
Abstract
Estrogens have been suggested for the treatment of neurodegenerative disorders, including stroke, because of their neuroprotective activities against various neurotoxic stimuli such as glutamate, glucose deprivation, iron, or β-amyloid. Here, the authors report that 17β-estradiol (0.3 to 30 mg/kg) and 2-OH-estradiol (0.003 to 30 mg/kg) reduced brain tissue damage after permanent occlusion of the middle cerebral artery in male NMRI mice. In vitro, 17β-estradiol (1 to 10 μmol/L) and 2-OH-estradiol (0.01 to 1 μmol/L) reduced the percentage of damaged chick embryonic neurons treated with FeSO4. In these primary neurons exposed to FeSO4, the authors also found reactive oxygen species to be diminished after treatment with 17β-estradiol (1 to 10 μmol/L) or 2-OH-estradiol (0.01 to 10 μmol/L), suggesting a strong antioxidant activity of the estrogens that were used. Neither the neuroprotective effect nor the free radical scavenging properties of the estrogens were influenced by the estrogen receptor antagonist tamoxifen. The authors conclude that estrogens protect neurons against damage by radical scavenging rather than through estrogen receptor activation.
Keywords
Oxidative damage and free radical-mediated cell death have been linked to neurodegenerative disorders such as Parkinson's disease (Jenner, 1996) and Alzheimer's disease (Mattson et al., 1994; Munch et al., 1998). Because antioxidants protect brain tissue from ischemic damage, reactive oxygen species (ROS) have been suggested to play a major role in the damaging cascade that leads to neuronal death induced by cerebral ischemia (McCord, 1985; Müller et al., 1995; Wolz et al., 1996). After ischemia, the generation of ROS could be induced by several mechanisms, including the excessive release of excitatory amino acids (Choi, 1988, 1994; Lafon-Cazal et al., 1993; Mattson, 1995), disturbed mitochondrial function (Keller et al., 1998), or the delocalization of intracellular iron (Oubidar et al., 1996). Neuroprotection by antioxidants is, therefore, a promising therapeutic strategy for the treatment of chronic neurodegenerative diseases and stroke (Hall, 1993; Wolz et al., 1996).
Estrogens show antioxidative activities because of the phenolic structure in the A ring of these steroids (Behl et al., 1997; Subbiah et al., 1993). Increasing data support the view that estrogens may be useful for the therapy of disorders that are linked to ROS-mediated cellular damage, including cardiovascular diseases (Keany et al., 1994; Schwartz et al., 1995) and Alzheimer's disease (Henderson et al., 1994; Tang et al., 1996). Estrogens could be useful in preventing or delaying the onset of Alzheimer's disease in postmenopausal women (Henderson et al., 1994; Paganini-Hill and Henderson, 1996; Simpkins et al., 1997). 17β-Estradiol was found to reduce low-density lipoprotein oxidation, suggesting a mechanism against atherosclerosis by preserving endothelium-dependent vascular relaxation (Keaney et al., 1994). Increasing evidence also suggests that estrogens improve the outcome after cerebral ischemia in rodents (Alkayed et al., 1998; Dubal et al., 1998; Pelligrino et al., 1998; Toung et al., 1998). These studies mainly investigated effects of the estrogen 17β-estradiol at physiologic doses on the cerebral infarction after a prolonged pretreatment.
In vitro, estrogens have been shown to protect neurons against insults induced by glutamate, glucose deprivation, iron, and β-amyloid (Behl et al., 1995; Goodman et al., 1996). Estrogens might affect neuronal damage by several mechanisms, such as the enhanced synthesis of neurotrophic factors (Singh et al., 1995), inhibition of N-methyl-d-aspartate receptors (Weaver et al., 1997), or through anti oxidative activities (Behl et al., 1995, 1997; Goodman et al., 1996). In most of the in vitro studies, micromolar concentrations of estrogens were used to demonstrate their neuroprotective effect against oxidative stress. Thus, it remained to be clarified whether estrogen derivatives also protect neurons against oxidative damage at nanomolar concentrations in vitro and then through estrogen receptor stimulation, or rather as a result of their antioxidative properties, as suggested by Behl et al. (1997). In the current study, we measured the effects of the 3-OH steroids 17β-estradiol and 2-OH-estradiol on the intracellular ROS level and the oxidative damage in primary chick neurons after incubation with FeSO4. Moreover, we evaluated whether the neuroprotective effects of these estrogens in vitro also could be verified in a mouse model of permanent focal cerebral ischemia through acute administration of low pharmacologic doses of estrogen derivatives.
