were measured with a Vickers image-splitting device (Vickers Instruments Inc., Maiden, MA, USA). Typically, in each window preparation a minimum of four arteriolar segments were evaluated. In this study, the vasodilator acetylcholine (ACh) was used in two different concentrations to assess vascular dilation after its topical application by the cranial window. Acetylcholine is well known to elicit endothelial-dependent vasodilatation (Kontos et al, 1988; Wei et al, 1992). Acetylcholine (Sigma, St Louis, MO, USA) was dissolved in artificial cerebrospinal fluid to achieve the final concentrations of 10−7 and 10−5 mol/L and then applied by the cranial window to induce a vasodilatory response. After each application of ACh in the space under the window for 2 to 4 mins, vessel diameter was measured. The vascular reactivity to ACh was expressed as a percent change from the baseline diameter at each measurement time point.
Traumatic Brain Injury
After the baseline vessel diameter and ACh-induced vessel diameter were measured, the cranial window was gently removed. The exposed brain was then covered with a piece of wet gauze. Next, a 10-mm, circular stainless steel helmet was installed over the sagittal suture between bregma and lambda and secured with dental acrylic. The respiratory tube was disconnected from tracheal tube, and the animal was rapidly placed prone on a foam pad and subjected to IAI, as described in detail in previous publications (Marmarou et al, 1994). Briefly, a weight of 450 g was dropped from a height of 2 m through a Plexiglas tube on the metal helmet, which was centered immediately under the lower end of the tube. The animal was returned to the respirator immediately after the injury with the continuation of blood gas and blood pressure monitoring. The helmet was then removed and the cranial window was reinstalled.
Experimental Design
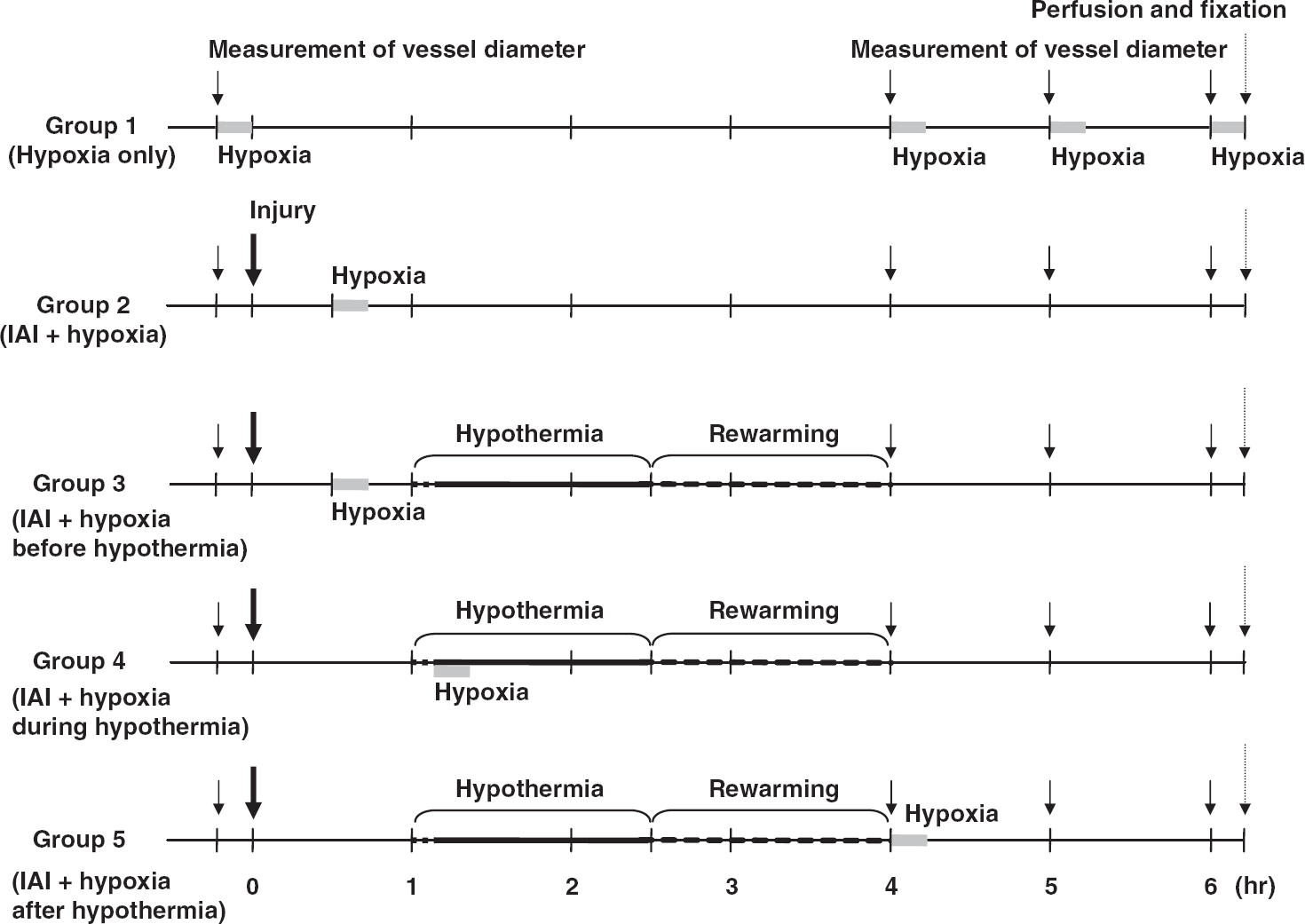
In these studies, both vascular function and the burden of axonal damage were assessed in the context of TBI followed by hypoxia and hypotension. To this end, we arbitrarily divided the animals into five groups. Each group contained five animals.
