
Abstract
It has been suggested that reactive oxygen species (ROS) play a role in the pathophysiology of brain damage. A number of therapeutic approaches, based on scavenging these radicals, have been attempted both in experimental models and in the clinical setting. In an experimental rat and mouse model of closed-head injury (CHI), we have studied the total tissue nonenzymatic antioxidant capacity to combat ROS. A major mechanism for neutralizing ROS uses endogenous low-molecular weight antioxidants (LMWA). This review deals with the source and nature of ROS in the brain, along with the endogenous defense mechanisms that fight ROS. Special emphasis is placed on LMWA such as ascorbate, urate, tocopherol, lipoic acid, and histidine-related compounds. A novel electrochemical method, using cyclic voltammetry for the determination of total tissue LMWA, is described. The temporal changes in brain LMWA after CHI, as part of the response of the tissue to high ROS levels, and the correlation between the ability of the brain to elevate LMWA and clinical outcome are addressed. We relate to the beneficial effects observed in heat-acclimated rats and the detrimental effects of injury found in apolipoprotein E-deficient mice. Finally, we summarize the effects of cerebroprotective pharmacological agents including the iron chelator desferal, superoxide dismutase, a stable radical from the nitroxide family, and HU-211, a nonpsychotoropic cannabinoid with antioxidant properties. We conclude that ROS play a key role in the pathophysiology of brain injury, and that their neutralization by endogenous or exogenous antioxidants has a protective effect. It is suggested, therefore, that the brain responds to ROS by increasing LMWA, and that the degree of this response is correlated with clinical recovery. The greater the response, the more favorable the outcome.
The physiological response to brain injury is extremely complex and involves the activation of an overlapping network of humoral, tissue, and cellular pathways. The initiating event (whether ischemia, mechanical trauma, or infection) triggers the release of endogenous mediators. These, in turn, via successive amplification steps, initiate a cascade of molecular, cellular, and tissue responses resulting in delayed tissue edema, necrosis, and impaired function (Faden and Salzman, 1992; Phillis, 1994; Siesjo et al., 1989, Siesjo, 1993, Povlishock and Christman, 1995; Gennarelli, 1997). For nearly two decades, reactive oxygen species (ROS) have been the focus of interest as possible candidates for the elicitation of various pathological responses in the pathogenesis of ischemia and trauma (Demopoulos et al., 1980; Kontos, 1989; Siesjo et al., 1989; Chan, 1989; Hall and Braughler, 1989; Yukido and Long, 1990; Hall et al., 1992; Helfaer et al., 1994). Studies on mechanisms underlying selective neuronal vulnerability implicate the neurotoxic action of glutamate through activation of the NMDA receptor, and blockade of this receptor is associated with neuroprotection. It has been suggested that the initial events in blunt head trauma lead to a final common pathway of neuronal death involving loss of cellular calcium homeostasis, production of free radicals, and tissue acidosis (for review, see Siesjo, 1993). A number of therapeutic approaches, based on intervention by scavenging ROS, have been attempted both in experimental models (for review see McIntosh, 1993, Hensley et al., 1997) and in the clinical setting (for review see Wahlgren, 1997). In addition, it was postulated that because of the synergism between excitotoxicity and pro-oxidant events, therapeutic modalities based on combined mechanisms of action (e.g., glutamate antagonists and antioxidants) should prove more effective than monotherapy (Kempski, 1994).
The brain is the tissue most vulnerable to oxidative damage because of its high rate of oxidative metabolic activity, intensive production of reactive oxygen metabolites, relatively low antioxidant capacity, low repair mechanism activity, nonreplicating nature of its neuronal cells, and the high membrane surface to cytoplasm ratio (Evans, 1993; Reiter, 1995). The high concentrations of polyunsaturated fatty acids in the membrane lipids of the brain are the source for the decomposition reactions termed “lipid peroxidation,” in which a single initiating free radical can precipitate the destruction of adjacent molecules (Watson, 1993). The brain also contains high levels of transition metals, such as iron, which are located in specific sites in the brain (e.g., substantia nigra). These redox-active metals are capable of catalyzing the production of highly toxic radicals via the metal-mediated Haber-Weiss reaction (Halliwell and Gutteridge, 1989). The presence of ascorbic acid in close proximity to the binding sites of the ferric ions may result in the pro-oxidant rather than in the antioxidant activity of ascorbic acid (Halliwell, 1996). Therefore, it is postulated that highly reactive oxygen radicals are continuously being produced in the brain as the result of the interaction between various transition metals and reducing equivalents. This phenomenon is extremely important in brain injury when mobilization of redox-active metals may occur, followed by their exposure to reducing agents. The outcome of such interactions may explain some of the oxidative damage detected after traumatic or ischemic injury to the brain.
In addition, several cell regulatory mechanisms are known or considered to be affected by oxidative stress (Hensley et al., 1997). Cytokine or growth factor-mediated signal transduction pathways that are activated by brain trauma include tyrosine phosphorylation and activation of the resident cytosolic protein complex NF-kB. Proteolytic cleavage and removal of the inhibitory peptide ikB results in translocation of the active species to the nucleus, its binding to kB-specific DNA sequences, and activation of gene transcription. Recently, Lander et al. (1995) suggested that a main oxidatively sensitive element in the NF-kB triggering cascade is a redox-modulated G protein encoded by the protooncogene p21ras. AP-1 is another transcription factor complex, composed of c-fos and c-jun heterodimers, whose mRNA are induced by hydrogen peroxide (H2O2) (Amstad et al., 1992), and it is implicated in programmed cell death. Oxidative damage to calmodulin-regulated enzymes, resulting in dysregulation of cellular homeostasis and intracellular transduction cascades, was reported by Yao et al. (1996). Thus, ROS have been shown to affect not only “classical” targets, such as lipids and proteins, but also gene transcriptional processes associated with pathological events. Therefore, antioxidants may help to preserve not only membranes and enzymes, but also to suppress destructive oxidatively-sensitive gene regulatory processes.
