
Abstract
Although interleukin-6 (IL-6) has various neuroprotective effects against cerebral ischemia, the topographic distribution and cellular source of IL-6 after cerebral ischemia remain unclear. In the current study, the localization of IL-6 protein was immunohistochemically examined in rats after 3.5, 12, 24, and 48 hours of reperfusion after 1.5 hours of middle cerebral artery occlusion. Middle cerebral artery occlusion was induced by the intraluminal suture method. The specificity of the anti–IL-6 antibody used in the current study was confirmed by Western blot analysis and an immunoabsorption test. To identify the cellular source, lectin histochemical study and immunohistochemical study with microtubule-associated protein-2, ED1, and glial fibrillary acidic protein also were carried out. The sham group did not show any clear IL-6 immunoreactivity. After 3.5 hours of reperfusion, IL-6 immunoreactivity was first detected on the reperfused side, and it was upregulated, especially in the periinfarct region, after 24 hours of reperfusion. Also, IL-6 was expressed after 3.5 hours of reperfusion in the contralateral cerebral cortex and bilateral hippocampi. Double staining showed that the cells containing IL-6 were neurons and round-type microglia, not astrocytes. The current findings suggest that IL-6 expression in ischemically threatened neurons and reactive microglia is closely associated with brain tissue neuroprotective mechanisms against cerebral ischemia.
Thrombolytic therapy with tissue plasminogen activator in the acute phase of cerebral infarction has been demonstrated to be effective (NINDS, 1995), although several problems remain unresolved. Reperfusion injury is a major problem that needs to be resolved, in addition to bleeding complications, and an additional regimen of treatment is required to reduce reperfusion injury of cerebral neurons. Reperfusion after ischemia induces inflammatory response, which is characterized by local expression of various cytokines in the brain (DeGraba, 1998). Tumor necrosis factor-α and interleukin (IL)-1β, detected as early as 1 hour after the onset of ischemia, have been suggested to induce neuronal injury (Saito et al., 1996; Uno et al., 1997; Zhang et al., 1998; Davies et al., 1999). Interleukin-1 receptor antagonist and blocking of tumor necrosis factor-α have been found to reduce infarct size in focal cerebral ischemia in rats and are thought to have neuron-protective effects (Loddick et al., 1996; Barone et al., 1997).
First identified as a B-cell-stimulating factor (Kishimoto, 1989), IL-6 is one of the major cytokines in the CNS and has been implicated in cellular responses in cerebral ischemia (Akira et al., 1993). Increases in serum and CSF levels of IL-6 have been reported in acute stroke patients (Tarkowski et al., 1995). Consistent with these clinical data, rapid and marked elevation of IL-6 bioactivity and mRNA has been detected in animal models of cerebral ischemia (Wang et al., 1995; Saito et al., 1996; Loddick et al., 1998). Recent in vivo and in vitro studies demonstrate that IL-6 exerts a neuroprotective effect against several types of brain injury, including cerebral ischemia (Hama et al., 1991; Toulmond et al., 1992; Yamada et al., 1994; Hirota et al., 1996). Exogenous IL-6 injection prevents learning disability and delayed neuronal loss in gerbils after transient forebrain ischemia (Matsuda et al., 1996). In a focal ischemia model in rats, exogenous IL-6 injection reduces infarct size (Loddick et al., 1998).
Despite these beneficial effects of exogenous IL-6, expression of IL-6 protein in the brain after cerebral ischemia is not fully understood. In the current study, we elucidate the chronologic and topographic distribution of IL-6 protein by immunohistochemical study in a transient focal cerebral ischemia model in rats.
