
Abstract
Reactive oxygen species have been implicated in brain injury after ischemic stroke. These oxidants can react and damage the cellular macromolecules by virtue of the reactivity that leads to cell injury and necrosis. Oxidants are also mediators in signaling involving mitochondria, DNA repair enzymes, and transcription factors that may lead to apoptosis after cerebral ischemia. Transgenic or knockout mice with cell- or site-specific prooxidant and antioxidant enzymes provide useful tools in dissecting the events involving oxidative stress in signaling and damage in ischemic brain injury.
Keywords
Reactive oxygen radicals have been implicated in the pathophysiology of many neurologic disorders and brain dysfunctions (Kontos, 1985; Siesjö et al., 1989; Chan, 1994). Evidence has accumulated during the past two decades showing that reactive oxygen radicals are involved in brain injuries such as cerebral ischemia and reperfusion. In focal or global cerebral ischemia, cerebral blood flow (CBF) is reduced in brain regions that are supplied with oxygen by the occluded vessels. Reoxygenation during spontaneous or thrombolytic reperfusion provides oxygen as a substrate for numerous enzymatic oxidation reactions in the cytosolic compartments or subcellular organelles and mitochondria. It has been demonstrated that approximately 2% to 5% of the electron flow in isolated brain mitochondria produces superoxide anion radicals (O2−) and hydrogen peroxide (H2O2) (Boveris and Chance, 1973). These constantly produced reactive oxygen species (ROS) are scavenged by superoxide dismutase (SOD), glutathione peroxidase (GSHPx), and catalase. Other small molecular antioxidants, including glutathione (GSH), ascorbic acid, and α-tocopherol, are also involved in the detoxification of free radicals. During reperfusion, these endogenous antioxidative defenses are likely to be perturbed as a result of overproduction of oxygen radicals by cytosolic prooxidant enzymes and mitochondria, inactivation of detoxification systems, consumption of antioxidants, and failure to adequately replenish antioxidants in ischemic brain tissue. It has been demonstrated in numerous studies that ROS are directly involved in oxidative damage with cellular macromolecules such as lipids, proteins, and nucleic acids in ischemic tissues, which lead to cell death. Recent studies have provided evidence that indirect signaling pathways by ROS can also cause cellular damage and death in cerebral ischemia and reperfusion. Despite many existing methodologies that allow investigators to quantify various oxygen radicals such as hydroxyl radical (Oliver et al., 1990) and nitric oxide radical (Malinski et al., 1993; Tominaga et al., 1993) in ischemic tissue and in neurons that are under oxidative stress (Dawson et al., 1992; Lafon-Cazal et al., 1993; Dugan et al., 1995; Bindokas et al., 1996; Patel et al., 1996), the causative role of ROS in ischemic brain injury remains elusive to stroke researchers. However, the recent development and availability of transgenic (Tg) and knockout mutant rodents that either overexpress or are deficient in antioxidant and prooxidant genes have provided powerful tools in dissecting the molecular and cellular mechanisms of direct oxidative damage, or signaling pathways, or both, involved in ischemic brain damage. This review will focus on the use of Tg and knockout animals with altered levels of oxidant and antioxidant enzymes in signaling and damaging pathways afforded by oxidative stress in cerebral ischemia and reperfusion. Oxygen radicals in central nervous system (CNS) injury are referred to in previous reviews (Chan, 1994, 1996).
PROOXIDANT AND ANTIOXIDANT ENZYMES IN CEREBRAL ISCHEMIA
Prooxidant enzymes
Many prooxidant and antioxidant enzymes are known to participate in oxidative stress-induced signaling and injury in cerebral ischemia (Table 1). Based on oxidant products, three major classes of prooxidant enzymes can be designated: (1) nitric oxide synthases; (2) cyclooxygenases, xanthine dehydrogenase, xanthine oxidase and NADPH oxidase; and (3) myeloperoxidase and monoamine oxidase. Nitric oxide synthases (NOS) use arginine and O2 as substrates and produce nitric oxide (NO) as an oxidant product. Three isoforms of NOS exist in CNS parenchyma: neuronal NOS (nNOS, NOS1), a constitutive isoform that is localized in neurons; an inducible isoform that is induced in microglia/macrophages and astrocytes and endothelial cells (iNOS, NOS2); and a constitutive form that is localized in the endothelium (eNOS, NOS3). Neuronal NOS and eNOS activities are Ca2+-dependent, whereas iNOS is Ca2+-independent. It should be noted that NO produced by nNOS and iNOS has been implicated in both in vitro cell culture injury and in vivo ischemic brain damage. Nitric oxide produced by eNOS is known to be neuroprotective because of its vasodilative effects. The distribution of nNOS (the most abundant form and widely distributed NOS) in the CNS was first described by Bredt and Snyder (1990). This enzyme is identical to NADPH-diaphorase, an enzyme known to be without function, but resistant to hypoxic ischemia (Ferriero et al., 1988). Subsequent extensive in vitro cell culture and in vivo ischemia studies have linked this Ca2+-dependent enzyme to cell death induced by N-methyl-