MATERIALS AND METHODS
Animals
Male NMRI mice (Charles River, Sulzfeld, Germany) were used for ischemia experiments. The animals were maintained under controlled light and environmental conditions (12:12 hour dark-light cycle, 23° ± 1°C, 55% relative humidity) and had free access to food (Altromin, Lage, Germany) and water.
Permanent focal cerebral ischemia in mice
Permanent middle cerebral artery (MCA) occlusion was performed in male NMRI mice (12 to 17 animals per group) according to the method described by Welsh et al. (1987). Briefly, after the mice were anesthetized with tribromoethanol (600 mg/kg intraperitoneally), a small hole was drilled in the skull to expose the middle cerebral artery. The stem of the middle cerebral artery and both branches were permanently occluded by electrocoagulation. Body temperature was maintained at 37° ± 1°C with a heating lamp during the surgical procedure. Afterward, the mice were kept at an environmental temperature of 30°C for 2 hours after middle cerebral artery occlusion. For histologic evaluation, the mice were anesthetized again with tribromoethanol and perfused intraperitoneally with a 1.5% solution of neutral red (0.5 mL) 2 days after middle cerebral artery occlusion. The brains were removed and stored in a fixative (4% formalin in phosphate buffer solution, pH 7.4) for 24 hours. As described previously, in this model of focal cerebral ischemia in mice, only cortical tissue was found to be infarcted, and furthermore, the infarct volume correlated with the infarct surface (Backhauβ et al., 1992; Culmsee et al., 1998). The tissue on the brain surface unstained by neutral red was determined as infarcted surface area (in square millimeters) by means of an image analyzing system (Kontron, Eching, Germany) according to Backhauss et al. (1992). Mice received 17β-estradiol (0.03 to 30 mg/kg) and 2-OH-estradiol (0.0003 to 30 mg/kg) subcutaneously 24 hours before induction of permanent focal ischemia. A second dose of the estrogens was administered intraperitoneally 3 hours before MCA occlusion. Control animals received vehicle (5% dimethylsulfoxide [DMSO] in castor oil) only.
Cell culture agents and other substances
Dulbecco's modified Eagle medium (DMEM), fetal bovine serum, and penicillin-streptomycin solution were obtained from Gibco (Eggenstein, Germany). Poly-l-lysine hydrobromide (molecular weight 70,000 to 100,000), FeSO4, 17β-estradiol, 2-OH-estradiol, DMSO, and castor oil were purchased from Sigma (Deisenhofen, Germany). The estrogens were dissolved in pure DMSO and then diluted in the cell culture medium to a final DMSO concentration of 1%.
Primary neuronal cultures and drug treatment
Primary neuronal cultures were prepared from 7-day-old chick embryo telenccphalons as described previously (Pettmann et al., 1979). The cerebral hemispheres were mechanically dissociated through nylon meshes with 48-μm mesh width. The homogenized cell suspension was plated into poly-l-lysine-coated dishes containing 15-mm glass coverslips or culture flasks (25 mm2) with a seeding density of 4 × 104 cells/cm2. The cells were cultured in DMEM supplemented with 20% fetal calf serum at 37°C, 5% CO2, and 95% relative humidity. Culture medium was replaced every 2 days, and the neuronal cells were used for experiments on day 5 after seeding. The percentage of neurons in the cultures was above 98%, as determined by an immunohistochemical demonstration of tetanus toxin binding sites (Pettmann et al., 1979).
To cause neuronal injury, the cells were incubated with 100 μmol/L FeSO4 for 24 hours in serum-free DMEM. Afterward, the percentage of damaged neurons was calculated by the trypan blue exclusion method.
