
Abstract
The uncoupling proteins (UCPs) are mitochondrial transporter proteins involved in proton conductance across inner mitochondrial membrane (IMM). UCP2, which is one of the members of this class of proteins, has a wide but restricted tissue distribution including brain. Its physiologic role according to emerging evidences, although still not clear, indicate that distribution of UCP2 may be related to regulation of mitochondria membrane potential (ΔΨm), production of reactive oxygen species (ROS), preservation of calcium homeostasis, modulation of neuronal activity, and eventually inhibition of cellular damage. These factors are very important in determining the fate of neurons and damage progression in the brain during various neurodegenerative diseases including cerebral stroke. Recent evidence indicates that an increased expression and activity of UCP2 are well correlated with neuronal survival after stroke and trauma. This review briefly covers the present understanding of UCP2, which eventually may be beneficial to understand the precise role of UCP2 to develop strategy to identify its potential therapeutic application.
Introduction
The uncoupling proteins (UCPs) are inner mitochondrial membrane (IMM) anion-carrier protein that transport protons (H+) to the mitochondrial matrix and in turn dissipates the proton motive force (Δ
Emerging evidence suggests that UCP2 may play an important role during cerebral stroke by regulating mitochondrial potential (ΔΨm) and energy balance, neuroendocrine and autonomic functions, reactive oxygen species (ROS) production and fatty acid anion transport, cell death and inflammation (Richard et al, 2001; Mattiasson et al, 2003; Mattiasson and Sullivan, 2006; Deierborg et al, 2008). In the brain, UCP2 is predominantly expressed in fast-firing neurons, microglia, neutrophils, residing and invading monocytes, cells of the choroids plexus as well as endothelial cells in several parts of the fore-, mid-, and hindbrain (Richard et al, 1998, 2001). Interestingly, UCP2 has varied mRNA expression and protein disproportion in various species (Richard et al, 2001). This may be because of different transcription and translation activities of UCP2, and which is influenced by metabolic rate, free fatty acid (FFA), nucleotides, coenzyme Q, superoxide anion (O2−), developmental stage, etc. The distinct role of UCPs, in particular UCP2, in pathophysiology at present is a matter of debate; this review article will look into the present understanding of UCP2 and emphasis will be made to highlight the potential neuroprotective role of UCP2 in stroke and diabetes.
Structure and action of uncoupling protein-2
The functional structure of UCP2 is a homodimer composed of 309 amino-acid residues. It is present on chromosomes 11 and 7 in the human and mouse, respectively. Uncoupling protein-2 exhibits 59%, 71%, 33%, and 38% structural similarities with UCPl, −3, −4, and −5, respectively (Fleury and Sanchis, 1999; Vidal-Puig et al, 1997). The rat UCP2 shares approximately 99% and 95% sequence homology with the UCP2 of the mouse and human, respectively, which emphasizes the remarkable conservation of UCP2 (Hidaka et al, 1998).
The primary function of UCPs in the mitochondria is translocation of protons, which are pumped out against concentration gradient during the flow of electrons through respiratory chain complexes I—IV (Figure 1, refer legend for details). The proton electrochemical gradient, the Δ
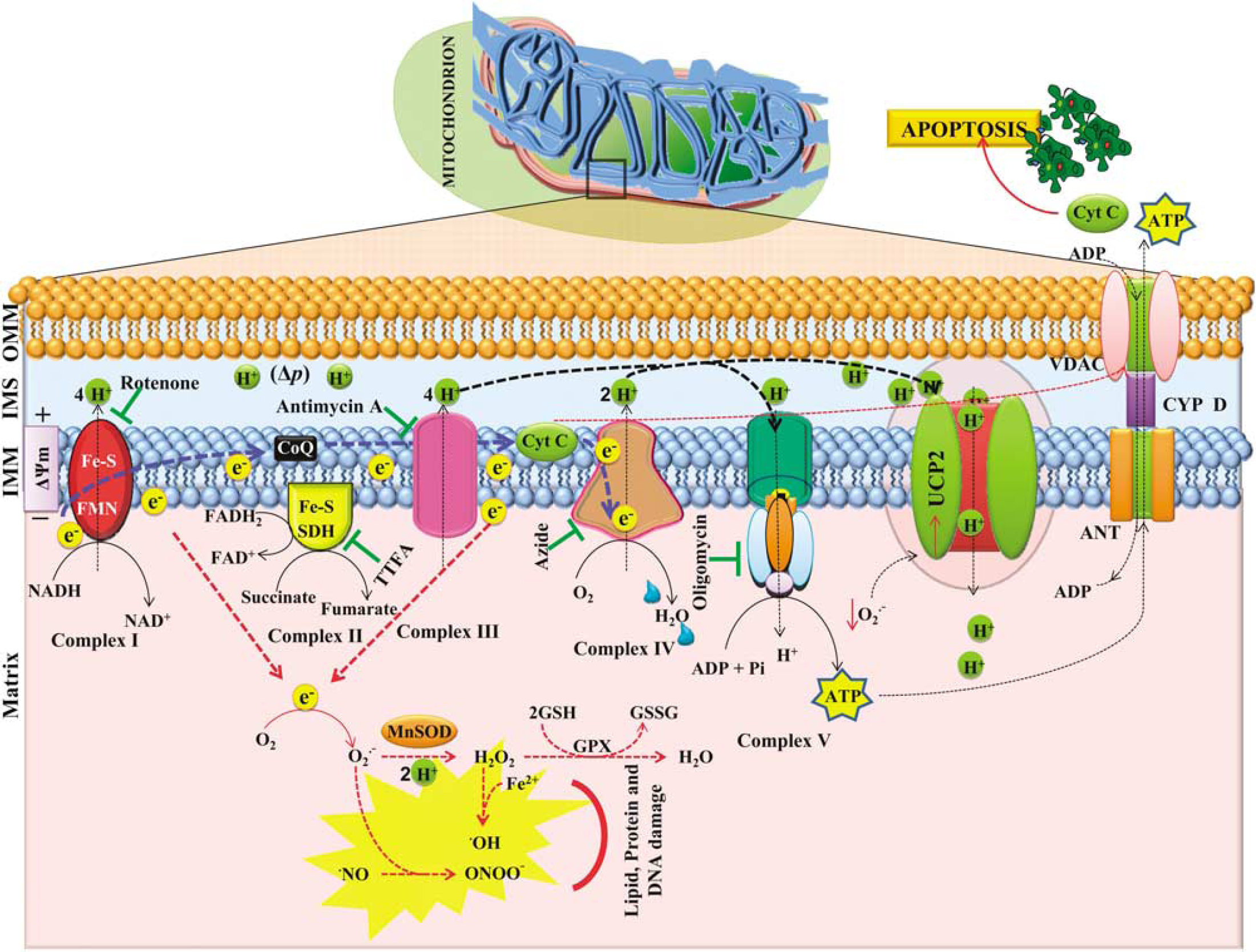
Schematic diagram of mitochondria, showing mitochondrial electron transport chain (ETC) complexes (I–V), UCP2, ANT, and VDAC. During the respiration, the free energy generated by oxidation of substrates is conserved in reduced molecules of nicotinamide adenine dinucleotide (NADH) and FADH2. These reducing equivalents donate the electron to protein complexes and the conserved energy in these molecules is used to transport protons to the intermembrane space. This leads to the establishment of a proton motive force (Δ