Prooxidant enzymes in ischemic brain
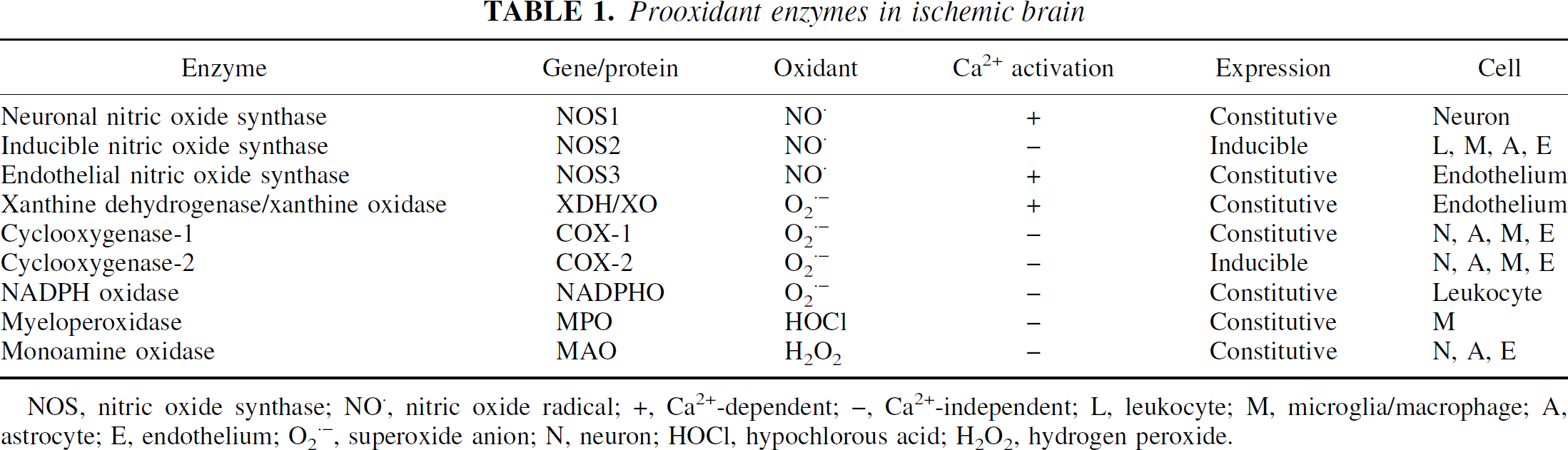
NOS, nitric oxide synthase; NO, nitric oxide radical; +, Ca2+-dependent; –, Ca2+-independent; L, leukocyte; M, microglia/macrophage; A, astrocyte; E, endothelium; O2−, superoxide anion; N, neuron; HOCl, hypochlorous acid; H2O2, hydrogen peroxide.
A rapid (1 hour after ischemia) and differential expression of eNOS in cerebral microvessels after middle cerebral artery occlusion (MCAO) in rats has led to the suggestion that increased expression of eNOS may protect neurons by increasing CBF in the penumbra area (Zhang et al., 1993). The level of constitutively expressed eNOS is up-regulated in the cerebral cortex and hippocampus after transient global cerebral ischemia and reperfusion in an indomethacin-sensitive mechanism (Beasley et al., 1998).
The differential role of nNOS and eNOS in NO generation in the brain after cerebral ischemia has recently been demonstrated (Wei et al., 1999). Other studies indicate that NO produced by iNOS in nonneuronal cells may contribute to cerebral ischemic damage (Iadecola, 1997). Inducible nitric oxide expression peaks 24 to 48 hours after ischemia and occurs in infiltrating neutrophils and cerebral vascular cells (Iadecola et al., 1996). Recent in vitro cell culture studies have demonstrated that the induction of iNOS and the formation of 3-nitrotyrosine under oxygen–glucose deprivation kill cerebral endothelial cells by apoptosis (Xu et al., 2000), suggesting the injurious role of iNOS expression in an ischemic setting.
Prooxidant enzymes, including cyclooxygenase-1, cyclooxygenase-2 (COX-2), xanthine dehydrogenase, xanthine oxidase, and NADPH oxidase, generate O2− as the main oxidant in various cell types within the parenchyma in ischemic brain. These are constitutively expressed enzymes with the exception of COX-2, which is highly inducible. It has been reported that NO produced by iNOS enhances COX-2 activity in the ischemic brain and that iNOS-positive neutrophils are in close proximity to COX-2–positive neurons (Nogawa et al., 1998).
The third group of prooxidant enzymes, myeloperoxidase and monoamine oxidase, generate hypochlorous acid and H2O2 as main oxidants in leukocytes and in parenchymal cells, respectively. One interesting note is that the expression of the prooxidant enzymes is cell-specific, in contrast to the subcellular site specificity of antioxidant enzyme expression (Table 1).
Antioxidant enzymes
Superoxide dismutases are specific antioxidant enzymes that detoxify O2− to H2O2. Three SODs, copper/zinc SOD (CuZnSOD, SOD1), manganese SOD (MnSOD, SOD2), and extracellular SOD (ECSOD, SOD3), are major antioxidant enzymes based on cellular distribution and localization (Table 2). CuZnSOD is a major cytosolic enzyme with a level constituted at approximately 0.1% of total proteins in mammalian cells. MnSOD is a mitochondrial enzyme, whereas ECSOD is an isoform that is localized in extracellular space, cerebrospinal fluid, and cerebral vessels (Marklund, 1982; Oberley, 1982). All three SOD isoforms dismutate O2−, forming H2O2, which is scavenged by peroxisomal catalase or GSHPx, at the expense of GSH. Glutathione is regenerated from oxidized GSH by GSH reductase in the presence of NADPH. Other lipid peroxides are also scavenged by GSHPx. As specified for superoxide radicals, CuZnSOD has been extensively used in experimental studies involving cerebral ischemia and reperfusion. Unfortunately, mixed and confusing results have been obtained when free nonmodified CuZnSOD was used. The extremely short half-life of CuZnSOD (6 minutes) in circulating blood, and its failure to pass the blood–brain barrier and to be taken up intracellularly, make it difficult to use for enzyme therapy in cerebral ischemia (Chan et al., 1993b). However, a modified enzyme with an increased half-life, such as polyethylene glycol-conjugated CuZnSOD, has been successfully used to reduce infarct volume in rats that were subjected to focal cerebral ischemia (He et al., 1993). Liposome-entrapped CuZnSOD has an increased half-life (up to 4.2 hours), blood–brain barrier permeability, and cellular uptake, and it has proven to be an effective treatment in reducing the severity of ischemic and traumatic brain injuries (Chan et al., 1987; Imaizumi et al., 1990). Yet in some instances, modified CuZnSOD (that is, polyethylene glycol-conjugated CuZnSOD) has been used with conflicting results. The fact of these mixed preclinical results makes it imperative to use other experimental strategies, such as Tg/knockout mutants, so that the pharmacologic role of antioxidant enzymes can be established in cerebral ischemia.