The present review addresses the following issues:
the source and nature of ROS in the brain;
the endogenous defense mechanisms that fight ROS, with special emphasis on low-molecular weight antioxidants (LMWA);
a novel method, based on cyclic-voltammetry, for determining total tissue reducing antioxidants;
a model of closed-head injury (CHI), and the temporal changes in brain LMWA after CHI;
the correlation between the ability of the brain to increase its level of LMWA and the clinical outcome of CHI
elevation of endogenous LMWA after CHI in rats that have been acclimated to heat and their better recovery from injury;
the impaired ability of apolipoprotein E (apoE)-deficient mice to augment LMWA after CHI, and their more sluggish recovery from injury;
the cerebroprotective effects of radical neutralizing agents, such as an iron chelator, superoxide dismutase, nitroxides and HU-211, a novel NMDA antagonist with antioxidant properties.
BRAIN REACTIVE OXYGEN SPECIES, DEFENSE MECHANISMS, AND THE CYCLICVOLTAMMETRY TECHNIQUE
Source and nature of reactive oxygen species in the brain
Reactive oxygen species include superoxide, hydroxyl, nitric oxide, peroxyl and alkoxyl radicals, as well as nonradical oxygen metabolites such as hydrogen peroxide, lipid hydroperoxide, singlet oxygen, and hypochlorous acid. The brain, a relatively small tissue, uses 20% of the inspired oxygen (Ames, 1983). Although most of the oxygen molecules are consumed for the production of energy in the form of ATP molecules, a small proportion (about 5%) is used in alternative pathways leading to the production of ROS. In the mitochondria, oxygen molecules are used in a complex of reactions by mitochondrial cytochrome oxidase. This enzyme complex adds four electrons to the oxygen molecule and transforms it into two water molecules. The process, which occurs in stages, is involved in the production of ROS such as superoxide radicals (O2܋), hydroxyl radicals (OH˙) and hydrogen peroxide (H2O2) (Halliwell and Gutteridge, 1989). The chain transport of electrons in the mitochondria is not absolutely efficient and, therefore, leakage of ROS from the mitochondrial organelles may occur, especially when there is an excess of oxygen (Reiter, 1995). Other endogenous sources of ROS in the brain include enzymatic processes in which ROS are generated directly or indirectly (e.g., monoamine oxidase, nitric oxide synthase, and cyclooxygenase) and various cells such as activated neutrophils. Excitatory amino acids also generate high levels of ROS on their massive release after injury (Dugan and Choi, 1994). The brain is also exposed to ROS from exogenous sources such as irradiation, pollutants that can enter the brain via blood carriers, as well as xenobiotics and drugs.
Polyunsaturated fatty acids, highly abundant in the brain, serve as a major biological target for the oxidative damage induced by ROS (Rice-Evans and Burdon, 1993; Watson 1993). Lipid peroxidation causes damage to biological membranes and nerves and leads to production of secondary reactive metabolites (e.g. aldehydes). Other biological targets may be proteins, which can sustain direct damage, or chain reaction analogues of fatty acids (Stadtman, 1992). DNA also serves as a major candidate for oxidative damage (Ames et al., 1993). In individuals suffering from Parkinson's disease, oxidative DNA damage is greater within brain regions rich in dopaminergic neurons (Ames et al., 1993; Halliwell and Gutterigde, 1990; Diplock, 1994, Youdim and Riederer, 1997).
Endogenous defense mechanisms of the brain
To contend with the continuous exposure to ROS, the living cell has developed several lines of defense. These include preventive mechanisms against oxidative damage, repair mechanisms, and an antioxidant defense system. The latter assists the living cell to cope directly with ROS and to prevent oxidative damage. It would be expected that the brain, which is particularly vulnerable to oxidative damage, possesses an efficient antioxidant activity. Indeed, brain tissue contains various types of antioxidants, some of them unique to the brain. The antioxidant system can be classified into two major groups: enzymes and LMWA. The enzymes include a limited number of proteins: superoxide dismutase (SOD), catalase and peroxidase, as well as some supporting enzymes. The levels of the enzymes vary in different brain regions and among various species. Whereas the activity of SOD, which dismutes superoxide radicals to hydrogen peroxide, is quite high, the activity of catalase, acting to degrade hydrogen peroxide to water and oxygen, is relatively low (Sinet et al., 1980; Reiter 1995). Rat brain contains high levels of glutathione peroxidase, which removes hydrogen peroxide. There are also some supporting enzymes that act indirectly as antioxidants. For example, glucose-6-phosphate dehydrogenase supplies NADH molecules which, in turn, can regenerate glutathione to its active form. The role of the endogenous antioxidant enzymes in acute central nervous injury was shown by Chan et al. in genetically manipulated mice. Transgenic mice that overexpress CuZn-SOD and anti-apoptotic Bcl-2 in the brain were found to be protected in a model of brain ischemia/reperfusion injury, whereas knockout mutants, deficient in SOD, were more vulnerable to the same type of injury (Chan, 1996).
The LMWA group of molecules can be further classified into indirect-acting (e.g., chelating agents) and direct-acting antioxidants (e.g., scavengers and chain-breaking antioxidants). The latter antioxidants are extremely important in combating against oxidative stress. This subgroup contains several hundred compounds that originate in a number of sources (both endogenous and exogenous). However, only a minority of these molecules, such as glutathione, NAD(P)H and carnosine, are synthesized by the cell itself. The majority of the antioxidants, including ascorbic acid, lipoic acid, polyphenols, and carotenoids, are derived from dietary sources. Another source of LMWA is the waste products of the living cell (such as uric acid), which were also shown to possess antioxidant properties (Ames et al., 1981, 1993).