Vascular Studies
Group 1: Sham surgery and hypoxia:
These animals underwent the same surgical procedures used in all other animals; however, they were not subjected to IAI. Vascular reactivity to the two concentrations of ACh was assessed before inhalation of a 10% O2 gas mixture for a duration of 10 mins (hypoxia), and again at 4 and 5 h before the induction of other hypoxic insults, with a final assessment at 6 h after hypoxia (Figure 1).
This chart shows the time course for each experimental group.
Group 2: IAI followed by early hypoxia:
Vascular reactivity to two concentrations of ACh was assessed before IAI and the inhalation of a 10% O2 gas mixture for a duration of 10 mins initiated 30 mins after the induction of IAI. In addition, vascular reactivity to two concentrations of ACh was again assessed at 4, 5, and 6 h after injury (Figure 1).
Group 3: IAI and early hypoxia followed by hypothermic intervention:
In these studies, vascular reactivity to two concentrations of Ach was assessed before the induction of IAI. Thereafter, all animals were injured and then 30 mins later, the animals were subjected to the inhalation of a 10% O2 gas mixture for a duration of 10 mins. In these same animals, at 1 h after the induction of injury, a 90-mins period of hypothermia (33°C) was induced by the use of ice packs followed by slow rewarming to normothermic levels over an additional 90 mins period. Thereafter, vascular reactivity to ACh was evaluated at 4, 5, and 6 h after injury (Figure 1).
Group 4: IAI followed by the delayed induction of hypoxia during hypothermic intervention:
Again, vascular reactivity to two concentrations of ACh was assessed before the induction of IAI. Consistent with the above protocols, a 90 mins period of hypothermia (33°C) was then induced 1 h after injury. However, in this group, once the rectal temperature reached 33°C, the animals were subjected to the inhalation of a 10% O2 gas mixture for a duration of 10 mins. After hypothermia and rewarming as detailed above, the vascular responses to ACh were again assessed at 4, 5, and 6 h after injury (Figure 1).
Group 5: IAI followed by delayed hypothermia and posthypothermic rewarming:
As with previous protocols, vascular reactivity to two concentrations of ACh was assessed before the induction of IAI. In this group, at 1 h after injury, a 90 mins period of hypothermia (33°C) was induced, followed by a 90 mins period of rewarming. On the completion of the rewarming period, the animals were subjected to inhalation of a 10% O2 gas mixture for a duration of 10 mins, with vascular reactivity to ACh assessed at 4, 5, and 6 h after injury (Figure 1).
Axonal Damage Assessments
For evaluating the burden of axonal damage after TBI and hypothermia/hypoxia, we analyzed axonal damage in brain stem projection axons using strategies previously used in our laboratory (Koizumi and Povlishock, 1998; Suehiro and Povlishock, 2001). In this approach, we used the same animals evaluated in groups 1 to 5 for the conduct of detailed axonal analyses. At 6 h after injury after the measurement of vascular reactivity to ACh (in groups 1 to 5), the rats were killed with an overdose of euthanasia solution under general anesthesia, and perfused with 4% paraformaldehyde, 0.1% glutaralaldehyde, and 0.1 mol/L Millonig's phosphate buffer. After perfusion, the brains were removed, transferred to fixative, and stored overnight. They were placed in a sagittal brain-blocking device with 2 mm of the brain cut from the brain's midsection followed by further sagittal blocking to include the medulla, pons, and midbrain. This blocking strategy was based on the fact that in this model system, numerous damaged axons occur in the pyramids at the medullospinal junction in the descending corticospinal tracts (Povlishock et al, 1997). After harvesting the 2-mm-wide sagittal block, the tissue section was flat-mounted and serially sectioned on the vibratome at a thickness of 40 μm. The sections were processed for visualization of an antibody targeting to amyloid precursor protein (APP), a marker of impaired axonal transport and axonal damage using a protocol adapted in our laboratory (Stone et al, 1999). Briefly, the sections were reacted with 0.3% H2O2 in phosphate-buffered saline for 30 mins to block endogenous peroxidase activity and microwaved twice in citric acid while maintaining a 45°C maximum temperature for 5 mins. After microwave processing, the sections were allowed to cool for 20 mins each time. The sections were incubated for 1 h with 0.2% Triton X in 10% normal goat serum in phosphate-buffered saline and then for 18 h with the rabbit anti-β-APP (Invitrogen, Carlsbad, CA, USA) diluted 1:1500 in 1% normal goat serum in phosphate-buffered saline. On the following day, the sections were incubated for 1 h with biotinylated rat-absorbed goat anti-rabbit immunoglobulin G (Vector Laboratories Inc., Burlingame, CA, USA) diluted 1:1200 in 1% normal goat serum in phosphate-buffered saline. Then, sections were visualized by incubation for 1 h in avidin biotinylated enzyme complex (Vectastain ABC kit; Vector Laboratories) followed by 0.05% diaminobenzidine, 0.01% H2O2, and 0.3% imidazole in 0.1% mol/L sodium phosphate buffer for 15 mins. The sections were mounted on 0.5% gelatin-coated glass slides, serially dehydrated, and coverslipped.