MATERIALS AND METHODS
Animal preparation
The protocol described here had received prior approval as meeting the Animal Experimental Guidelines of Keio University School of Medicine. Adult male Sprague-Dawley rats (Japan Laboratory Animals, Tokyo, Japan) weighing 290 to 330 g were divided into two groups: a middle cerebral artery occlusion (MCAO) group (n = 36) and a sham group (n = 7). The animals in the MCAO group were anesthetized with a mixture of 1.0% to 1.5% halothane and 30% oxygen170% nitrogen during the operation. A temperature probe (TD-300, Shibaura Electronics, Tokyo, Japan) was inserted into the rectum, and a heat lamp was used to maintain rectal temperature at 37.0° to 37.5°C. Middle cerebral artery occlusion was induced by the intraluminal filament technique of (Koizumi et al. 1986), as modified by Belayev et al. (1996). In brief, a midline surgical incision was made to expose the right common, internal, and external carotid arteries. The distal portion of the external carotid artery was ligated, and the common and internal carotid arteries were temporarily occluded with clips (Sugita aneurysm clips, Mizuho Ikakogyo, Tokyo, Japan). A small incision then was made in the proximal portion of the external carotid artery, and a 3-0 nylon monofilament suture (Matsuda Ikakogyo, Tokyo, Japan) was inserted into the internal carotid artery. The distal segment of the suture had been coated with poly-l-lysine solution (Sigma, St. Louis, MO, U.S.A.) to ensure consistent ischemia in the brain. The filament was advanced until it obstructed the origin of the right middle cerebral artery (MCA). After the intraluminal filament had been fixed in position, the neck incision was closed with silk thread. The animals then were allowed to recover from the anesthesia and were evaluated for the extent of their neurologic deficits 1 hour after MCAO. The deficits were graded as severe, moderate, mild, or absent according to the method of neurologic examination devised by Bederson et al.(1996). In brief, animals that consistently circled toward the left (paretic) side were graded as 3. Rats that consistently showed reduced resistance to lateral push toward the left side with no circling movement were graded as 2. Rats with any amount of consistent forelimb flexion and no other abnormalities were graded as 1. Rats with no neurologic deficits were graded as 0. Only animals with grade 2 or 3 deficits were used for further study in the form of the MCAO group (n = 25), since these animals were expected to invariably have a large infarct area in the territory of the MCA (Bederson et al., 1986). After 90 minutes of MCAO, the rats were reanesthetized with the same anesthetic combination as described earlier, and the intraluminal suture was carefully removed. The common and internal carotid arteries then were inspected to ensure the return of good pulsation. The neck incision was closed with a silk suture, and the animals were allowed to survive with free access to food and water. In the sham group, the neck incision was made only to expose the right common, external, and internal carotid arteries, and a suture was not inserted into the artery. Other procedures were identical to those in the MCAO group.
Western blot analysis
The animals were decapitated after 24 hours of reperfusion (n = 3) or 24 hours after the sham operation (n = 2). Samples were taken from the entire cerebral cortex on the occluded side. Tissue was homogenized in a Teflon-coated homogenizer containing buffer (0.32 mol/L sucrose, 1 mmol/L ethylenediamine tetraacetic acid, 5 mmol/L Tris-HCl [pH 7.4], 0.1 mmol/L phenylmethylsulfonylfloride, 10 μmol/L leupepsin, 10 μmol/L pepstatin A, and 1 mmol/L beta-mercaptoethanol). Samples containing 50 μg of protein each or recombinant IL-6 protein (Upstate Biotechnology, Lake Placid, NY, U.S.A.) were subjected to 10% Polyacrylamide gel electrophoresis in the presence of 0.1% sodium dodecyl sulfate. Protein bands were transferred from the gel to a nitrocellulose membrane (Hybond ECL, Amersham, Buckinghamshire, England) by using a transfer apparatus and a blotting buffer, including 0.192 mol/L glycine and 5% methanol (pH 8.4). The membrane then was incubated with 1:200 dilution of an affinity purified polyclonal rabbit anti-mouse IL-6 antibody (Genzyme Corporation, Cambridge, MA, U.S.A.). Next, the membrane was reacted with horseradish peroxidase–linked anti-rabbit antibody (Amersham). Protein bands were detected by an enhanced chemiluminesence method (ECL kit, Amersham). The protein concentration of each sample was determined with a Bio-Rad protein assay kit (Bio-Rad Laboratories, Hercules, CA, U.S.A.) using bovine serum albumin as the standard.