Major low-molecular weight antioxidants in the brain.
The reducing antioxidant capacity of the brain
One of the most important tools for evaluating the involvement of ROS in pathological disorders is quantification of the antioxidant activity of the tested biological tissue. Most of the methods used to evaluate such antioxidant activity determine specific scavengers, or are specific for the reactive species involved, and do not indicate the overall antioxidant status of the tissue (Rice-Evans and Miller, 1994). To overcome this difficulty, several methods designed to evaluate the overall antioxidant activity of a biological tissue were developed. However, these methods suffer from severe disadvantages (Rice-Evans and Miller, 1994). Measurement of antioxidant enzyme activity is relatively easy because of the availability of well-characterized techniques for the determination of the limited number of enzymes involved. However, evaluation of the total antioxidant activity derived from LMWA is more difficult because of the large number of scavengers and the need for advanced laboratory techniques for their quantification.
As previously described, LMWA that interact directly with ROS play a major role in defending the brain against oxidative damage and they possess several advantages over antioxidant enzymes. These molecules are relatively small and readily penetrate the cells, localizing at specific sites of oxidative stress, thereby supplying site-specific protection against site-specific oxidative damage. Low-molecular weight antioxidants display a wide spectrum of activity against a variety of ROS and they can be regenerated in the brain to their active form. A review of the literature shows that there is no comprehensive review on the role of the overall activity of the direct-acting LMWA after traumatic brain injury and in pathological disorders.
The cyclic-voltammetry technique. The cyclic-voltammetry technique has been used in chemistry to study electron transfer between molecules for a long time. Kissinger et al. (1973) suggested that this technique could be used to evaluate levels of antioxidants such as ascorbic acid, and by implantation of microelectrodes in rat brain they have showed that this methodology can be applied in vivo. Recently, we developed a new method that enables evaluation of the overall antioxidant activity derived from the LMWA in the brain as well as in other biological tissues and fluids. The new technique is based on a common chemical property of the direct-acting LMWA. Examination of the chemical nature of these compounds revealed that these agents are reducing equivalents that donate their electron(s) to the ROS. To date, the cyclic-voltammetry technique has been used in chemistry and biochemistry for measuring overall antioxidant capacity. Therefore, we hypothesized that determination of the total reducing power of a biological fluid or tissue may indicate its antioxidant status, which is derived from LMWA. We have indeed verified this assumption in many biological systems, including brain tissue (Kohen 1993; Kohen et al., 1992; 1996; 1997; Chevion et al., 1997). To measure total reducing power we used a cyclic voltammeter. The measurements allowed us to draw conclusions regarding the types of LMWA present in the brain (indicated by the oxidation potentials of the anodic waves, Fig. 1) and the total concentration of the LMWA (indicated by the anodic current, Fig. 1) without measuring the specific compounds. Although specificity is an important issue when dealing with antioxidants, the brain, as other tissues or body fluids, contains a large number of such molecules, which may act in concert and synergistically with each other. Thus, by measuring levels of individual antioxidants one may obtain only a partial picture and overlook the whole tissue-reducing power, derived from LMWA. The sensitivity limit of the cyclic-voltammetry methodology is in the range of 1 μmol/L, allowing physiological levels of antioxidants to be readily determined (ascorbate 50 μmol/L, uric acid- 300 μmol/L). Using cyclic-voltammetry methodology, we were able to evaluate both hydrophilic LMWA such as ascorbic acid, uric acid (first anodic wave), and NADH, carnosine (part of the second anodic wave) (Fig. 1), as well as lypophylic LMWA, including tocopherols and carotene (manuscript in preparation). In a complementary assay performed to confirm and quantify the cyclic voltameter measurements, we characterized specific LMWA composing the anodic waves by using an HPLC apparatus equipped with an electrochemical detector.