Quantitative Analysis of the Axonal Damage
After completion of the APP immunocytochemical procedures, the slides were transferred to an Eclipse 800 microscope (Nikon, Tokyo, Japan) interfaced with a computer-assisted imaging system DP Controller, Version 3.2 (Olympus Corporation, Tokyo, Japan). Consistent regions of the medulla at the medullospinal junction were enlarged to a magnification of × 10 and saved as a tagged image file format. On the basis of our previous experience with this model (Koizumi and Povlishock, 1998; Suehiro and Povlishock, 2001), the image was viewed on a monitor using image analysis software IPLab, version 3.7 (BD Biosciences Bioimaging, Rockville, MD, USA) and changed to gray scale (Figure 2A and 2B). The APP-immunoreactive axonal profiles were outlined and overlaid with cyan color, a strategy used to suppress background immunoreactivity (Figure 2C). The sampling area was delineated by a rectangle measuring 500 × 200 μm that was superimposed over the specified region (Figure 2C). The number of damaged APP-immunoreactive axonal profiles within this rectangle that exceeded 20 μm2 was then counted. This number was expressed as the density of damaged axons per unit area. For the corticospinal tract, eight alternate serial sections from the same tissue block were analyzed in this manner. These eight sections were randomly selected to prevent bias in their analysis.
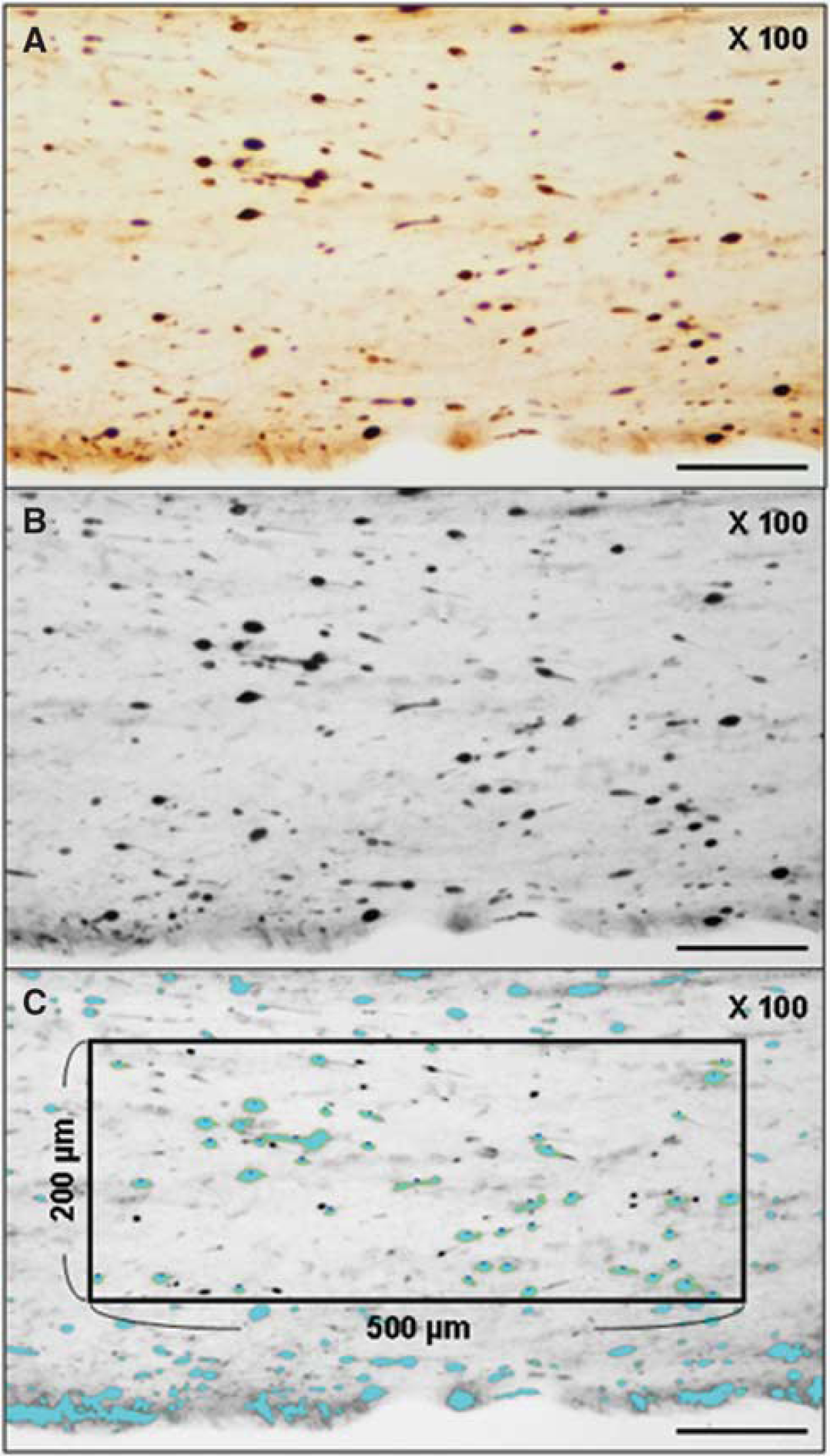
The photomicrograph of the representative section of the rat brain in group 3 shows numerous damaged APP-immunoreactive axons within the corticospinal tract at the level of the medullospinal junction (
General Statistical Analysis
Statistical analysis was performed using the statistical software SPSS, version 16.0 (SPSS Inc., Chicago, IL, USA). All data were presented as mean ± s.e.m. The physiologic parameters, laboratory data, and vascular reactivity to ACh, which were normally distributed, were analyzed by oneway analysis of variance. When a significant difference was found, multiple comparisons among time points in same group and groups at each time point were performed using Bonferroni test and Scheffe test, respectively. The number of damaged axons, which were not normally distributed, was analyzed by the Kruskal–Wallis test followed by the Bonferroni test for multiple comparisons. A value of
Results
General Physiologic Observations
A total of 25 rats (group 2 to 5,
There were no significant differences in baseline body weight and hematocrit between groups. The rectal temperature in the normothermic groups (groups 1 and 2) was maintained at 37°C over the entire experimental period (Figure 3). In the hypothermic groups (groups 3, 4, and 5), the rats were cooled to the target temperature of 33°C within 19 mins (Figure 3). No significant differences were found in the intervals needed to reach this temperature between groups 3, 4, and 5 (12.6 ± 0.4, 12.8 ± 0.4, and 13.2 ± 1.5 mins, respectively). The rats were maintained at this level for 90 mins, including the induction of hypothermia. After the completion of hypothermia, they were rewarmed to a normothermia over 90 mins. There were no significant differences in the rewarming rates between groups 3, 4, and 5 (26.0 ± 0.9, 23.9 ± 0.8, and 23.5 ± 0.8 mins/°C, respectively). The mean arterial blood pressure and arterial oxygen tension during hypoxic period were significantly lower than those during pre-TBI period or other measurement points (Table 1). During the hypothermic period, the mean arterial blood pressure was lower and the arterial oxygen tension was higher than that seen in pre-TBI period (Table 1). Although the mean value of arterial carbon dioxide pressure during the hypoxic period in group 4 was lower than that in other groups, there were no significant differences between all groups. Other than these physiologic changes, all the other physiologic variables reported in Table 1 were within normal physiologic limits.