Determination of mitochondrial ROS production
Mitochondrial ROS production was measured as described before (Ahlemeyer et al., 1998; Ravati et al., 1999). Briefly, ROS production was monitored 3 hours after incubation with 100 μmol/L FeSO4 using the lipophilic non fluorescent dye dihydrorhodamine 123, which accumulated in mitochondria and was oxidized by ROS to the positively charged fluorescent rhodamine 123 (Dugan et al., 1995). To record fluorescence, cells were stained with 5 μmol/L dihydrorhodamine 123 for 15 minutes and then washed three times with phosphate-buffered saline. Digital video imaging of rhodamine 123 fluorescence was conducted using a fluorescence microscope (Axiovert 100 inverted-stage microscope, Zeiss, Germany) with attenuated ultraviolet illumination from a 75-W xenon lamp. An electronic shutter, which opened during image acquisition, minimized photobleaching and phototoxicity. Fluorescence intensity was measured at an excitation wavelength of 490 nm and an emission wavelength of 510 nm. The values of the fluorescence intensity were determined in an arbitrary scale as fluorescence units. Images were collected by the use of a CCD-camera (C 2400-87, Hamamatsu, Germany) and were digitalized to 256 × 256 pixels. Before measurement of the fluorescent values, the background was determined to be subtracted from the images. Data were analyzed using Argus 50 software (Hamamatsu, Germany).
Statistics
All values were calculated as means ± SD. One-way analysis of variance combined with Scheffé's test or Duncan's analysis were used for in vitro and in vivo data, respectively.
RESULTS
Effect of 17β-estradiol and 2-OH-estradiol on Fe2+-induced neuronal damage