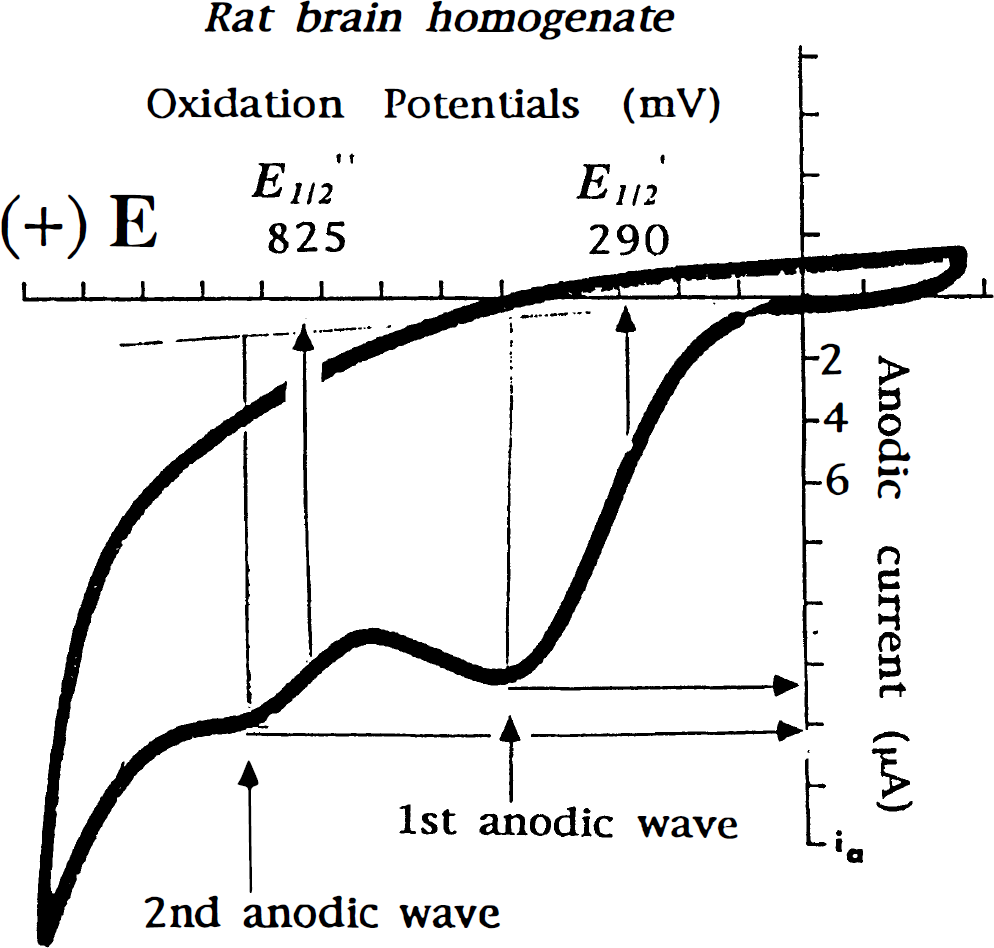
A typical tracing of brain cyclic voltameter. E1\2\ denotes the oxidation potential (mV), and the anodic current denotes the total concentration of the molecules being oxidized at the particular potential.
CLOSED-HEAD INJURY MODELS AND STUDIES ON LOW-MOLECULAR WEIGHT ANTIOXIDANTS
In the following sections we describe the results of our own studies on an experimental model of CHI developed in our laboratory for the rat (Shapira et al., 1988) and, recently, for the mouse (Chen et al., 1996). This model can reproduce the posttraumatic sequelae observed in humans and some of the data obtained in this model are essentially similar to those observed in human head injury (edema formation, BBB breakdown, motor and cognitive dysfunction, etc.). In the posttraumatic period, we also showed activation of the arachidonic acid metabolic cascade (Shohami et al., 1987, 1989) that produces free radicals, as well as enhanced cytokine production (Shohami et al., 1994a), a typical inflammatory response involving ROS. However, because of the extremely short half-lives of oxygen radicals, and methodological problems, there is no direct evidence for the production of ROS.
Reactive oxygen species in the pathophysiology of closed-head injury
Two approaches were applied in our CHI model to the study of the role of ROS in posttraumatic events: The first is based on measuring of the total reducing capacity of brain tissue, using the cyclic-voltammetry technique (as described above). Here, changes in total tissue levels of LMWA reflect the consumption and/or recruitment of these molecules as an endogenous defense mechanism for neutralization of the ROS continuously produced after injury. We used this approach to compare the changes in brain LMWA in response to CHI in two models in which basic physiological changes might affect the ability to recruit LMWA: heat acclimation of rats, and apoE deficiency in mice. In both models, CHI was induced, and the clinical outcome was compared with that of their matched controls. The second approach is based on pharmacological intervention aimed at neutralizing ROS. The beneficial effects on the outcome of CHI of (1) an iron-chelator, (2) SOD, (3) a radical-neutralizing agent from the nitroxide family, and (4) a novel NMDA antagonist (HU-211), which possesses antioxidants properties, have been described and imply that cerebroprotection is, at least in part, caused by the prevention of oxidative damage.
In cyclic-voltammetry tracings of brain extracts of water-soluble LMWA, two anodic, component-specific waves, representing two classes of reducing equivalents, were identified at a potential of 350 ± 50 and 750 ± 50 mV. The first class consisted of ascorbate and urate and other unknown scavengers, and the second probably included imidazole-containing molecules such as NADH, histidine, carnosine, and other histidine-related compounds. After CHI, the nature of the LMWA was not altered, because the two anodic potentials remained basically unchanged. However, a nonsignificant shift was observed caused by variations in the relative contributions of the individual LMWA comprising the anodic wave. However, the anodic current, Ia, which represents the concentration of LMWA at any particular potential, was markedly affected. A biphasic response to CHI was measured in the traumatized rats, with an initial decrease (∼40% at 5 minutes) followed by a transient increase (at 4 hours) and a second decrease at 24 hours (of ∼60%), followed by a return to the basal, pre-CHI level at 48 hours, which persisted up to 7 days. The significant decrease in the concentration of the reducing equivalents 5 minutes after CHI may be explained by their immediate utilization. This hypothesis is supported by the observation that ROS-neutralizing agents are cerebroprotective when administered soon after CHI (see below, and Beit-Yannai et al., 1996a). At 4 hours, the disruption of the BBB is maximal (Shapira et al., 1993), and high levels of LMWA originating in the plasma may accumulate in the damaged area. Because these compounds can then react with the ROS being produced via processes activated in the posttraumatic period (e.g., arachidonic acid metabolism, inflammation), it is likely that their levels again decrease up to 24 hours after CHI. Similar biphasic changes in brain-reducing capacity levels were also found in the right (contralateral) hemisphere, suggesting that the trauma exerted a global effect on the reducing capacity of the brain.
The correlation between the ability of the brain to increase low-molecular weight antioxidants and the clinical outcome of closed-head injury
Effect of heat acclimation on the endogenous low-molecular weight antioxidants profile.