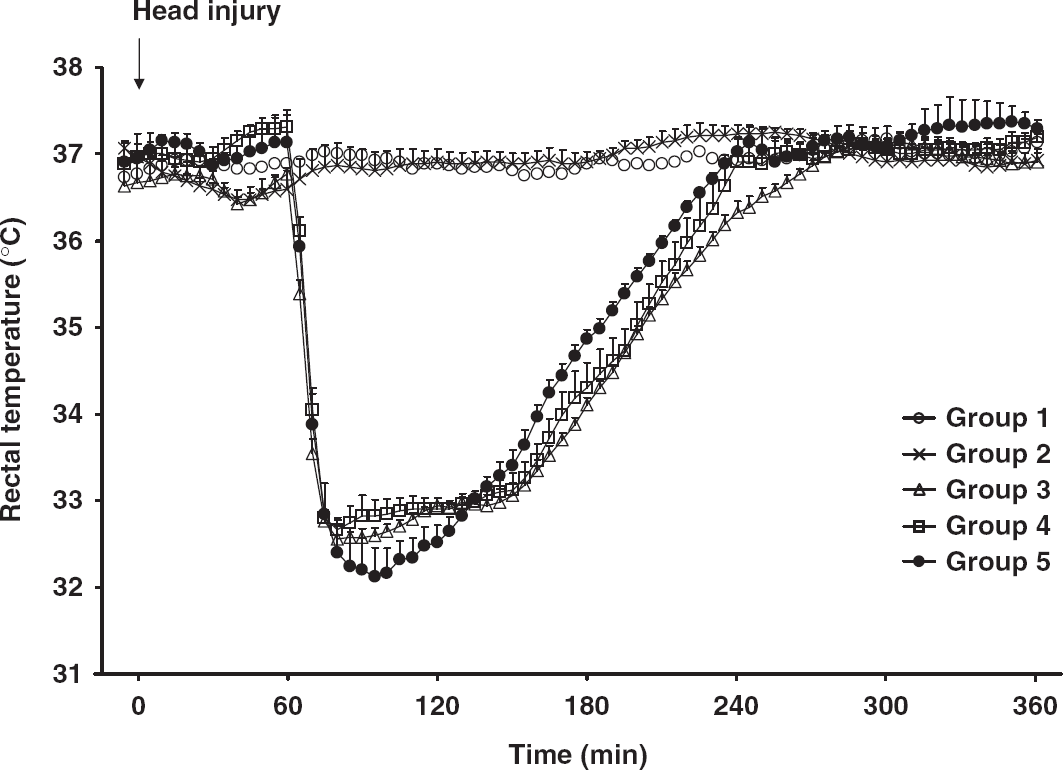
This graph shows the changes of the mean rectal temperature throughout the duration of this study. The data points represent 5 mins intervals. The values are expressed as mean± s.e.m.
Physiological parameters
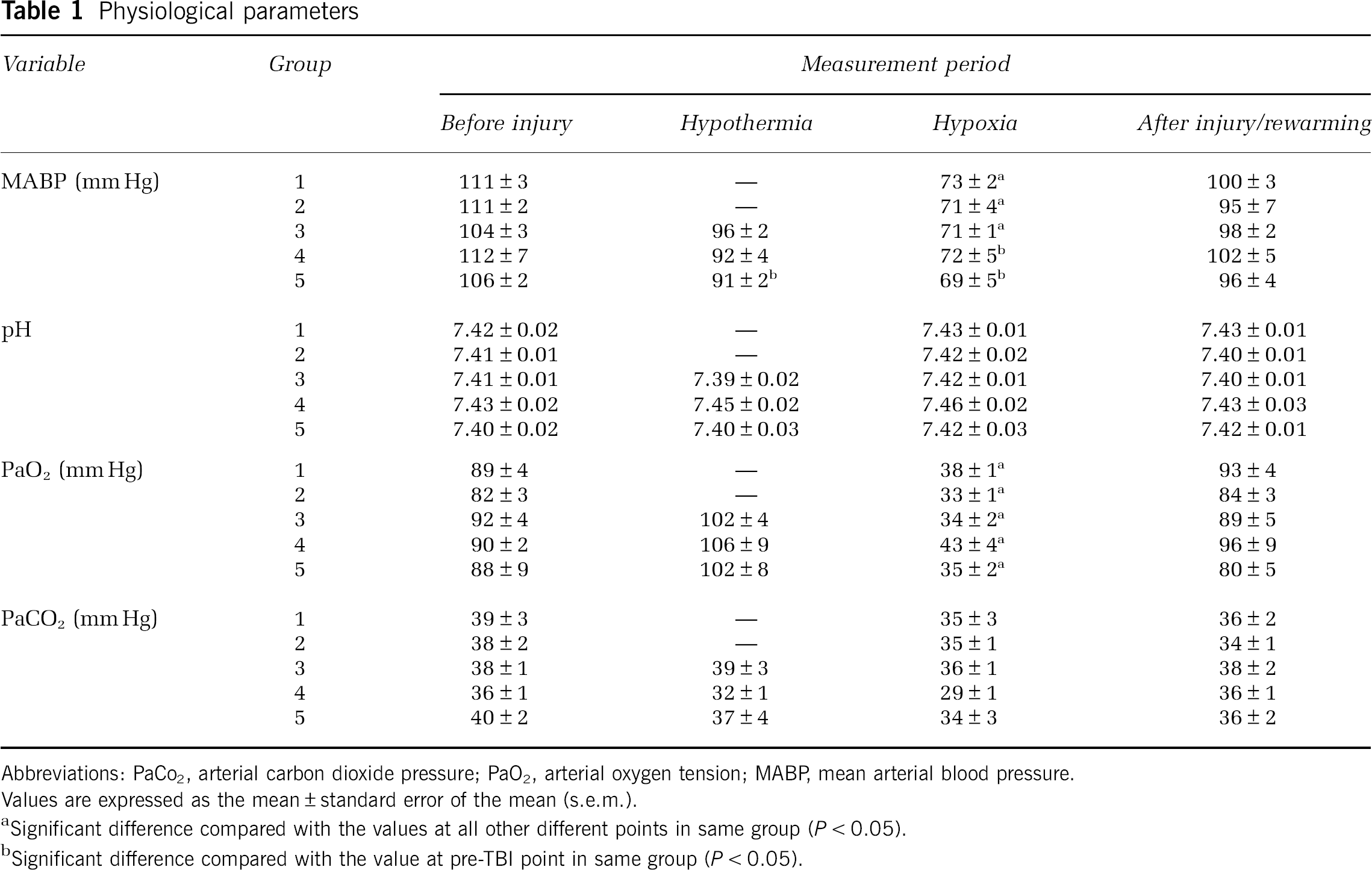
Abbreviations: PaCo2, arterial carbon dioxide pressure; PaO2, arterial oxygen tension; MABP mean arterial blood pressure. Values are expressed as the mean ± standard error of the mean (s.e.m.).
Significant difference compared with the values at all other different points in same group (
Significant difference compared with the value at pre-TBI point in same group (
Brain Arteriolar Reactivity After Hypoxia Alone or Impact Acceleration Injury Hypoxia
The arteriolar reactivity in sham-injured animals (group 1) was measured separately to reveal any injury-induced change. In group 1, before injury, the vasodilator responses to ACh at 10−7 and 10−5 mol/L elicited increases of 7.9% ± 0.5% and 17.6% ± 0.6% in vessel diameter, respectively (Figures 4 and 5). The consecutive measurements within the same segments taken at the 4, 5, and 6 h after the hypoxic insult showed no significant differences compared with the preinjury reactivity (Figures 4 and 5). In group 2, which included animals subjected to injury and hypoxia, the preinjury vasodilator response to ACh at 10−7 and 10−5 mol/L elicited increases of 9.2% ± 0.6% and 18.3% ± 0.8% in vessel diameter, respectively. The vascular reactivity at 4, 5 and 6 h after injury was significantly decreased almost to the point of extinction compared with the values observed before injury (Figures 4 and 5).
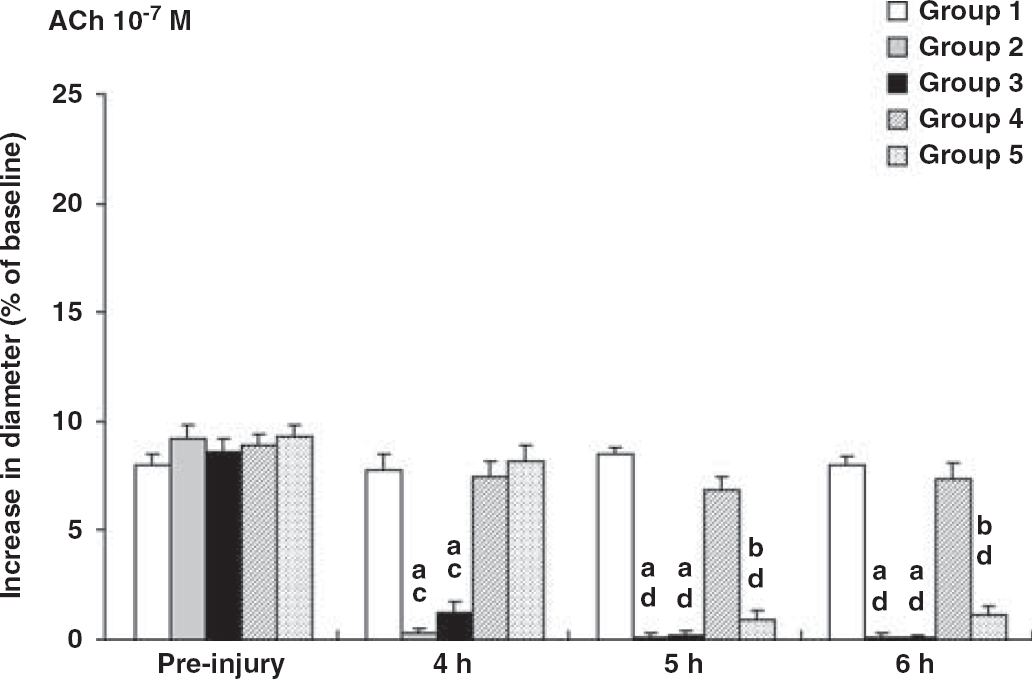