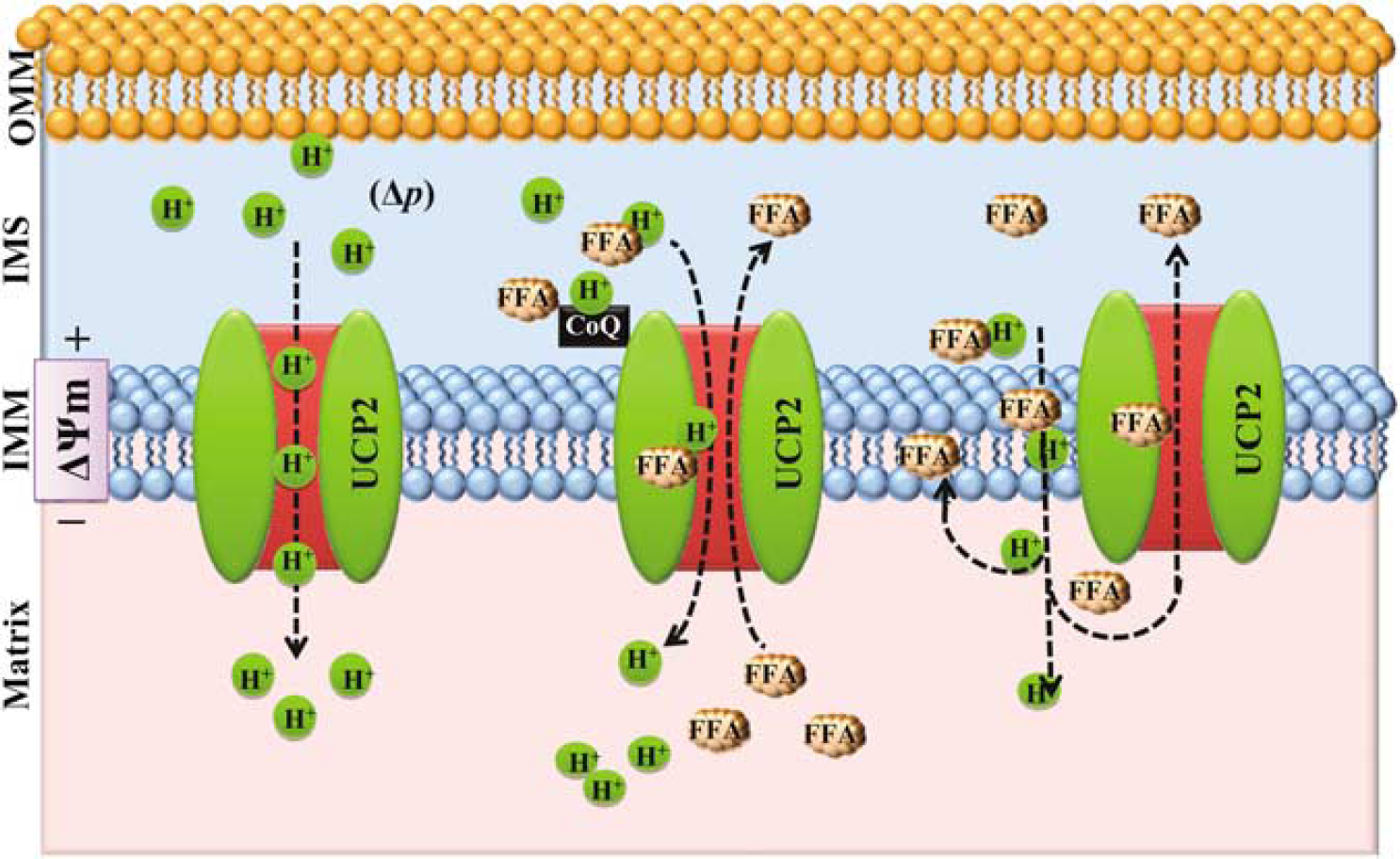
Drawing shows proposed mechanism of uncoupling protein-2 (UCP2)-mediated uncoupling. UCP2 transport protons directly or with the help of free fatty acid (FFA) to the matrix from intermembrane space (IMS). FFA carries protons through UCPs with or without coenzyme Q (CoQ) and then FFA returns back. However, electroneutral, protonated fatty acids flip across the inner mitochondrial membrane (IMM) and release the protons in the matrix. The fatty acid anion is transported back by UCP2 and above cycle is repeated to sustain the mitochondria function.
Neuroprotective role of uncoupling protein-2 in cerebral stroke
Cerebral Stroke and Mitochondria
Cerebral stroke has been increasingly recognized as a medical emergency because of sudden onset, and activates several intricate and overlapping cell-survival/damaging mechanisms. The damaging mechanisms may proceed through necrotic or apoptotic cell death and dependent on the dynamic interplay among the various cells in affected territory (Mehta et al, 2007). The mitochondrion has emerged as a crucial organelle in regulating cell death during cerebral ischemia, besides playing a major role in the regulation of energy metabolism, ROS production, and Ca2+ homeostasis. Numerous reports suggest that mitochondria is the major source of ROS (Kudin et al, 2008) and produce O2− and hydrogen peroxide (H2O2) radicals (please refer to Figure 1 and its legend for more details about ROS production in the mitochondria) during transient/ permanent focal and global cerebral ischemia (Peters et al, 1998; Murakami et al, 1998; Kim et al, 2002; Dirnagl et al, 1995). This can be supported by the evidence that suggest superoxide dismutase, SOD2−/+ (MnSOD) mice have increased mitochondrial dysfunction, infarct size, neural apoptosis, and neurologic deficit, whereas, SOD2 overexpression have increased mitochondrial tolerance (Silva et al, 2005).