When CHI was induced in heat-acclimated rats, they showed faster and better recovery of motor functions, as compared with that of normothermic rats subjected to a similar severity of injury (Shohami et al., 1994b). In addition, edema and BBB disruption were also significantly lower in the heat-acclimated rats (Fig. 2). Hadas et al. reported a reduction in the production of prostagladins by acclimated rats (1989). Because the arachidonic acid pathway is activated by CHI and is known to produce free radicals, we speculated that attenuated production of ROS is part of the benefits gained by heat acclimation and results in a more favorable clinical outcome after CHI. Therefore we examined the total tissue-reducing equivalents in the brains of acclimated rats, in an attempt to correlate the observed protection with the ability of the brain to neutralize ROS.
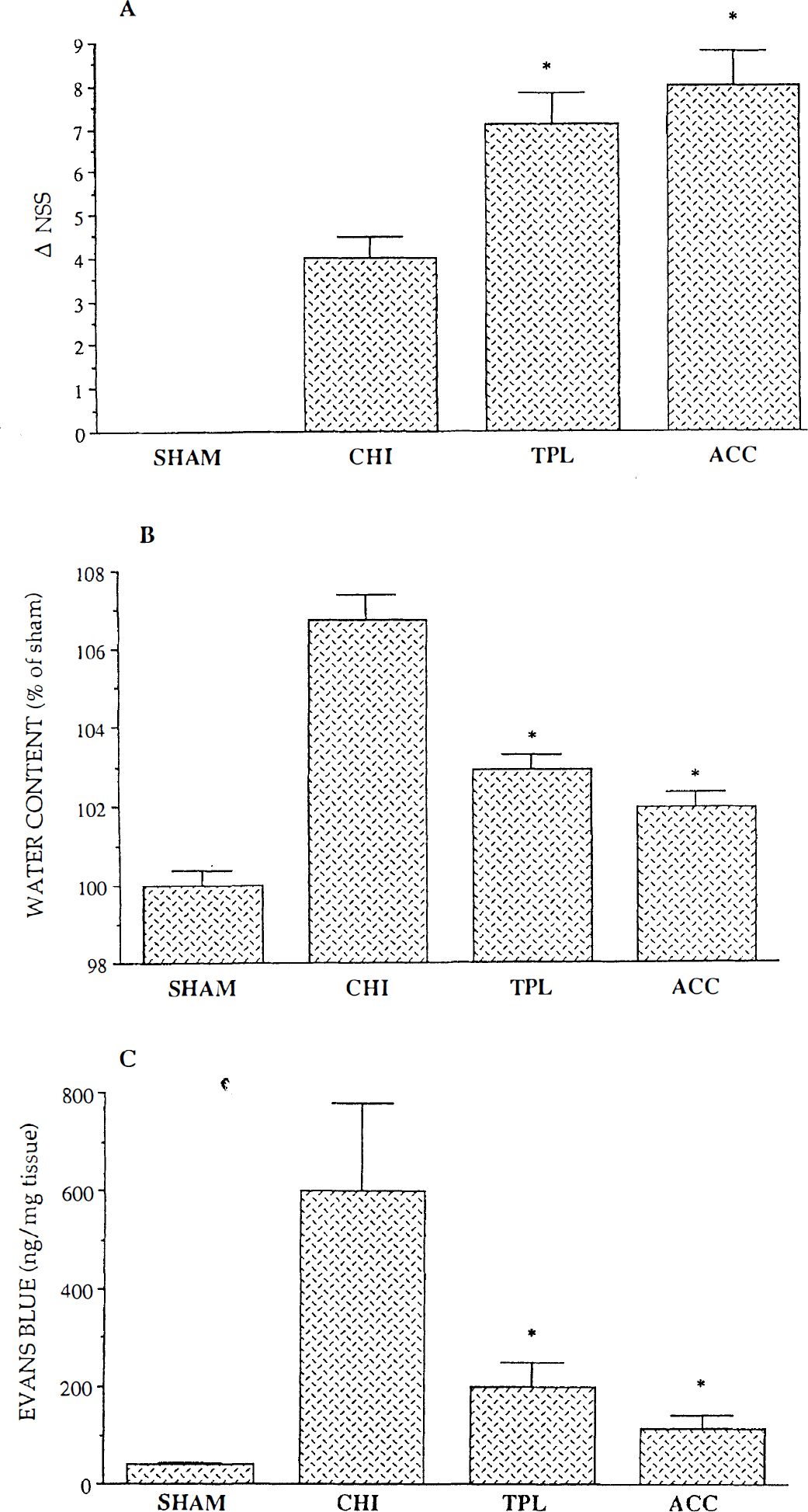
The outcomes of closed-head injury (CHI) on clinical recovery, edema, and blood-brain barrier (BBB) integrity. Sham rats were anesthetized with ether, their scalp skin was incised, but trauma was not induced. Closed-head injury rats were subjected to closed-head injury as described by Shapira et al., 1988. TPL-4-hydroxy-2,2,6,6,-tetramethylpiperidine-1-N-oxyl, a radical neutralizing agent of the nitroxide family was administered within 5 minutes of CHI (50 mg/kg, intravenously) as described (Beit-Yannai et al., 1996a). Heat acclimated rats were maintained for 30 days at 34°C, before being subjected to CHI, as described by Shohami et al., 1994. *P < 0.05 as compared with CHI.
A cyclic voltammetry study of the water-soluble fraction of LMWA revealed that the acclimated rats displayed anodic potentials similar to those of the normothermic rats. Hence, the acclimation process had not changed the chemical nature of the LMWA present in the brain. However, the anodic current (Ia, which is proportional to the concentration of reducing equivalents) of both potentials was lower (∼ 60% of the control), i.e., the levels were significantly reduced.