This bar graph shows the vascular reactivity to acetylcholine (ACh) at 10−7 mol/L. The vascular reactivity to ACh before injury is similar in each group, with an approximate 10% increase in diameter. The vascular reactivity after injury in groups 1 and 4 was preserved, with no significant differences compared with vascular reactivity before injury. However, in contrast to the values before injury, the vascular reactivity to ACh before injury is significantly decreased at 4, 5, and 6 h after injury in groups 2 and 3. The preserved vascular responses at 4 h after injury in group 5 are also significantly diminished at 5 and 6 h after the initiation of the hypoxic insult. These decreased values are significantly lower than those in groups 1 and 4 at each time point. Valuesare expressed as mean ± s.e.m. aSignificant differences compared with corresponding preinjury value (
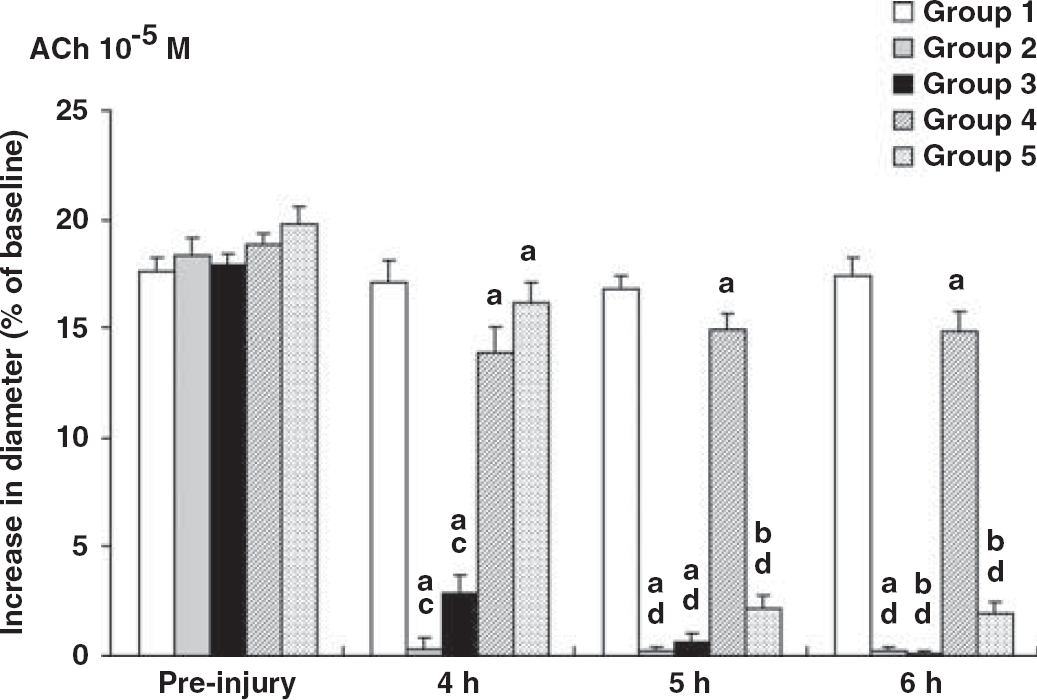
This bar graph reveals the vascular reactivity to acetylcholine (ACh) at 10−5 mol/L. Note that the vascular reactivity to ACh before injury reveals a 20% increase in each group. Although ACh-induced vasodilation in group 1 shows no significant change, ACh-induced vasodilation after injury in group 4 shows a modest decrease in comparison to the preinjury values. Postinjury ACh-induced vasodilatation in groups 2, 3, and 5 is reduced, showing vascular reactivity comparable to that seen with the use of 10−7 mol/L ACh. Values are expressed as mean ± standard error of the mean (s.e.m.). Significant differences compared with corresponding preinjury value (
Brain Arteriolar Reactivity After Impact Acceleration Injury and Hypothermia, with Different Epochs of Hypoxia
The brain arteriolar responses after IAI followed by hypothermia and hypoxic insult are shown in Figures 4 and 5. The preinjury vascular reactivity in groups 3, 4, and 5 were similar to those in groups 1 and 2, with the vascular diameters increasing 10% and 20% after the application ACh of 10−7 and 10−5 mol/L, respectively (Figures 4 and 5). In groups 3 and 5, the vascular reactivity after injury was significantly