Mammalian superoxide dismutases

CuZn, cooper, zinc; SOD, superoxide dismutase; Mn, manganese; EC, extracellular; K-SOD, rate-constant SOD.
GENETIC MANIPULATION IN RODENT STUDIES OF OXIDATIVE STRESS IN ISCHEMIA
One strategy is to use Tg and knockout technology to alter the levels of prooxidants, antioxidants, and oxidant-related enzymes or proteins, and to use these genetically modified animals to study the role of a specific oxidant or antioxidant in ischemic brain injury.
The most commonly used method for creating Tg mice is the microinjection of the DNA of interest (transgene) into the pronuclei of fertilized mouse oocytes (Gordon et al., 1980). This technique is used to introduce a linear DNA fragment (genomic or cDNA with proper promoter) that contains sequences required for directing correct transcriptional initiation and for encoding a gene product (protein, enzyme) that can be readily identified using various biochemical assays. Pronuclear microinjection of DNA leads to random integration of various copies of the injected DNA into a single site on a mouse chromosome. The integrated DNA can be passed on to future generations. Another way to produce Tg mice is to introduce foreign genes to embryonic stem cells (Thomas and Capecchi, 1987). The embryonic stem cells are subsequently screened for the integration of transgenes. These embryonic stem cells with the transgene (active or inactive knockout) can then be microinjected into the blastocytes to further produce chimeric mice. The mating of chimeric mice with wild type mice produces heterozygous offspring (+/−) that contain the transgene. Furthermore, breeding among heterozygous mice generates homozygous animals (+/+ for overexpression, –/– for knockout). By use of this Tg technology, several mice strains with various genotypes that relate to oxidant/antioxidant enzymes/proteins have been successfully developed. Most of these Tg and knockout mutant animals have been used in the study of the role of oxidants in ischemic brain injury (Table 3).
Transgenic and knockout mutant mice in the study of the role of free radicals in CNS injury
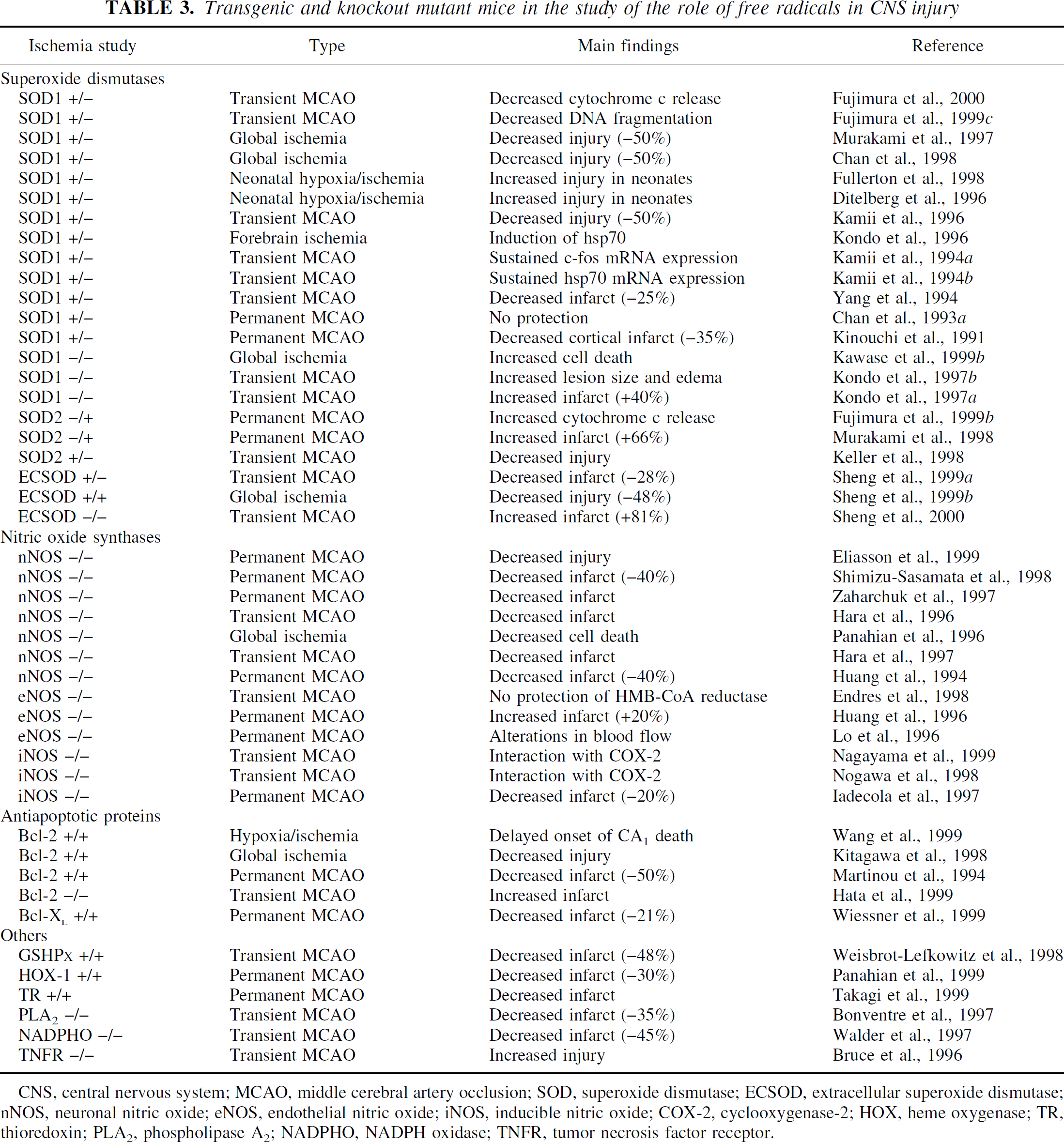
CNS, central nervous system; MCAO, middle cerebral artery occlusion; SOD, superoxide dismutase; ECSOD, extracellular superoxide dismutase; nNOS, neuronal nitric oxide; eNOS, endothelial nitric oxide; iNOS, inducible nitric oxide; COX-2, cyclooxygenase-2; HOX, heme oxygenase; TR, thioredoxin; PLA2, phospholipase A2; NADPHO, NADPH oxidase; TNFR, tumor necrosis factor receptor.