In view of the abundance of uric acid and its dual activity, as anti- and pro-oxidant, we determined the levels of uric acid (which appears at the first anodic potential in the voltagram), along with the activity of the enzyme catalyzing its production, xanthine oxidase, in brain extracts and in the serum of normothermic and heat-acclimated, nontraumatized rats. Unexpectedly, lower levels of uric acid were found in the brains as well as in the sera of acclimated rats as compared with those in the control rats. The brain xanthine oxidase activity of acclimated rats was also lower than in the control rats, indicating a lower rate of uric acid synthesis after heat acclimation (Beit-Yannai et al., 1996b).
Effect of apolipoprotein E deficiency on the endogenous low-molecular weight antioxidant profile after closed-head injury. Apolipoprotein E is a 36-KD plasma glycolipoprotein that plays a key role in lipoprotein metabolism by facilitating the cellular uptake of remnants of triglyceride-rich chylomicrons and very low-density lipoproteins (Brewer et al., 1989, Mahley, 1988). Apart from its role in facilitating plasma cholesterol transport, apoE is thought to participate in the mobilization and redistribution of lipids during the normal development of the nervous system and to play a key role in the regulation of lipid metabolism after peripheral injury (Ignatius et al., 1987). Lomnitski et al. (1997) studied the antioxidant capacity of apoE by incubating Cu+2 with β-very low-density lipoproteins in the presence of recombinant human apoE3. They reported that Cu+2-induced lipid-peroxidation was retarded by apoE. Thus, apoE knockout mice might possess defective endogenous antioxidant mechanisms in addition to their impaired cholinergic function (Gordon et al., 1995). Epidemiological studies on Alzheimer's disease, the major cause of dementia in the elderly, have revealed the role of genetic and environmental factors. These include the allele ε4 of apoE (Roses, 1994) and head trauma (Gentleman et al., 1993), and there seems to be a synergistic relationship between the two genetic and environmental risk factors. Therefore, apoE-deficient mice could serve as models for investigating the role of endogenous antioxidants in the recovery from CHI. Thus, the induction of CHI on genetically susceptible mice might be a relevant model for Alzheimer's disease studies.
Indeed, using apoE-deficient mice, we recently showed that the recovery of the cognitive and neurological functions of these mice after CHI was slower than that of control mice (Chen et al., 1997). These derangements were associated with more pronounced neuronal cell death in the hippocampus of the apoE-deficient mice. Therefore, we speculated that the increased susceptibility of the apoE-deficient mice to CHI could be caused by the dysfunction of their neuronal, and probably other, brain-repair mechanisms, suggesting that these mice lack, at least in part, an endogenous antioxidant mechanism(s). In line with this supposition, Lomnitski et al. (1997) showed, using cyclic voltammetry, that although nontraumatized mice, both control and apoE-deficient, have similar levels of reducing equivalents, they differ markedly in their response to CHI. Although a biphasic response to CHI was measured in both groups of traumatized mice, the initial decrease (at 5 minutes after injury) and the subsequent increase (at 4 hours) were significantly less pronounced in the apoE-deficient mice. Under similar conditions, these mice were unable to mobilize antioxidants to the same extent as the controls. The lower antioxidative response of the brain to CHI in apoE-deficient mice suggests that this phenomenon may play a role in their impaired ability to recover from brain injury. Furthermore, these findings support the notion of a synergistic relationship between genetic and environmental risk factors in neurodegenerative diseases such as Alzheimer's disease.
Pharmacological approach: effect of reactive oxygen species-neutralizing agents on the outcome of closed-head injury
Therapeutic strategies aimed at decreasing brain injury have included the administration of radical scavengers at the time of reperfusion. The rationale for this treatment is that if ROS are instrumental in mediating biological damage, their rapid removal should afford protection. A LMWA, like tirilazad mesylate (U-74006F), one of the most efficacious inhibitors of lipid peroxidation, was shown to ameliorate postischemic neurologic deficits and to protect against ischemic brain damage (Xue et al., 1992). Spin traps, such as nitrones, have also been reported to be effective neuroprotectants in a variety of brain injury models (Hensley et al., 1997). In contrast, the efficacy of traditional oxygen radical scavenger enzymes such as superoxide dismutase (SOD) and catalase is limited by their inability to cross the BBB and penetrate the cells (Chan et al., 1987; Imaizumi et al., 1990).
Desferal: an iron-chelating agent. Desferal, a potent chelator of redox-active metals, was shown to alleviate brain edema in a variety of brain injury models (e.g., Ikeda et al., 1989). In our model of CHI, when administered within 5 minutes after injury, desferal facilitated the clinical recovery of traumatized rats and markedly reduced edema (Zhang et al., 1997). By specific iron staining of brain sections, we recently found that tissue injury leads to iron mobilization and to an increase in free chelateble iron pools (unpublished finding). Thus, our observations on the protective effect of an iron-chelating agent in the present model of CHI agree with those reported by Ikeda et al. (1989) and by White and Krause (1993), and support the role played by redox-active transition metals in the pathophysiology of CHI.
Superoxide dismutase. The administration of recombinant human SOD (r-h-MnSOD, a gift from Biotechnology General. Ltd. Rehovot, Israel) to traumatized rats within minutes of CHI, had some beneficial effects, mainly on survival and clinical recovery. However, the effect on edema or on BBB was moderate and insignificant (Zhang et al., 1997). It should be noted that the nonpenetrable enzyme SOD could enter the brain parenchyma because in our CHI model, BBB is disrupted within minutes of injury (Shapira et al., 1993). Therefore, our findings suggest that extracellular O2 is involved in the pathological events after CHI, and that SOD, by neutralizing these radicals, facilitates clinical recovery. However, the effect of exogenous SOD is limited as it cannot penetrate the cells. To overcome this limitation, polyethylene glycol-conjugated SOD was prepared and used in humans with head injuries (Muizelaar et al., 1994) and experimental animals (Hamm et al., 1996; Muir et al., 1995). These studies showed that neutralization of superoxide radicals has a beneficial effect on functional outcome and cerebral blood flow after traumatic injury, implicating that these radicals play a role in the pathophysiology of brain trauma.