decreased after either the prehypothermic (group 3) or posthypothermic (group 5) induction of the hypoxic insult. Vascular reactivity to ACh at different concentrations (10−7 and 10−5 mol/L) was significantly reduced to 1.3% ± 0.5% and 2.9% ± 0.8% compared with 4 h after injury in group 3, and to 0.9% ± 0.4% and 2.2% ± 0.6% respectively at 5 h after injury in group 5, with no improvement at 5 and/or 6 h after injury in either group (Figures 4 and 5). In contrast, in group 4, when the hypoxic insult was initiated during the hypothermic period, the vascular reactivity was maintained, with the vascular reactivity to ACh of 10−7 mol/L at 4, 5, and 6 h showing no statistically significant decrease compared with their preinjury values (Figure 4). In fact, the vascular reactivity to Ach of 10−5 mol/L at 4, 5, and 6 h was virtually preserved (Figure 5).
Axonal Injury
Immunocytochemical findings:
By quantitative analyses, the number of damaged axons (17 ± 4 per mm2, mean ± s.e.m.) in group 1, sustaining hypoxia alone, was significantly lower than that in any other experimental group, indicating that hypoxia alone exerted no damaging axonal effects (Figure 6). However, in group 2 that had sustained IAI with early hypoxia, the numbers of damaged axons were striking and equaled 463 ± 60 per mm2 (Figure 6). This dramatic burden of axonal damage was in contrast to the significantly decreased numbers of axons found in group 4 (194 ± 28 per mm2), which involved the combination of IAI with hypoxia during the hypothermic intervention (Figure 6). This finding in group 4 (194 ± 28 per mm2), wherein the hypoxia was embedded in the hypothermic intervention, also stood in distinct contrast to the numbers of damaged axons observed in groups 3 and 5 (369 ± 54 and 290 ± 33 per mm2, respectively), wherein the hypoxic insult was initiated either before or after hypothermic treatment (Figure 6).