The use of Tg and knockout mice to study gene-related functions in cerebral ischemia was first reported by Kinouchi et al. (1991). These authors used Tg mice of the Tg/SF-218 strain carrying the human SOD1 gene developed 4 years earlier (Epstein et al., 1987). These mice were subsequently bred with CD-1 mice to produce transgenic offspring that carry the SOD1 gene. There are no observable phenotypic differences between the Tg mice and non-Tg normal littermates. In heterozygous SOD1 Tg mice, a threefold increase in SOD1 activity has been observed in all brain regions, whereas in the homozygous SOD1 Tg mice, a fivefold increase in SOD1 activity was achieved (Chan et al., 1994). In these mice, a 35% decrease in infarct volume was observed after permanent focal ischemia involving coagulation of the distal MCA and bilateral common carotid artery occlusion (Kinouchi et al., 1991). This model of MCAO preserved collateral blood flow, which may explain the discrepancy in the results obtained in the suture model of permanent MCAO in which no protection was observed in the same SOD1 Tg animals (Chan et al., 1993a). After transient MCAO, significant neuronal protection (25% to 50%) was observed at various reperfusion times (Yang et al., 1994; Kamii et al., 1996) (Table 3). In global ischemia, SOD1 overexpression is neuroprotective with a 50% reduction in hippocampal CA1 cell death (Murakami et al., 1997). Recently, rats that overexpress SOD1 have also been developed, which show a 50% reduction in hippocampal CA1 neuronal cell death after global ischemia (Chan et al., 1998).
In the neonatal mouse brains, SOD1 overexpression is deleterious and there is increased cell death after hypoxic–ischemic injury. This result is explained by the fact that higher levels of H2O2 exist in SOD1 Tg neonates because the neonatal brain cannot compensate for increasing levels of downstream enzymes, catalase and GSHPx (Ditelberg et al., 1996; Fullerton et al., 1998). This study underscores the importance of the antioxidative status of the CNS, which might determine the outcome of ischemic injury or protection in SOD1 Tg mice.
The role of SOD1 in cerebral ischemia is further confirmed by the use of Sod1-deficient mice. These Sod1 knockout mice (Sod1 −/+ and Sod1 –/–) had increased cell death and edema after transient MCAO and global cerebral ischemia (Kondo et al., 1997a,b; Kawase et al., 1999b).
The importance of mitochondrial production of oxygen radicals and the protective role of Sod2 after permanent ischemia have been demonstrated in Sod2 knockout mice. These mutant mice show exacerbated infarct volume after permanent MCAO (Murakami et al., 1998), and increased mitochondrial cytochrome c release and subsequent DNA fragmentation after permanent focal cerebral ischemia (Fujimura et al., 1999b). However, mice that overexpress Sod2 showed neuronal protection against oxidative stress after transient focal cerebral ischemia (Keller et al., 1998).
The level of ECSOD (Sod3) in brain parenchyma is quite low compared with other systemic organs. However, recent studies have clearly demonstrated that Sod3, when overexpressed in mice, provides neuronal protection in focal stroke (Sheng et al., 1999a) and in global cerebral ischemia (Sheng et al., 2000), whereas Sod3 –/– mice have a worsened outcome from focal cerebral ischemia (Sheng et al., 1999b).
The important role of GSHPx in H2O2, or lipid peroxide-induced ischemic brain damage, or both, has been documented in a study using GSHPx +/+ mice (Weisbrot-Lefkowitz et al., 1998), and in a study showing the neuroprotection afforded by ebselen, a mimic of GSHPx (Dawson et al., 1995).
Another major area of oxidative injury using knockout mutants in cerebral ischemia involves NOS isoforms. Homozygous knockout mice of nNOS and eNOS were developed by Huang and colleagues (Lo et al., 1996; Huang, 1999). The deleterious role of nNOS after ischemic injury has been clearly established in nNOS –/– mutant mice. A 40% reduction in lesion size was observed after permanent focal cerebral ischemia in nNOS –/– mice (Huang et al., 1994; Zaharchuk et al., 1997; Shimizu-Sasamata et al., 1998; Eliasson et al., 1999). An even greater neuroprotection was seen after transient ischemia (Hara et al., 1996, 1997). The role of eNOS and NO in maintaining local blood flow is likely responsible for the increased lesion volume in eNOS –/– animals after cerebral ischemia (Huang et al., 1996; Endres et al., 1998). In contrast, there is ischemic injury in iNOS –/–knockout mutants after permanent focal cerebral ischemia (Iadecola et al., 1997), suggesting the damaging role of NO produced by these isoforms (Fig. 1).