Nitroxides as antioxidants. A complementary strategy of intervention with the overproduction of ROS involves the use of stable nitroxide radicals and has been applied to the protection of cells (Krishna et al., 1994; Samuni et al., 1991a; 1991b), organs (Gelvan et al., 1990), and whole animals (Hahn et al., 1992) from diverse insults. Nitroxides, which are cell-permeable, nontoxic, nonimmunogenic stable radicals, have been used as biophysical probes for monitoring membrane stability, cellular pH, oxygen concentration, and intracellular redox reactions, or as contrast agents for nuclear magnetic resonance imaging. These compounds undergo a one electron redox reaction and catalyze the dismutation of O2 radicals. They can also reduce hypervalent metals and catalytically facilitate H2O2 dismutation by hemeproteins (Krishna et al., 1996a, 1996b). Previous studies showed that nitroxides are capable of preventing lipid peroxidation, (possibly occurring as a result of CHI), which is mediated by ROS (Pogrebniak et al., 1991), to selectively detoxify paramagnetic species including radicals and transition metals. Studies of bacterial and cultured mammalian cells revealed that nitroxides provide cytoprotection from the toxicity induced by H2O2 and by tumor necrosis factor (Samuni et al., 1991a; 1991b). In our model of CHI, nitroxides were also found to markedly improve the outcome, as shown by the facilitated clinical recovery, reduced edema and protection of the BBB (Fig. 2), and to have a therapeutic window in the range of 4 hours after CHI (Beit-Yannai et al., 1996a, Zhang et al. 1997).
Dexanabinol (HU-211), an NMDA antagonist and anti-oxidant. Dexanabinol (HU-211), a synthetic cannabinoid (+)-(3S,4S)-7-hydroxy-Δ-6 tetrahydrocannabinol 1,1-dimethylheptyl, is inactive as a cannabimimetic but shows pharmacological properties characteristic of N-methyl-D-aspartate (NMDA)-receptor antagonists. In binding studies with 3H-TCP and 3H-MK-801, it has been shown to block NMDA-receptors stereospecifically by interacting with a site close to but distinct from that of noncompetitive NMDA-receptor antagonists and from the recognition sites of glutamate, glycine and poly-amines (for review see Shohami et al., 1996). In addition to the antagonistic properties of HU-211 to the NMDA receptor, Eshhar et al. (1995) reported that the drug rescued neurons in culture from injury evoked by sodium nitroprusside, hydrogen peroxide, and oxygen-glucose deprivation. It also reduced protein oxidation initiated by gamma irradiation and scavenged ROS. Based on these findings, it was suggested that in the presence of radicals, HU-211 can also act as an antioxidant or as a radical scavenger, and not purely as a noncompetitive antagonist of the NMDA receptor. Moreover, we recently showed that HU-211 also inhibits the production of tumor necrosis factor (Shohami et al., 1997), thus also blocking the signal transduction pathway that activates the NFκB. In a series of studies we and other investigators have shown that HU-211 is a very potent cerebroprotectant in models of cerebral ischemia, CHI, experimental meningitis, and optic nerve crush (for review see Shohami et al., 1996). A comparison of the cerebroprotective effect of HU-211 with that of another well-studied NMDA antagonist (MK-801), revealed that in our model of CHI, MK-801 is less effective and its sedative effects are too pronounced to allow it to be considered as a potential therapeutic agent (Shapira et al., 1990), whereas HU-211 is devoid of any psychotropic influence, and exerts long-term protective effects (Shohami et al., 1993). In all the above-mentioned models, the activation of four deleterious pathways is involved in the delayed posttraumatic damage: glutamate excitotoxicity, intracellular calcium accumulation, cytokine production, and free radical formation. These pathways are probably interconnected. Therefore, we suggested that the cerebroprotective activity of HU-211, stems from the synergism between its multiple mechanisms of action and inhibitory effect on all four pathways.
SUMMARY
The present review focuses on a novel approach for investigating the temporal changes in endogenous LMWA in terms of total tissue response to injury. Two mechanisms are concomitantly activated in the brain following CHI: one involves the production of ROS; the other is a tissue response to the enhanced production of these destructive mediators. “Which is the most damaging ROS” or “which is the most active endogenous antioxidant” may not be the questions at issue in the context of posttraumatic mechanisms because these molecules may act synergistically with each other. In addition, there is an interconversion between the reactive species, as well as between the endogenous antioxidants. Therefore, we believe that the present approach, which, to the best of our knowledge, is the first attempt to measure the total capacity of brain tissue to neutralize ROS, is valid under normal, and even to a larger extent, under pathological conditions. Further studies on the temporal changes of the individual antioxidants after CHI, are in progress in our laboratory.
Figure 3A summarizes the “radical approach” to the role of ROS and of the LMWA endogenous defense mechanism in determining the clinical outcome of CHI. The extent of the final damage, comprising a number of physiological (BBB disruption, edema etc.) and behavioral (motor and cognitive dysfunction) parameters, depends on a delicate balance between the autodestructive and protective mechanisms triggered by the initial injury. The simplified scheme suggests that the production of ROS is common to a number of biochemical routes that have been shown to be activated in a variety of traumatic and ischemic brain injury models. These involve the eicosanoid cascade (via phospholipase A2), activation of the NMDA receptor, accumulation of intracellular Ca, and the release of cytokines. Each of these pathways has been found to be activated in brain injury, and numerous attempts to develop agents that block one or more of these pathways have been reported. Nevertheless, because the cascade of events is not linear, and all these pathways are activated in parallel, and result in the production of ROS, it is likely that inhibition of ROS would prove effective in clinical trials.