Immunohistochemistry
Animals were anesthetized by intraperitoneal injection of pentobarbital 3.5 hours (n = 5), 12 hours (n = 5),24 hours (n = 7), and 48 hours (n = 5) after the start of reperfusion or 24 hours after the sham surgery (n = 5). They then were perfused with 200 mL of 50 mmol/L phosphate-buffered saline (PBS), pH 7.4, followed by 200 mL of 4% freshly depolymerized paraformaldehyde in 0.1 mol/L phosphate buffer, pH 7.4. The brains were removed and postfixed for 6 hours in 4% paraformaldehyde in phosphate buffer at 4°C before cryoprotection by sequential bathing in 10%, 15%, 20%, and 30% sucrose. The brains then were frozen in powdered dry ice, and consecutive 20-μm thick coronal sections (16-μm thick ones for the double staining) were prepared on a cryostat (Cryocut 1800, Leica Instruments GmbH, Nussloch, Germany). The sections were thaw-mounted onto gelatin-chrome alum-coated slides and stored at −30°C. They subsequently were rehydrated and washed in PBS (pH 7.4) for 5 minutes, quenched with 0.3% H2O2 in PBS for 20 minutes to block endogenous peroxidase, and washed three times for 5 minutes each PBS. Next, they were preincubated for 30 minutes in a solution containing skimmed milk to block nonspecific binding, and the sections were incubated overnight at 4°C with a 1:100 dilution of polyclonal rabbit anti-mouse IL-6 antibody, which was the same as that used in the Western blot study. The slides were stained with an avidin-biotinylated enzyme complex system (Vectastain ABC Elite Kit, Vector Laboratories, Burlingame, CA, U.S.A.). The immunoreactive product was visualized with the diaminobenzidine reaction. To examine the specificity of the immunoreactivity, the primary antibody was omitted to provide a nonspecific control. In addition, immunoabsorption testing was carried out by using a mixture of primary antibody and recombinant mouse IL-6 control protein (concentration 5 ng/μL).
Absolute immunoreactive cell counts (per 0.15 mm2) were made in a blind manner by one of the authors (K. T.) with an ocular micrometer (Nikon, Tokyo, Japan) attached to a light microscope at × 160 magnification. Based on a rat brain atlas (Paxinos and Watson, 1986), the ischemic core (parietal cortex area 2), and the periischemia region (hindlimb area of the cortex) in the cerebral cortex, the contralateral cerebral cortex (parital cortex area 2, corresponding to the ischemic core on the surgery side) and the corresponding areas of the sham group at the level of the globus pallidus (bregma −0.8 mm) were selected for cell counting. Counting was performed on three consecutive slices, and the mean value for each area was calculated.
All data are expressed as mean ± SD. The data were tested by analysis of variance followed by post hoc analysis with the Bonferroni correction for multiple simultaneous comparisons.
In additional histochemical studies, the following antibodies were applied to adjacent sections and detected with diaminobenzidine to identify the cellular source of IL-6. Monoclonal anti-microtubule associated protcin 2 (MAP2) antibody (dilution 1:500, Boehringer Mannheim, Philadelphia, PA, U.S.A.) and anti-glial fibrillary acidic protein (GFAP) (dilution 1:300, Dako, Glostrup, Denmark) were used to identify neurons and astrocytes, respectively. Monoclonal anti-ED1 antibody (dilution 1:500; Serotec, Oxford, England) was used as a panmacrophage/monocyte maker. Microglia also were histochemically identified by using isolectin-B4 from Griffonia simplicifolia seeds (GSAI-B4, Sigma), as described by Streit et al. (1987, 1990). Briefly, the sections were incubated overnight at 4°C with isolectin (concentration 20 μg/mL) in PBS containing divalent cations. Adjacent sections also were stained with cresyl violet for conventional histologic examination.
Double-staining immunohistochemistry
Double staining with GSAI-B4/IL-6 and GFAP/IL-6 was performed overnight at 4°C by using a mixture of two corresponding primary antibodies having the same concentration as stated earlier. A fluorescein isothiocyanate–conjugated anti-mouse IgG antibody (Amersham) was used as the secondary antibody for GSAI-B4 and GFAP. On the other hand, IL-6 was detected by a Texas Red–conjugated anti-rabbit IgG antibody (Amersham). Double staining with MAP2/IL-6 was performed by first treating with anti-MAP2 antibody and then staining with fluorescein isothiocyanate. The sections then were washed, and anti-IL-6 antibody was applied and stained with diaminobenzidine. These sections were examined by fluorescent microscopic study to detect fluorescein isothiocyanate and Texas Red.
RESULTS
Western blot analysis
A single band with a molecular mass of 21 kD was detected for recombinant IL-6 protein (Fig. 1A). A similar band having the same molecular mass was detected in the sample taken from the animals subjected to 24 hours of reperfusion after ischemia, but not in the sample from the sham group. The identical band also was detected in the sample taken from the contralateral cortex at 24 hours of reperfusion.