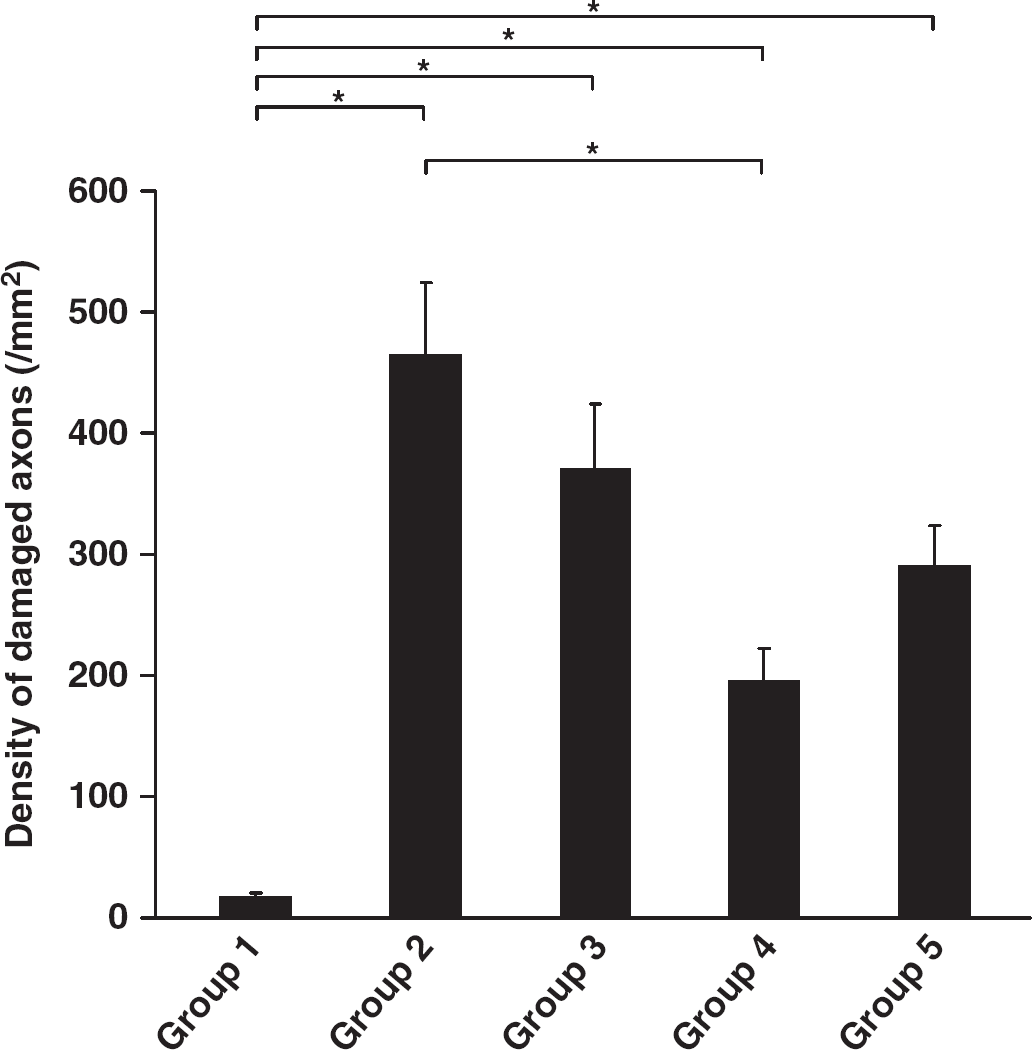
Bar graph shows a comparison of the mean density of APP-immunoreactive damaged axons in the corticospinal tract among groups. Note that hypoxia alone (group 1) exerted no significant axonal damage in comparison to the other groups. Also, note that when hypoxia occurs during hypothermia (group 4) the numbers of damaged axons were significantly decreased in relation to IAI with early hypoxia (group 2). Values represent the means ± s.e.m. Statistical differences were analyzed by the Kruskal–Wallis test followed by the Bonferroni test for multiple comparisons (*
Discussion
The results of this study show that the induction of hypoxia alone (10% oxygen for 10 mins duration) does not impair either brain vascular function or axonal structure in sham/noninjured animals. However, when hypoxia alone occurs early after TBI, it significantly alters vasoreactivity, while also resulting in a significant burden of axonal damage. Although this study included no direct assessment of the vascular reactivity or axonal burden after TBI alone, this was unnecessary in light of our previously published works. Specifically, in terms of the axonal burden of TBI followed by hypoxia, the currently reported numbers of damaged axons per unit area (500 per mm2) significantly exceeded the numbers of damaged axons (400 per mm2) seen after TBI alone after comparable posttraumatic survival periods (Marmarou et al, 2005). Similarly, the vasoreactive responses to TBI and hypoxia were markedly exacerbated in comparison to those responses seen with brain injury alone. Although our previously published work consistently showed that TBI alone significantly impaired vascular reactivity to ACh over a prolonged posttraumatic period (Suehiro et al, 2003; Wei et al, 1980), this study revealed that the addition of posttraumatic hypoxia, particularly early hypoxia, caused a virtual extinction of any vasoreactive response, further showing the adverse consequences of post-TBI hypoxia. Additional evidence of the adverse consequences of posttraumatic hypoxia is also found in our excluded animal population in which the addition of early post-TBI hypoxia generated brain herniation through the cranial window in response to sustained vasodilation and vascular engorgement. Of further note is the fact that when the same hypoxic insult was also imposed on brain-injured animals before or after posttraumatic hypothermic intervention, brain arteriolar function was significantly altered in terms of its reactivity to different concentrations of ACh, while there was also an increase in the numbers of damaged axons. In this paradigm, the hypoxic insult was induced before or after the brain temperature either had recovered from or had reached hypothermic levels, showing that under normothermic conditions, hypoxia exerts a damaging influence, at least, in terms of the cerebrovascular reactivity and the burden of axonal damage.