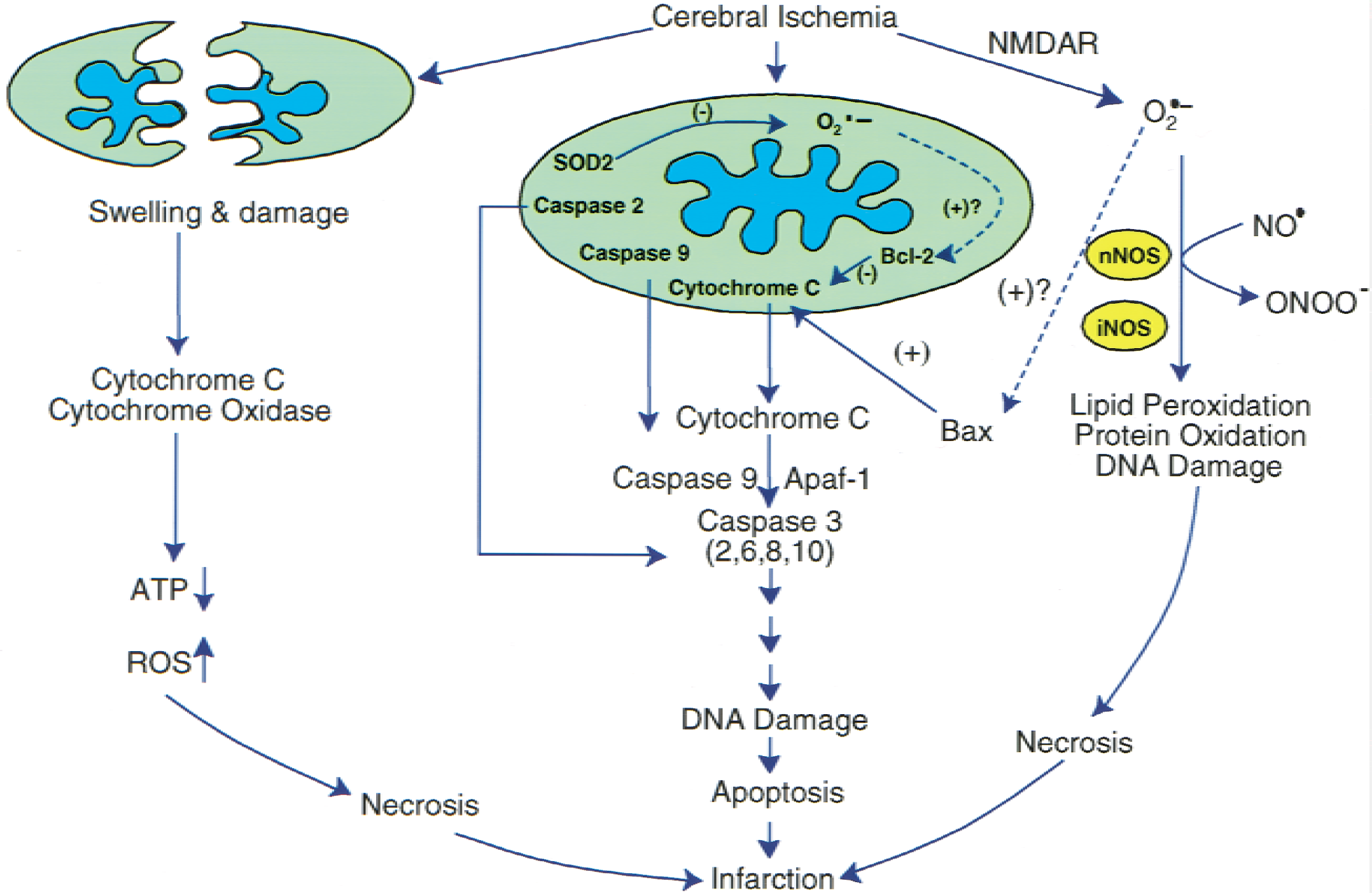
Mitochondria as targets for oxidative stress signaling after cerebral ischemia. Cerebral ischemia and reperfusion generated reactive oxygen species (ROS) within the mitochondria, which then signal the release of cytochrome c by mechanisms that may be related to Bcl-2 and translocation of Bax. Cytochrome c, once released, binds to Apaf-1 followed by caspase-9 to form a complex that subsequently activates caspase-3 and other caspases, such as −2, −6, −8 and −10. Activated caspase-3 is known to cleave to many nuclear DNA repair enzymes, which then leads to nuclear DNA damage without repair, resulting in apoptosis. The activation of the N-methyl-
In addition to SOD and NOS isoforms, many new studies have involved prooxidant and antioxidant enzymes in Tg or knockout mutant animals in cerebral ischemia. A general rule of thumb is that mice with increased expression of the antioxidant have neuroprotection, whereas mice deficient in antioxidant enzymes are sensitive to ischemic injury (Table 3).
Overexpression of the mitochondrial antiapoptosis protein Bcl-2 is believed to suppress superoxide production and prevent the release of cytochrome c and the subsequent caspase apoptosis cascade (Hockenbery et al., 1990; Kane et al., 1993; Cai and Jones, 1998). Thus, overexpression of neuronal Bcl-2 in Tg mice has been shown to be neuroprotective (Martinou et al., 1994), whereas Bcl-2 knockout mice show increased injury after transient focal cerebral ischemia (Hata et al., 1999).