In addition, mitochondria can functions as calcium-buffering organelle. It can efficiently take up Ca2+ to avoid Ca2+ fluctuations and maintain cellular bioenergetics (White and Reynolds, 1997). However, after glutamate excitotoxicity and cerebral ischemia, the intracellular Ca2+ is increased (Castilho et al, 1998; Kristian and Siesjo, 1998). The Ca2+ influx through glutamate-dependent
Possible Role of Uncoupling Protein-2 in Regulation of Reactive Oxygen Species
The brain, owing to its high lipid content and possibly lesser antioxidants, is highly prone to ROS induced damage during cerebral ischemia and reperfusion. The ROS is generated when ETC slows down or stalls during the inadequate supply of adinosine diphosphate (ADP) or dysfunction of ATP synthase. In such conditions, the electron carrying intermediates have an increased chance to transfer single electron to molecular oxygen (O2) and the protons pumped out of the matrix can no longer return back (Mattiasson and Sullivan, 2006). This build up the ΔΨm and the increased ΔΨm reported during reoxygenation is linked to the ROS generation (Skulachev, 1998; Gergely et al, 2002). Interestingly, it has been suggested that uncoupled or noncoupled respiration decreases the intracellular oxygen, and thereby, prevent nonenzymatic one-electron reduction of O2 to O2− (Skulachev, 1996). This hypothesis received attention when it was found that macrophages from
Recently, Mattiasson et al (2003) has proposed that UCP2 could be one of the targets to provide neuroprotection in stroke by alleviating the mitochondrial ROS production and shifting the ROS from mitochondrial to the extramitochondrial space (Andrews et al, 2008). This facilitates the change in cellular redox state, and thus, UCP2 may contribute to redox signaling by direct interaction with the ETC. This can be further supported by the evidence suggesting a significant and specific increase in the ΔΨm and intracellular ROS level in endothelial cells pretreated with antisense oligonucleotides directed against
The UCP2-dependent modulation of ROS production is directly linked to ΔΨm, suggesting that small reduction in the ΔΨm because of mild uncoupling has significant effect on ROS generation (Teshima et al, 2003). Thus, UCP2 appears to decrease the redox pressure on the mitochondria and offer intriguing target to develop potential strategy to provide neuroprotection.
Uncoupling Protein-2 and Mitochondrial Potential
The proton gradient established on transfer of protons during the ETC generates the ΔΨm and provides the driving force to synthesize ATP along with the back transport of protons to the matrix. However, under limiting supply of ADP required for synthesis of ATP through FOF1-ATPase, increased mitochondrial respiration because of supply of nicotinamide adenine dinucleotide from glycolytic pathway, inability of FOF1-ATPase to cope up with the energy demand, and intracellular alkalization as during postischemic reoxygenation may lead to mitochondrial membrane hyperpolarization. (Di Lisa and Bernardi, 1998; Gergely et al, 2002; Back et al, 2000; Iijima et al, 2003b; McLeod et al, 2004; Iijima, 2006). The hyperpolarization during ischemic and long-term hypoxic condition is associated with increased ROS production (Nicholls and Ward, 2000) and mitochondrial Ca2+ accumulation (Smith et al, 2003). Interestingly, the mild uncoupling because of FCCP reduced the ROS generation besides decreases the ΔΨm (Yu et al, 2006). In addition, the pharmacologic induction of mitochondrial uncoupling by 2,4-dinitrophenol 1 h after reperfusion reduced infarct volume by 40% after 2 h of focal cerebral ischemia (Maragos and Korde, 2004). Emerging evidence suggest that UCP2 decreases the ΔΨm by transporting the protons back to the matrix. This has been linked to reduction in ROS generation (Arsenijevic et al, 2000; Mattiasson et al, 2003). Besides decreasing ΔΨm, UCP2 maintained the ΔΨm after oxygen glucose deprivation in primary cortical neurons overexpressing UCP2 in comparison to depolarization in control cells (Mattiasson et al, 2003). In addition, the induction of UCP2 in subpopulations of neuron and microglia after brain lesion is inversely correlated with caspase-3 activation (Bechmann et al, 2002), which suggests the role of UCP2 in regulation of cell death by stabilizing ΔΨm (Nicholls and Ward, 2000; Sullivan et al, 2003; Diano et al, 2003). These evidence, therefore, provide justification to target UCP2 for neuroprotection in excitotoxicity, stroke, and related conditions.