The results of this study also indicate that the protective effects of hypothermia are closely correlated with the timing of hypoxic insult in relation to the hypothermic intervention. As noted above, the hypoxic insult in groups 3 and 5 was initiated at 30 mins or 4 h after injury, respectively, showing that in these groups the posttraumatic hypothermic intervention failed to provide any protective effect for cerebral vascular reactivity as well as any attenuation of axonal damage. In contrast, however, in group 4, when the hypoxic insult was initiated at approximately 75 mins after injury, during the period of hypothermic intervention, the vascular reactivity was now preserved as was the attenuation of the hypothermic-induced axonal protection. Taken together, these findings confirm the damaging effects of posttraumatic hypoxia, while showing that hypothermia protects against hypoxic damage as long as the hypoxic insult occurs during hypothermic intervention. In that hypoxic insults are common events in the clinical course of many patients with TBI, these findings possibly suggest the more prolonged use of hypothermic intervention to provide more complete protection. In humans, this premise is partially supported by the clinical studies of Jiang et al (2006) who have showed the benefits of more prolonged (4 to 5 days) hypothermic intervention, a strategy whose benefits may, in part, be related to the attenuation of the damaging consequences of secondary insults described above.
In previous studies, we have shown that TBI alters brain vascular function to different known vasodilators evaluated by the same cranial window technique, detailed in this investigation (Kontos and Wei, 1992; Povlishock et al, 1980). Further, we also observed that 1 h delayed mild hypothermia followed by slow rewarming after injury also partially protects vascular function (Suehiro et al, 2003). Additional evidence from these previous experiments also suggests that the generation of free radicals after TBI can adversely influence cerebrovascular function (Kontos and Wei, 1992). Similar findings were also reported by Globus et al, 1995 who noted that oxygen radicals as well as glutamate are elevated after injury in the pericontusional domains and that these elevated oxygen radical and glutamate levels can be attenuated through the use of posttraumatic hypothermic intervention.
In this study, our data suggest that brain arterioles are vulnerable after the primary injury and that they are further compromised by the hypoxic insult. This is consistent with published data showing that hypoxic insults worsen outcome in animal models as well as in patients with TBI (Reed and Welsh, 2002; Signorini et al, 1999).
To date, many have showed that hypoxic insult alters the outcome of TBI. Ishige
Although it is well recognized that patients with TBI must be zealously protected from hypoxic insult, it is also appropriate to explore various therapeutic approaches to attenuate the damaging consequences of hypoxia. Among those therapies targeting TBI, hypothermia has been shown to provide multiple neuroprotective benefits in both animals and humans (Dietrich, 1992; Globus et al, 1995; Jiang et al, 2004; Koizumi and Povlishock, 1998; Peterson et al, 2008; Schneider et al, 2008). How hypothermia exerts its protective effects in terms of hypoxia is unknown. Matsushita et al (2001) noted that a 4 h of hypothermic intervention followed by slow rewarming after injury complicated by a 30 mins of hypoxic insult is beneficial, reducing contusion volume in comparison to normothermic or rapidly rewarmed animals. In contrast, Robertson et al (2000) examining the long-term effects of hypothermia after severe TBI after hypoxic insult suggested that the 4 h of 32°C hypothermia provides no long-term protection in terms of either motor or cognitive performance as well as histologic preservation.
The reasons for these discrepant results are not entirely clear; however, they most likely reflect differences in animal modeling, the severity of the traumatic insult, differences in the rate of posthypothermic rewarming, and/or the end points evaluated, each of which require more rigorous assessment in the context of the hypoxic insult used. In this regard, it is of note in this investigation that we relied exclusively on target rectal temperature rather than temporalis muscle temperature that best approximates brain temperature (Jiang et al, 1991). This was carried out because of the extensive hardware requirements of the cranial window that created a relatively large and bulky field, precluding the easy placement of the temporalis thermistor.
Although we have showed the protective effects of hypothermia on hypoxic insults after TBI, a key question remains as to what mechanism(s) are linked to the hypothermic attenuation of the hypoxic insult. As noted previously, oxygen free radicals have been implicated in the process of traumatically induced brain vascular malfunction (Wei et al, 1980, 1981), as well as other damaging CNS sequelae (Globus et al, 1995), with the suggestion that hypothermia attenuates free-radical-related processes. However, it remains to be determined if hypoxia itself can further adversely modulate posttraumatic radical production, which then can be attenuated through the use of hypothermia.
Conclusions
The results of this study show that a hypoxic insult after TBI can exacerbate brain vascular function and axonal injury, even when the hypoxic insult occurs either before or after the use of hypothermic intervention. Importantly, however, data from this study also reveals the protective effects of hypothermic intervention in terms of blunting the damaging consequences of a hypoxic insult when it occurs during the course of hypothermic intervention. These results indirectly support the potential benefits of more prolonged hypothermic intervention in patients with TBI, who most likely sustain multiple, hypoxic insults after injury, with the caveat that these patients may remain at risk to hypoxic insult after the cessation of hypothermic treatment.
Footnotes
The authors declare no conflict of interest.