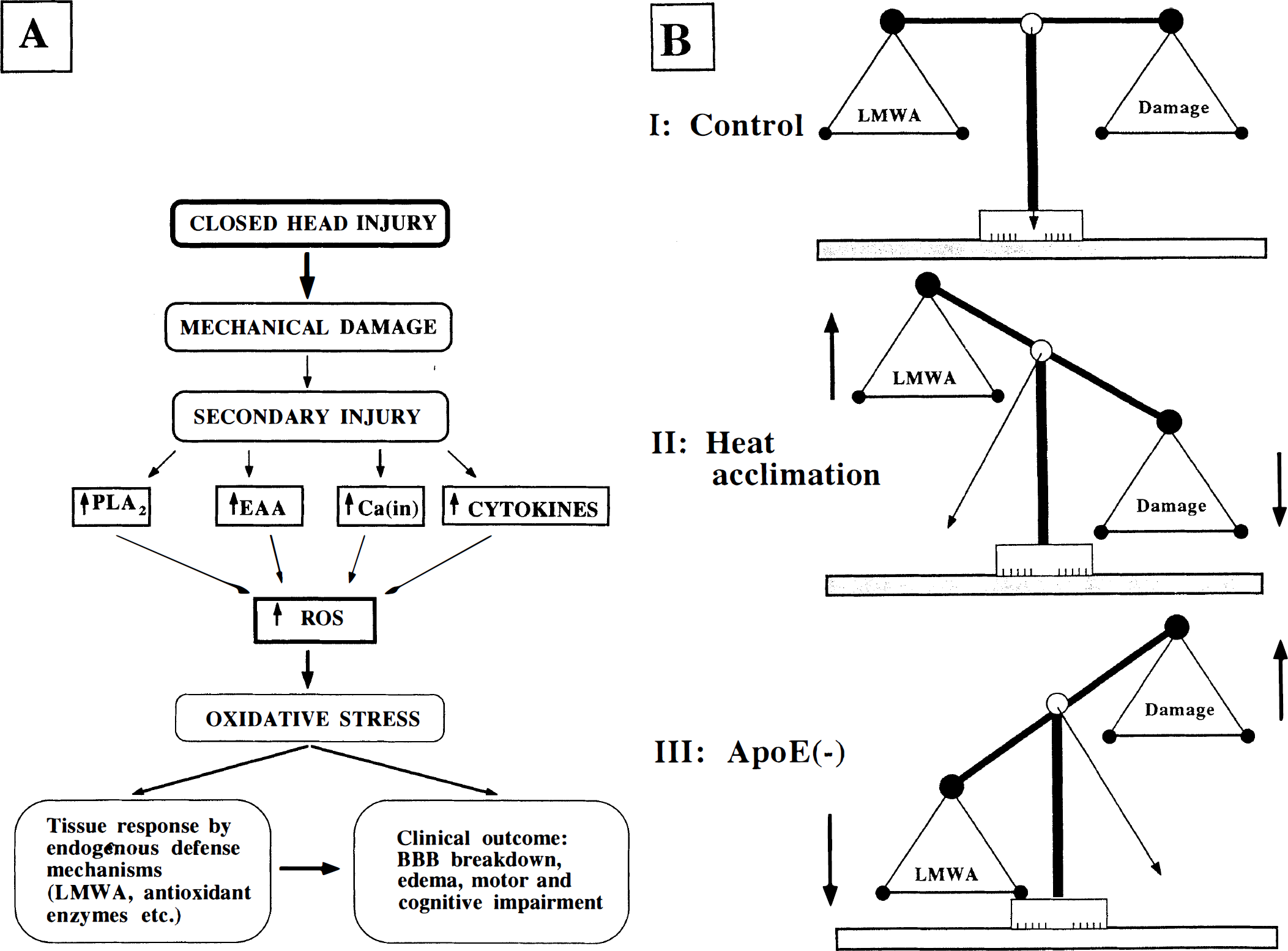
We have tried to shed some light on the role of LMWA as one of the responses of the brain to overproduction of ROS. Antioxidant enzymes have not been discussed in this context, although they may play an important role in combating ROS, as was shown by Chan (1996). The biphasic pattern of changes in LMWA supports the hypothesis that injured animals indeed use their stores of LMWA in an attempt to neutralize ROS and to protect the injured tissue. Two physiological manipulations were used in the studies reviewed here to support the notion that the more favorable outcome of CHI is associated with a better ability to neutralize ROS, namely to elevate endogenous LMWA after trauma. As shown in Fig. 3B, in heat-acclimated rats, higher levels of LMWA were found after CHI, than in their matched controls, and the incurred damage was accordingly reduced. The apoE knockout mice, which displayed impaired recruitment of LMWA, showed slower recovery and greater damage. In short, the greater the LMWA response, the better the outcome after CHI.
Whereas nitroxides act only by terminating radical-chain reactions, HU-211 displays multiple mechanisms of action, and heat acclimation triggers physiological adaptive mechanisms, some involving LMWA. The beneficial effects of heat acclimation, HU-211, and nitroxide indeed suggest that in our CHI model the most favorable outcome is achieved when treatment is based on more than one mode of action. Our findings indicate that clinical therapy could prove more effective by combining antioxidant therapeutic principles with other protective mechanisms.