Uncoupling Protein-2, Adinosine Triphosphate, and Mitochondrial Proliferation
The UCP2-dependent transport of protons dissociate oxidation from phosphorylation (ATP production) and dissipates energy as heat. This makes the condition a little more complicated after cerebral ischemia where ATP levels are reported to decrease (26%), although, likely recover to normal levels (80%) during the initial 4 h of reperfusion (Sun et al, 1995). The depletion of ATP during cerebral ischemia depends on the duration and severity of ischemic stress. Thus, profound ischemia lasting for a certain time, impair the ionic homeostasis and consecutively affect the cellular activity. Interestingly, it has been reported that despite the large increases in UCP expression, ATP production was enhanced in oxidative tissues (Short et al, 2001) and decreased intracellular ATP levels were accompanied by decreased
Uncoupling Protein-2 and Calcium
The excitotoxic elevation of intraneuronal Ca2+ is one of the major events during glutamate excitotoxicity and cerebral ischemia in mediating brain injury. The elevated intracellular Ca2+ is readily taken up by mitochondria through the electrophoretic channel called uniporter, when electrochemical gradient is established across IMM. The mitochondrial Ca2+ is effluxed by Ca2+/2H+ and/or Ca2+ /3Na+ exchanger depending on the ΔΨm as decrease of ΔΨm promotes Ca2+ release, whereas elevation of ΔΨm favors Ca2+ influx. However, vast mitochondria Ca2+ sequestration contributes to the formation of MPTP under cerebral ischemic condition. Moreover, mitochondrial Ca2+ elevation also stimulate the ROS production and FFA, which also promote the opening of MPTP. The MPTP allows free movement of apoptogenic molecules and as a result can activate cell-death cascades. Therefore, Ca2+ signals that are converged in mitochondria seem to regulate mitochondrial-dependent apoptotic cell death.
It is proposed that UCP2-mediated decrease of ΔΨm may be involved in the reduction of mitochondrial Ca2+ and production of ROS (Horvath et al, 2003), which subsequently can reduce the probability of MPTP and activation of cell-death cascades (Richard et al, 2001; Mattiasson et al, 2003). The UCP2-mediated potential drop, although may not be significant enough to affect the Ca2+ sequestration but may be advantageous to reduce the rate of Ca2+ uptake (Nicholls, 2002; Mattiasson and Sullivan, 2006). Therefore, it appears reasonable that UCP2-induced decrease of ΔΨm could limit the overloading of mitochondria with Ca2+, and hence decrease the potential for apoptotic events (Horvath et al, 2003). However, contradictory results existed (Trenker et al, 2007). Obviously, additional studies are necessary to further approve the importance of UCP2 in cellular signaling. Nevertheless, modulation of UCP2 may seem imperative in limiting the mitochondrial dysfunction and subsequent cell death in various neurodegenerative diseases including cerebral ischemia (Mattiasson et al, 2003).