SIGNAL TRANSDUCTION OF OXYGEN RADICALS IN CEREBRAL ISCHEMIA
Reactive oxygen species are known to cause macro-molecular damage that includes lipid peroxidation, protein oxidation, and DNA oxidation, which contribute to ischemic brain injury (Hall and Braugher, 1989; Chan, 1994, 1996). In addition to the direct biochemical interaction between ROS and cellular macromolecules that leads to cellular damage after cerebral ischemia, increasing evidence supports the notion that ROS exact their effects on ischemic CNS tissue through redox signal transduction pathways. Recent studies suggest that mitochondria, apurinic/apyrimidinic endonuclease/redox factor-1 (APE/Ref-1), a DNA repair enzyme, and transcription factors are major targets for redox signaling that leads to ischemic cell death (Fujimura et al., 1998, 1999c).
Mitochondria as targets for oxygen radical signaling in cerebral ischemia
The mitochondrion is the powerhouse of the cell. Under normal physiologic conditions, brain mitochondria generate adenosine triphosphate (ATP) through the electron transport chain, which contains five multi-subunit enzyme complexes, I to V. During mitochondrial respiration, O2− and H2O2 are produced in complex I (NADPH-dehydrogenase) and in complex III (ubiqui-none-cytochrome b-C1) (Boveris and Chance, 1973). The current concepts are that mitochondrial function is relevant to normal bioenergetic function, Ca2+ sequestration, free radical production, membrane permeability and transition, and regulation of necrotic or apoptotic pathways, or both, in acute stroke and chronic neurodegenerative diseases (Fiskum et al., 1999; Murphy et al., 1999). Reactive oxygen species signaling in mitochondria recently has been demonstrated in ischemic brain with the release of mitochondrial cytochrome c, a water-soluble peripheral membrane protein of mitochondria and an essential component of the mitochondrial respiratory chain (Boyer et al., 1977). It has been demonstrated that cytochrome c is translocated from mitochondria to the cytosolic compartment after transient focal cerebral ischemia in rats (Fujimura et al., 1998), in brain slices that are subjected to hypoxia–ischemia (Pérez-Pinzón et al., 1999), and in vulnerable hippocampal CA1 neurons after transient global cerebral ischemia (Sugawara et al., 1999). Mitochondria are known to be involved in both necrosis and apoptosis cell pathways (Fig. 1), which depend on the severity of insults or the nature of the signaling pathways (Ankarcrona et al., 1995; Bonfoco et al., 1995; Green and Reed, 1998; Pettmann and Henderson, 1998; Fujimura et al., 2000). In most instances, severe cerebral ischemia renders the mitochondria completely dysfunctional for making ATP, which ensures necrotic cell death. However, various cellular or biochemical signaling pathways involve mitochondria in apoptosis by releasing cytochrome c to the cytoplasm where it activates caspases by interacting with some cytosolic factors, including Apaf-1 and caspase-9 (Zou et al., 1997; Yoshida et al., 1998; Saleh et al., 1999), by further activating caspase-3 and its subsequent cleavage to many substrate proteins including poly(ADP-ribose) polymerase (Endres et al., 1997; Chen et al., 1998; Namura et al., 1998) (Fig. 1).
Direct evidence suggesting the involvement of oxidative stress signaling in mitochondrial release is as follows: first, deficiency in MnSOD (a mitochondrial antioxidant enzyme) in mice caused exacerbation in neuronal apoptosis after permanent focal cerebral ischemia (Murakami et al., 1998); second, the release of cytochrome c occurs before DNA fragmentation and is exacerbated in Sod2 −/+ mice after permanent focal cerebral ischemia (Fujimura et al., 1999b). Together, these studies suggest that an increased O2− level within the mitochondria signals cytochrome c release and subsequent DNA fragmentation. However, overexpression of Sod2 prevents neuronal apoptosis and reduces ischemic brain injury by suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction (Keller et al., 1998). Overexpression of Sod2 also provides protection against NMDA and NO toxicity in cortical cultures, whereas nNOS neurons from Sod2 –/– mice are markedly sensitive to NMDA toxicity (Gonzalez-Zulueta et al., 1998). The mechanism by which mitochondrial O2− signals cytochrome c release is not clear. It is also not clear whether the mitochondrial membrane permeability transition pore is involved in this signaling pathway because the latter involvement in cytochrome c release is being questioned (vander Heiden et al., 1997; Andreyev et al., 1998; Bossy-Wetzel et al., 1998). In addition to this intrinsic signaling, extrinsic mitochondrial signaling is also involved with oxidative stress. Recent studies have demonstrated that overexpression of cytosolic SOD1 activity ameliorated the early release of cytochrome c from mitochondria and subsequent neuronal apoptosis in Tg mice after transient focal cerebral ischemia (Fujimura et al., 2000). The mechanism underlying the down-regulation of cytosolic ROS signaling in mitochondrial release of cytochrome c after cerebral ischemia is unknown and requires further elucidation. One possibility is that increased cytosolic antioxidants may alter the redox potential of the cell that includes subcellular organelles such as mitochondria.
Bcl-2, a known antiapoptotic protein localized in the mitochondrial outer membrane, blocks apoptosis by inhibiting cytochrome c release translocation by blocking the mitochondrial voltage-dependent anion channel (Shimizu et al., 1999), by shifting the cellular oxidation-reduction potential (Ellerby et al., 1996), or by preventing mitochondrial superoxide radical production (Cai and Jones, 1998). A study of the role of Bcl-2 on cytochrome c release in vivo in Bcl-2 Tg mice after cerebral ischemia and reperfusion will provide the signaling link of Bcl-2 with oxidative stress in mitochondrial cytochrome c-dependent apoptosis.