Neuronal damage was induced by an incubation with 100 μmol/L FeSO4 for 24 hours to a level of 42.8 ± 4.0% trypan blue-stained neurons compared with 13.3 ± 3.9% in sister control cultures. Neurotoxicity induced by Fe2+ was significantly attenuated by 17β-estradiol (1 to 10 μmol/L) (Fig. 1A). In a second series of experiments, concentrations of 0.01 to 1 μmol/L 2-OH-estradiol reduced the percentage of damaged neurons from 43% in Fe2+ -treated cultures to a maximum of 22%. In contrast, concentrations of 100 μmol/L 17β-estradiol showed no neuroprotective effect, and 10 μmol/L 2-OH-estradiol markedly enhanced the Fe2+ -induced neuronal damage (Fig. 1). Tamoxifen (1 μmol/L) added simultaneously with the estrogens did not block the effects of 17β-estradiol and 2-OH-estradiol against neuronal damage (Fig. 1). Tamoxifen (1 μmol/L), 17β-estradiol (0.001 to 100 μmol/L), and 2-OH-estradiol (0.001 to 1 μmol/L) alone did not influence neuronal viability, whereas 2-OH-estradiol concentrations of 10 μmol/L and higher markedly enhanced the percentage of trypan blue-stained neurons.
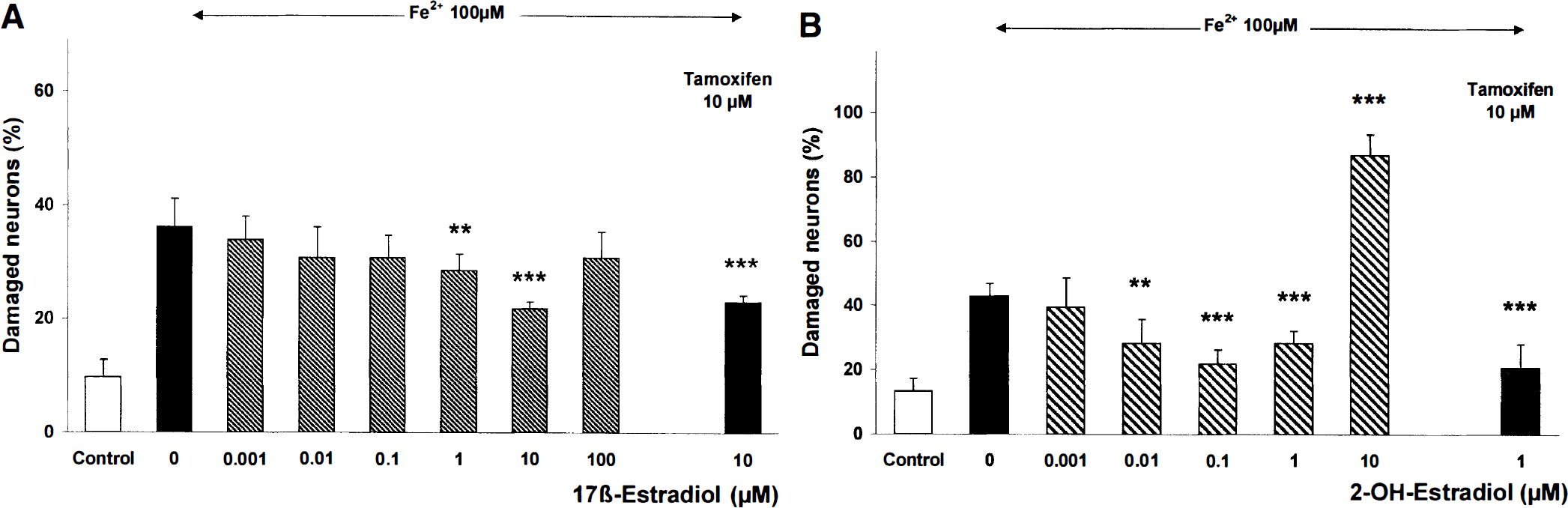
Effect of the estrogens 17β-estradiol and 2-OH-estradiol on Fe2+-induced neuronal damage. Chick embryonic neurons were treated with 100 μmol/L FeSO4. Cellular viability was determined 24 hours after Fe2+ treatment by trypan blue exclusion method. 171-Estradiol
Radical scavenging properties of the estrogens 17β-estradiol and 2-OH-estradiol
To quantify the production of ROS, a fluorescent microscope combined with a digital video imaging system was used, which allowed a measurement within single neurons. After exposing the cultures to 100 μmol/L Fe2+ for 3 hours, there was an increase in fluorescence from 6 fluorescence units in controls to 47 fluorescence units (Fig. 2). Both 17β-estradiol and 2-OH-estradiol decreased the Fe2+ -induced production of intracellular ROS fluorescence in a concentration-dependent manner. The lowest concentration of 17β-estradiol