Uncoupling Protein-2 and Neuronal Activity
In addition to the regulation of various cellular aspects of autonomic, endocrine, and metabolic processes, it has been proposed that the uncoupling property of UCP2 may be important in temperature regulation in the microenvironment of presynaptic terminals (Fleury et al, 1997; Horvath et al, 1999). This appears notable as UCP2 was reported to be localized at axons and axon terminals that are actively involved in synaptic vesicle formation, trafficking, and finally neurotransmitter release (Horvath et al, 1999). Moreover, the significant increase in the number of neuronal axons in the ischemic periphery of multiple infarction cases together with increase in UCP2 expression highlight the importance of UCP2 in such mechanisms (Nakase et al, 2007). The thermogenic capability of UCP2 although is not clear, however, the cells expressing c-fos after cold exposure are innervated by UCP2 containing axons (Horvath et al, 1999). Uncoupling protein-2 expression might be under the control of peroxisome proliferator-activated receptor-β (PPARβ; Aubert et al, 1997; Wang et al, 2003) as the expression of UCP2 was not upregulated in PPARβ knockout mice after cerebral ischemia (Arsenijevic et al, 2006). PPARs are ligand-activated transcription factors that regulate the expression of genes involved in fatty acid uptake and oxidation, lipid metabolism, and inflammation by binding to specific response elements within promoters (Kersten et al, 2000). Among the different types (α, β, and γ) of PPARs, PPARβ is the only one widely expressed in neurons and oligodendrocytes (Cullingford et al, 1998; Vanden Heuvel, 2007). The deletion of
Cerebral ischemia and diabetes
The consequence of stroke is often enhanced by risk factors such as diabetes, which increases the incidence of stroke in particular the ischemic stroke by 4 to 12 folds (Bemeur et al, 2007). In addition, between 20% and 50% of all patients presenting with stroke may be hyperglycemic at the time of admission (Scott et al, 1999). The neuronal glucose level in diabetic hyperglycemia is suggested to increase up by fourfold (Tomlinson and Gardiner, 2008), and increased glucose level or preischemic hyperglycemia is negatively related to stroke outcome (Li et al, 1994, 2000; Parsons et al, 2002). Studies have suggested that acute hyperglycemia and chronic diabetes can aggravate brain damage because of transient focal or forebrain ischemia (Nedergaard, 1987; Siesjo et al, 1996; Li and Siesjo, 1997; Li et al, 1998; Gisselsson et al, 1999) by glutamate excitotoxicity (Gaitonde et al, 1987; Li et al, 2000) and increased ROS generation in metabolically challenged tissue (Nelson et al, 1992, Nishikawa et al, 2000; Muranyi et al, 2006). The ROS generation is suggested to be a connecting link between high glucose, mitochondrial dysfunction, and apoptosis (Russell et al, 2002). Similarly, the excessive glucose supply is suggested to upregulate the glucose transporters and ΔΨm (hyperpolarization), which might be detrimental to the brain cells (Mastrocola et al, 2005). In addition, an evolution of infarction is accompanied by repeated waves of spreading depression. The consequence of these effects is the extension of core region or conversion of penumbra into the core in a short time.
Uncoupling Protein-2 and Diabetic Cerebral Ischemia
The brain, owing to high energy demands, is exclusively dependent on glucose for energy production. However, the persistent or regular episodic increase in glucose level, as in the case of diabetic hyperglycemic, may be damaging to neurons (Tomlinson and Gardiner, 2008). Additionally, the restoration of blood circulation after cerebral ischemia under hyperglycemic condition, leads to pathophysiologic manifestation of increased glucose such as increased ROS generation and mitochondrial dysfunction in the neurons. It has been suggested that cellular energy and mitochondrial membrane function in response to glucose are regulated by UCPs (Marti et al, 2001; Brown et al, 2002). The property of UCP2 to leak protons across the IMM may reduce ROS and mitochondrial potential. These mechanisms (ROS and potential alteration) appear to be involved in damage progression in cerebral ischemia under diabetic condition (Bemeur et al, 2007). The overexpression of UCP2 negatively regulates glucose-stimulated insulin secretion in β-cell of pancreas and the amount of ATP generated from glucose (Zhang et al, 2001). The expression of UCP2 is stimulated
Although UCP2 may offer neuroprotective effect after stroke with or without preexisting hyperglycemic condition, glucose sensing negatively controlled by UCP2, reduces release of insulin from β-cell and may increase the blood glucose level. Recently, it has been reported that an increase in UCP2 in the brain makes the glucose-excited neurons less sensitive to glucose (Parton et al, 2007). The study conducted on wild-type and
Conclusions
The emerging evidence suggests that UCPs play a very important role in mitochondrial functioning by acting as proton transporters. Their expression, activity, and distribution in various types of tissue appear to be a critical determinant in number of diseases including cerebral stroke. Recent studies have proposed the role for UCP2 in regulation of ΔΨm, ROS, calcium, and neuronal activity, which seems to have significant impact on cellular homeostasis and neuroprotection. These factors support the fact that exploring the potential role of UCP2 might be beneficial to develop the appropriate strategy to reduce progression of damage after stroke and other neurodegenerative diseases. In addition, continuous research is imperative to translate the promising role of UCP2 into suitable therapeutic strategy.