In addition, oxidants like superoxide can signal directly to components of the biochemical cascade involving cytochrome c-dependent apoptosis. In some limited studies, it has been shown that overexpression of CuZnSOD in SOD1 mice reduced the levels of activated caspase-9 (Noshita et al., 2000) and activated caspase-3 (Chan et al., unpublished data, 2000) after transient focal cerebral ischemia, suggesting multiple operating steps involving superoxide signaling in this apoptosis pathway (Fig. 1).
DNA repair enzyme as target for oxygen radical signaling in cerebral ischemia
APE/Ref-1, a constitutive multifunctional protein mainly expressed in the nucleus, is known to be involved in DNA base excision repair by removing the oxygen radical-induced AP site and by regulation of many other transcriptional factors, such as AP-1, that are sensitive to redox regulation (Bennett et al., 1997). Although the direct evidence between AP site repair and apoptosis is lacking, incomplete repair of AP sites has been reported to cause mutagenesis and genetic instability. Apurinic/apyrimidinic endonuclease (APE) is known to be associated with oxidative stress, and in some cases down-regulation of APE expression is associated with apoptosis in cells of the myeloid lineage (Robertson et al., 1997). It has been demonstrated (immunocytochemistry and Western blot) that the levels of constitutively expressed APE are rapidly reduced in neurons (that is, within 30 minutes) after transient focal cerebral ischemia (Fujimura et al., 1999a). The early rapid reduction of APE is also seen in vulnerable hippocampal CA1 neurons in rodents after transient global cerebral ischemia (Edwards et al., 1998; Kawase et al., 1999a). The rapid reduction of APE appears to be sensitive to oxidative stress because overexpression of CuZnSOD in SOD1 mice prevents the early reduction of APE in the ischemic brain (Fujimura et al., 1999c). The early reduction of APE in the ischemic brain after photothrombotic cerebral ischemia can also be prevented by treatment with the antioxidant 21-aminosteroid, suggesting that redox signaling may play a role in APE reduction after cerebral ischemia (Chang et al., 1999) (Fig. 2). APE/Ref-1 is also known as a redox factor for AP-1 transcription factors (Xanthoudakis et al., 1992). Further studies are required to elucidate the mechanisms of the redox signaling of APE and DNA repair in cerebral ischemia.
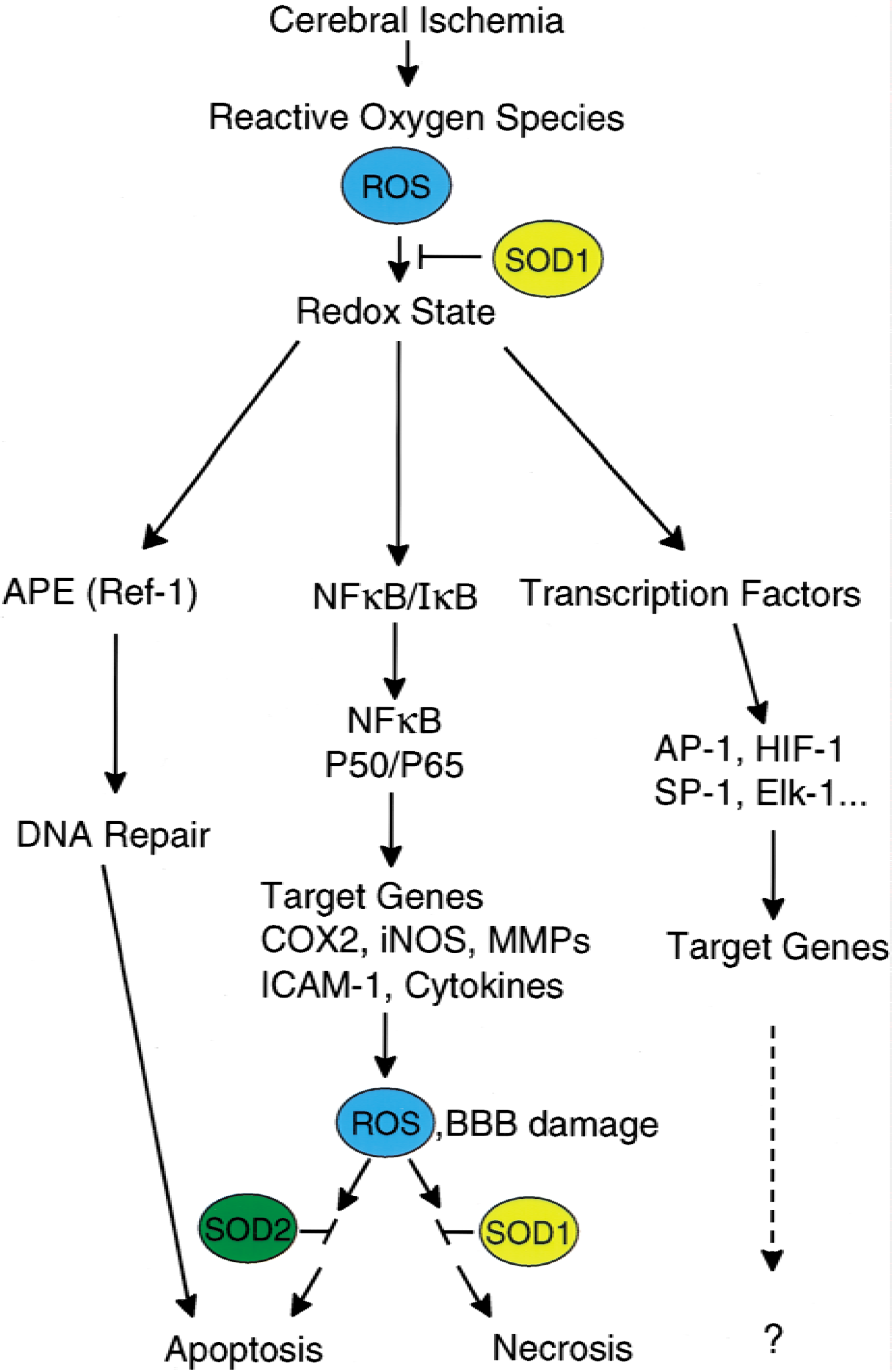
Oxygen radical stress signal transduction in pathways involving DNA repair and transcription factors. Oxygen radicals, once formed after cerebral ischemia and reperfusion, are known to alter the redox state of the cell (both cytosolic and mitochondrial components). This redox leads to rapid reduction of the constitutively expressed DNA repair enzyme APE/Ref-1 and the DNA repair machinery that may cause apoptosis. The major signaling pathway of oxygen radicals involves the transcription factors, NF-κB in particular. The activation of NF-κB causes its translocation into the nucleus and binding to the NF-κB site of many inducible genes, including but not limited to cyclooxygenase-2 (COX-2), inducible nitric oxide (iNOS), metalloproteinases (MMPs), intercellular adhesion molecules (ICAM-1), and cytokines. The expression of these genes may lead to formation of reactive oxygen species (ROS) and blood-brain barrier (BBB) breakdown, which lead to apoptosis, or necrosis, or both.
Transcription nuclear factor-κ B as an oxidative stress target in cerebral ischemia
Activation of many transcription factors, nuclear factor-κ B (NF-κB) in particular, is known to be regulated by the redox state of the cell (Dalton et al., 1999). A transcriptionally active NF-κB is a dimer of p50/p65 proteins. An inactive NF-κB complex is formed by two p50 homodimers or a p50/p65 heterodimer bound to a member of the Iκ B family.