required to significantly diminish ROS fluorescence was 1 μmol/L (Figs. 2 and 3A). A significant reduction of ROS fluorescence by 2-OH-estradiol was achieved already at 0.01 μmol/L (Figs. 2 and 3B). Tamoxifen (1μmol/L) did not block the effect of the estrogens on the production of ROS after Fe2+ treatment. Tamoxifen (1 μmol/L), 17 β-estradiol (0.001 to 10 μmol/L), or 2-OH-estradiol (0.001 to 10 μmol/L) alone did not affect ROS fluorescence compared with controls (data not shown).
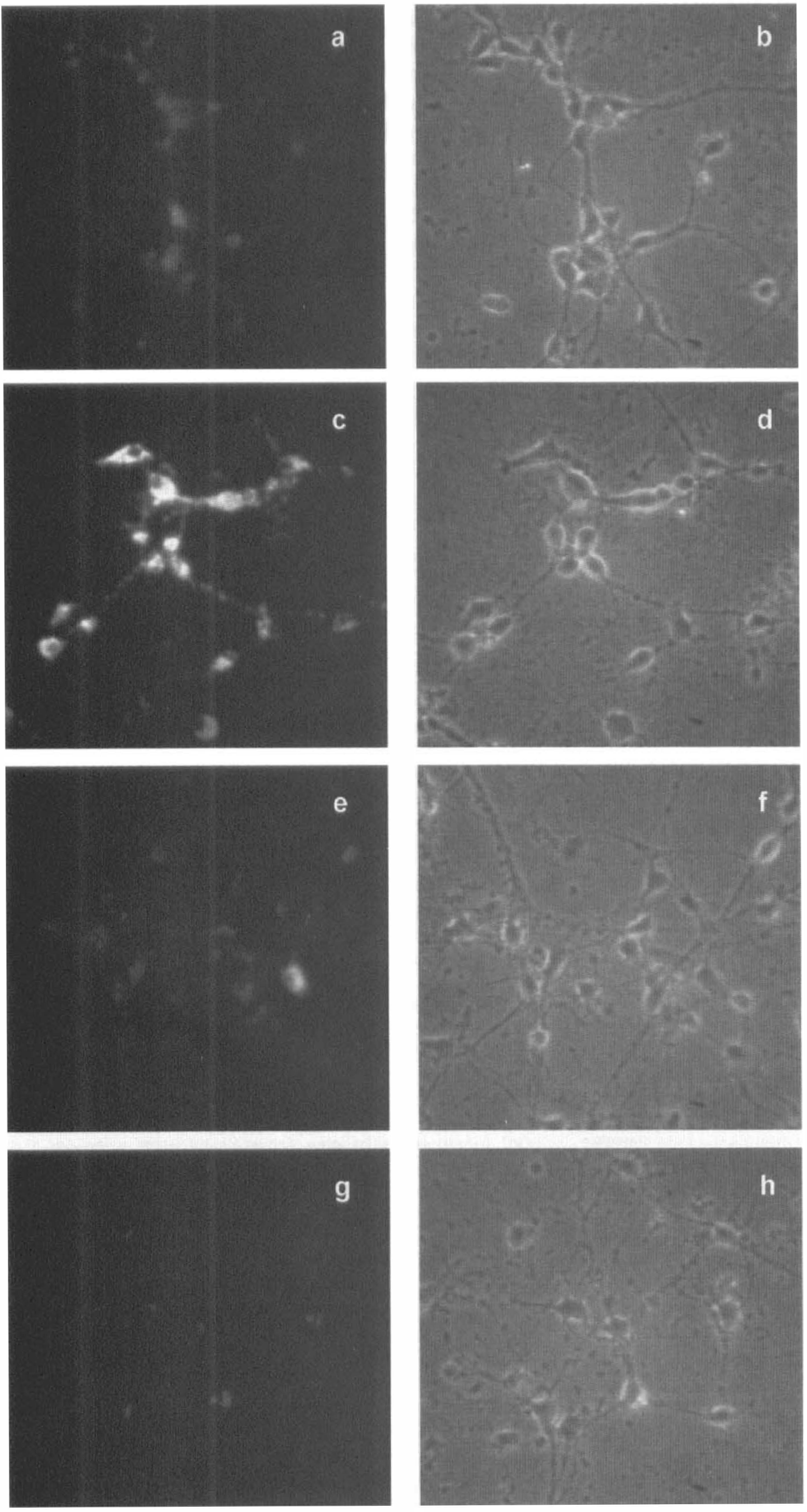
Effect of the estrogens 17β-estradiol and 2-OH-estradiol on Fe2+ - induced production of reactive oxygen species. Five days after seeding, primary neuronal cultures were treated with 100 μmol/L FeSO4 for 3 hours. 171-Estradiol and 2-OH-estradiol were added simultaneously with Fe2+. The cultures were incubated with the dye dihydrorhodamine 123 (5 μmol/L, 15 minutes) to determine changes in the fluorescence caused by mitochondrial production of reactive oxygen species. Representative photomicrographs show rhodamine 123 fluorescence of controls
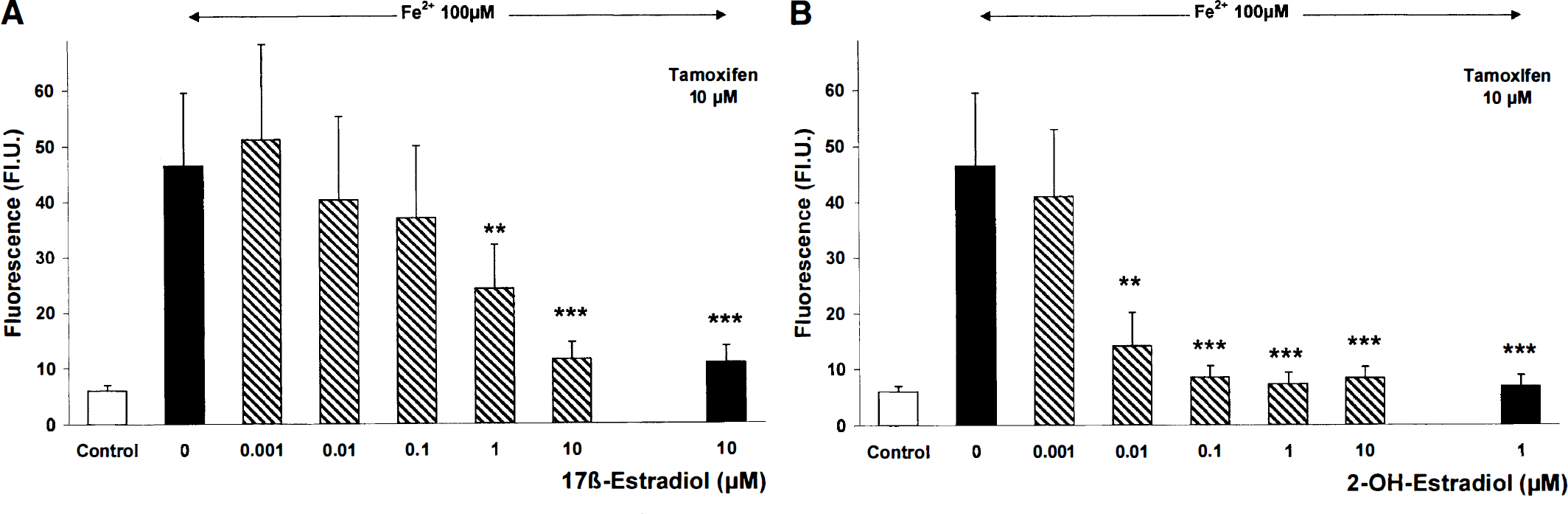
Effect of 17β-estradiol and 2-OH-estradiol on Fe2+-induced production of reactive oxygen species. Chick embryonic neurons were treated with Fe2+ (100 μmol/L, 3 hours). 17β-Estradiol
Focal cerebral ischemia in mice
Four series of ischemia experiments were performed in mice to test the protective capacities of 17 β-estradiol and 2-OH-estradiol against brain damage caused by permanent MCA occlusion. Both steroids investigated were able to reduce the ischemia-induced brain damage in the model of focal cerebral ischemia in mice. 17β-Estradiol (0.3 to 3 mg/kg) significantly reduced the infarct area on the mouse brain surface (Table 1). Doses of 17β-estradiol lower than 0.3 mg/kg showed no effect. 