Many diseases and proinflammatory stimuli, as well as ROS, activate NF-κB (Dalton et al., 1999). In contrast, antioxidants block NF-κB activation by inhibiting the phosphorylation of Iκ B in the serine residue (Traenckner et al., 1995). Notably, oxidative stress induces NF-κB nuclear translocation without degradation of Iκ B (Canty et al., 1999).
It has been shown in a preliminary study that overexpression of SOD1 in mice reduces the activation of NF-κB, as shown by electrophoretic mobility shift assay and by supershift assay and reduction of infarction after transient focal cerebral ischemia (Huang et al., 1999). These data suggest that activation of NF-κB by oxidative stress during reperfusion is detrimental to the ischemic brain. However, an opposite view exists because p50 knockout mice exhibited increased vulnerability of hippocampal neurons to excitotoxic injury (Yu et al., 1999). Thus, the effects of oxidative stress on NF-κB activation and subsequent neuronal damage or protection after cerebral ischemia should be under scrutiny based on these conflicting observations. To clarify this issue, it is necessary to access the downstream target genes that are known to be regulated by NF-κB. These downstream inducible genes include but are not limited to iNOS, COX-2, matrix metalloproteinase-9, intercellular adhesion molecules, HOX genes and cytokines, which are known to be involved in neuronal injury, and blood-brain barrier breakdown and inflammatory response after cerebral ischemia. The 5' region of the mouse iNOS gene contains redox-sensitive elements such as the antioxidant-responsive element and the NF-κB-responsive element (Kuo et al., 2000). The highly inducible iNOS gene may suggest the importance of oxidative stress in NF-κB signaling after cerebral ischemia and reperfusion. The fact that overexpression of CuZnSOD in SOD1 mice reduces expression of MMP-9 and COX-2 after brain injury or spreading depression may implicate the role of NF-κB activation in cerebral ischemia (Gasche et al., 1999; Morita-Fujimura et al., 1999, 2000; Yrjänheikki et al., 2000) (Fig. 2).
In addition to NF-κB, many transcription factors like AP-1, HIF-1, SP-1, and EIK-1 are known to be redox-sensitive proteins (Sen, 1998) and their regulation on gene expression by oxidative stress in cerebral ischemia has yet to be determined (Sharp et al., 2000). Other proteins that are known to be associated with inflammation and cell death and life signaling after stroke are possible molecular targets for oxidative stress signaling and have yet to be investigated (Sen, 1998).
CONCLUSION
Based on numerous results accumulated over the past decade, it is clear that oxidative stress is involved in cell death after cerebral ischemia. Tg and knockout animals that either overexpress or are deficient in pro- or antioxidant enzymes have provided unique tools to dissect out the molecular mechanisms of cell death, either necrosis or apoptosis, or both. Although oxygen radicals, by virtue of their reactivity, can injure neurons and other brain cells directly, increased evidence has pointed to the role of redox signaling of oxygen radicals. Such redox signaling targets mitochondrial cytochrome c release, DNA repair enzymes, and transcriptor NF-κB, which might lead to neuronal apoptosis. A better understanding of the mechanisms of these molecular signaling targets by oxidative stress may provide unique therapeutic opportunities in clinical stroke.
Footnotes
Acknowledgments
I thank my past and current postdoctoral fellows, graduate students, and research associates for their invaluable contributions to the data provided in this article. I also thank Cheryl Christensen for editorial assistance and Beth Houle for preparation of the figures.