2-OH-Estradiol reduced the infarct area at doses of 0.03 to 30 mg/kg (Table 1). The lowest effective dose of 2-OH-estradiol was determined at 0.03 mg/kg, although in one series of experiments, this steroid protected brain tissue from ischemic damage already at 0.003 mg/kg (data not shown). In the four series of ischemia experiments performed in the current study, 8 of 55 vehicle-treated mice died after MCA occlusion. In contrast, only 4 of 57 mice and 4 of 122 animals died in the groups treated with 17β-estradiol and 2-OH-estradiol, respectively.
17β-estradiol and 2-OH-estradiol reduce the infarct size after middle cerebral artery occlusion in mice
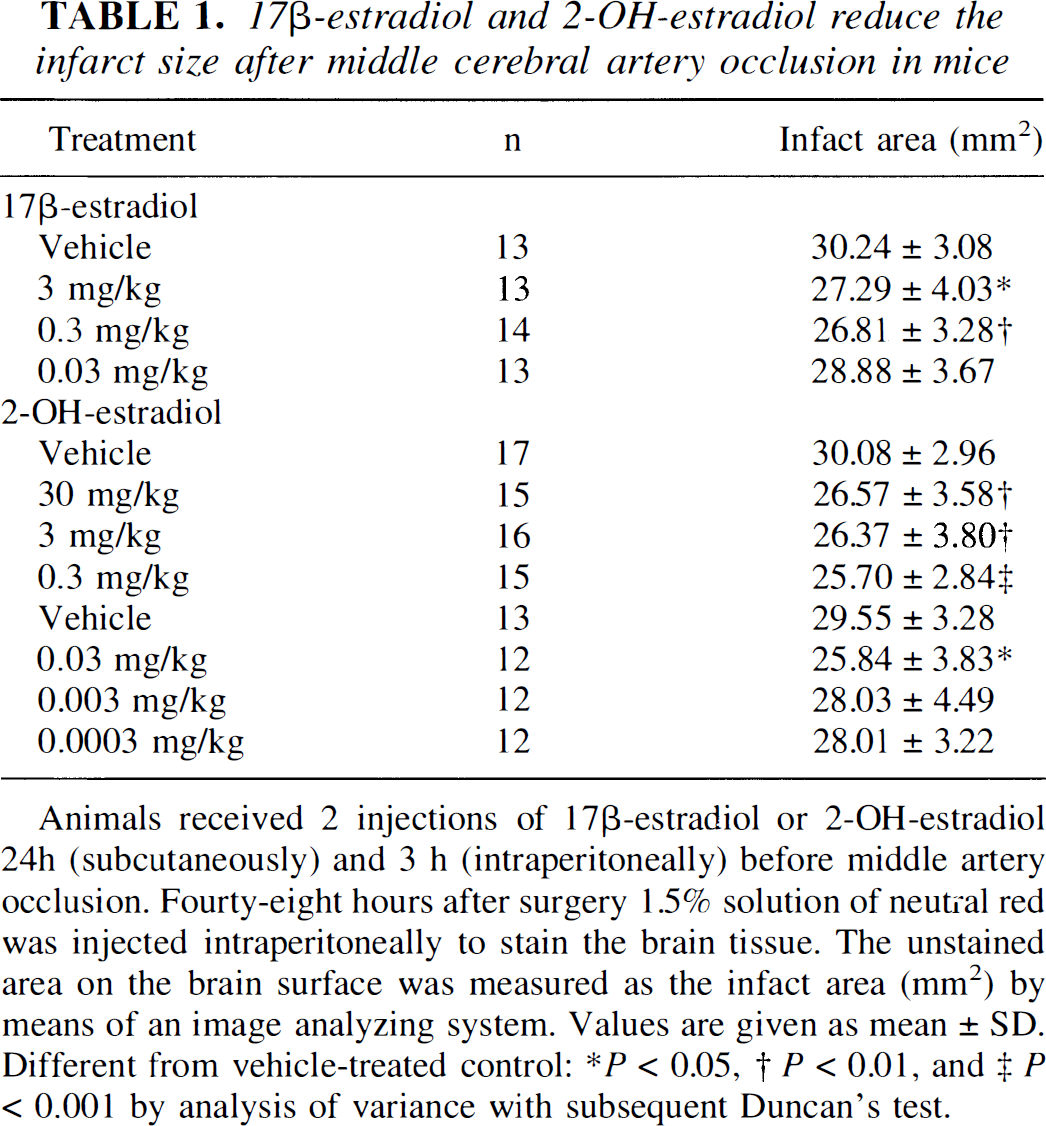
Animals received 2 injections of 17β-estradiol or 2-OH-estradiol 24h (subcutaneously) and 3 h (intraperitoneally) before middle artery occlusion. Fourty-eight hours after surgery 1.5% solution of neutral red was injected intraperitoneally to stain the brain tissue. The unstained area on the brain surface was measured as the infarct area (mm2) by means of an image analyzing system. Values are given as mean ± SD. Different from vehicle-treated control:
P < 0.05
P < 0.01
P < 0.001 by analysis of variance with subsequent Duncan's test.
DISCUSSION
In the current study, we demonstrated a protective activity of the estrogens 17β-estradiol and 2-OH-estradiol against iron-induced neuronal cell death in primary cultures of chick embryonic neurons. In addition, we showed that the suppression of mitochondrial ROS production and the neuroprotective effect of these estrogens were mediated receptor-independently through their antioxidative activity. These data are consistent with previous studies showing that, because of their antioxidative capacities, estrogens suppress iron-induced lipid peroxidation of liver microsomes (Ruiz-Larrea et al., 1994), reduce oxidative damage in rat cortical synaptosomes (Keller et al., 1997), and preserve vascular relaxation resulting from reduced oxidation of low-density lipoprotein (Keaney et al., 1994). In most of the previous studies, micromolar concentrations of the estrogens were used to demonstrate neuroprotection against trophic factor withdrawal and cytotoxic stimuli, including β-amyloid, iron, glutamate, or hydrogen peroxide (Behl et al., 1997; Goodman et al., 1996; Mattson et al., 1997). The concentrations used in earlier investigations were thus always higher than physiologic, nanomolar blood levels. From these in vitro experiments, the capacity of estrogens to scavenge cytotoxic free radicals at high concentrations was suggested as the underlying mechanism of neuroprotection. By measuring neuronal damage and production of ROS after exposure to iron, we demonstrate that 2-OH-estradiol is capable of protecting neurons from oxidative stress already at nanomolar concentrations (10 nmol/L). In contrast, 17β-estradiol showed antioxidative effects only if added to the cell culture medium at micromolar concentrations (1 to 10 μmol/L), which is in line with previous reports. The concentrations of both estrogens required for protection against iron-induced oxidative stress in vitro were in the order of magnitude expected to activate their receptors (10 to 1000 nmol/L). However, the estrogen receptor antagonist tamoxifen influenced neither the neuroprotective effect nor the antioxidative capacity of the estrogens, indicating that the effects were mediated by the inherent antioxidant properties of the steroids investigated. This finding confirms the results of a structure-activity study showing that the hydroxyl group at the C3 position of the steroid A ring confers the neuroprotective antioxidant activity of estrogens (Behl et al., 1997).
Although the precise mechanism of action has yet to be defined, tamoxifen is widely used to inhibit estradiol binding to the estrogen receptor. Under our experimental conditions, tamoxifen alone or in combination with estrogens did not affect cell survival or the cellular concentration of free radicals. Evidence shows that neuroprotection by estrogens does not necessarily depend on activation of estrogen receptors because higher concentrations than expected for estrogen receptor stimulation were needed to obtain neuroprotective effects in previous studies (Behl et al., 1995, 1997; Goodman et al., 1996). In a neuroblastoma cell line, 17α-estradiol, which does not bind to the estrogen receptor, prevented cell death induced by serum deprivation similar to 17β-estradiol (Green et al., 1997). Furthermore, 17β-estradiol prevented iron-induced membrane oxidation independent of estrogen receptor-mediated transcriptional activation in a synaptosome preparation (Keller et al., 1997). On the other hand, these authors did not exclude the stimulation of membrane receptors by 17β-estradiol at nanomolar concentrations as a possible mechanism of protection. Furthermore, estrogen receptor-mediated neuroprotection was suggested to include upregulation of neurotrophins such as nerve growth factor (Singh et al., 1995) and the high-affinity tyrosine kinase receptor (Gibbs, 1998). 17β-Estradiol protected human neuroblastoma cells against β-amyloid toxicity already at nanomolar concentrations, ameliorating both lipid peroxidation and persistent lactate production (Green et al., 1996; Gridley et al., 1997). Although neuronal death may be prevented by estrogens through receptor-dependent and receptor-independent mechanisms, we suggest from our data that estrogens reduce oxidative stress independent from binding to their receptors.
In addition, we were able to demonstrate that 17β-estradiol and 2-OH-estradiol protected male mice from ischemic brain damage at doses as low as 0.3 mg/kg and 0.03 mg/kg, respectively. Our finding indicates that the neuroprotective effects observed in cultured neurons also were relevant for estrogen effects obtained in vivo. Strong evidence demonstrates that scavenging free radicals exerts protection of brain tissue against damage caused by MCA occlusion (Müller et al., 1994; Wolz and Krieglstein, 1996). Furthermore, recently published data show that gender differences in response to focal cerebral ischemia in rats result from the different estrogen levels in female versus male animals (Alkayed et al., 1998; Zhang et al., 1998). In these studies, female rats showed smaller infarct sizes and higher CBF than male animals after transient MCA occlusion. Moreover, the loss of female sex steroids in ovarectomized female rats was demonstrated to increase the infarct volume compared with control female animals (Hum et al., 1998). Chronically administered 17β-estradiol abolished the increased susceptibility to ischemic brain damage in ovarectomized rats (Zhang et al., 1998). In the current study, we demonstrated that 17β-estradiol protected brain tissue from ischemic damage in male mice when administration was started 24 hours before ischemia. Moreover, we demonstrated that 2-OH-estradiol reduced the ischemic brain damage after MCA occlusion already at doses 10 times lower than the 17β-estradiol doses needed for protection. A similar finding was observed in vitro as far as 2-OH-estradiol reduced intracellular ROS concentration and neuronal cell death at concentrations 100 times lower than that of 17β-estradiol. Our results confirm the view that the structure-related neuroprotective effect of estrogens against oxidative stress in vitro (Behl et al., 1997) also helps to predict cerebroprotective activities in vivo. Under our experimental conditions, pharmacologic doses of the estrogens were necessary to achieve protection against ischemic brain damage. This finding suggests that, also in vivo, the antioxidative properties of the estrogens led to neuroprotection rather than a receptor-mediated mechanism.
In conclusion, we demonstrated that neuroprotection against oxidative damage by estrogens was most likely caused by the antioxidant properties of these steroids. The reduced amount of reactive oxygen species in vitro occurred at concentrations that were relevant for the cerebroprotective activities of the estrogens observed in the mouse model of focal cerebral ischemia. Thus, estrogens, and especially the catechol estrogen 2-OH-estradiol, may prove efficacious as neuroprotectants in the acute therapy of ischemic stroke.