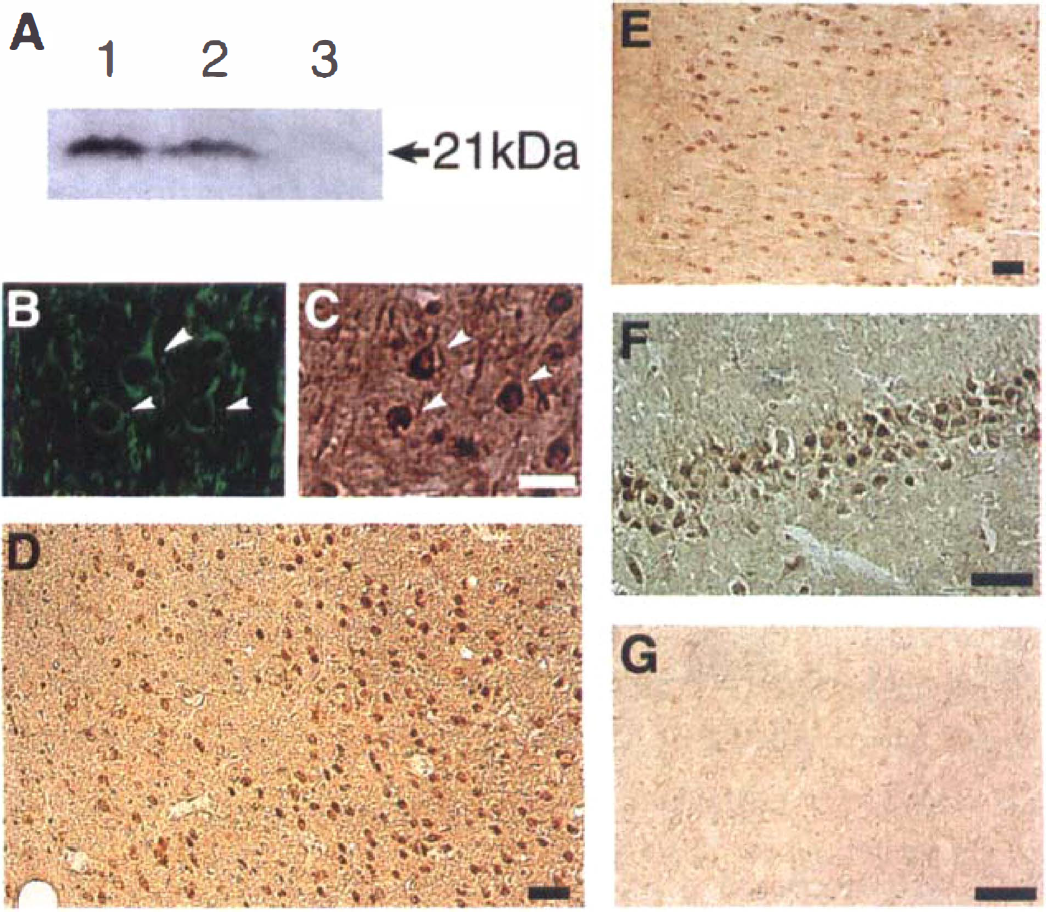
(A) Western blot analysis of interleukin (IL)-6. The samples were prepared from recombinant IL-6 (lane 1), the cerebral cortex on the surgery side at 24 hours of reperfusion after 1.5 hours of middle cerebral artery occlusion (MCAO) (lane 2), and 24 hours after sham-operation (lane 3).
Cresyl violet staining
Cresyl violet staining clearly demonstrated mild ischemic changes, such as shrunken cell bodies with triangular cytoplasm, in most of the neurons in the ischemic core after 12 hours of reperfusion. A consistent histologic infarct pattern subsequently was observed in the frontoparietal somatosensory cortex and the caudate-putamen on the occluded side, as reported by Belayev et al. (1996). After 24 hours of reperfusion, clear neuronal damage with necrotic process was observed in the infarct region, and after 48 hours of reperfusion, complete necrotic changes and edema were accompanied by many cells with small nuclei, indicative of glial cells.
Immunohistochemistry
Representative photomicrographs of sections stained with anti-IL-6 antibody are shown in Figs. 1 and 2. No definite IL-6 protein immunoreactivity was detected in any regions of the cerebral cortex in the sham group (Figs. 2A and 2F). In contrast, IL-6 immunoreactivity was clearly detected after 3.5 hours of reperfusion in both the ischemic core and the periischemia region (Figs. 2B and 2G), so that the boundary zone between the ischemic core and the periischemia region was unclear. In the periischemia region and the contralateral side, IL-6 immunoreactivity subsequently increased slightly and peaked at 12 to 24 hours of reperfusion (Figs. 1E and 2G through 2J). In the ischemic core, on the other hand, IL-6 immunoreactivity peaked at 3.5 hours of reperfusion and decreased subsequently until 48 hours (Figs. 2B through 2E). The boundary zone between the infarct and the periischemia regions was most prominent at 24 hours of reperfusion (Fig. 1D).
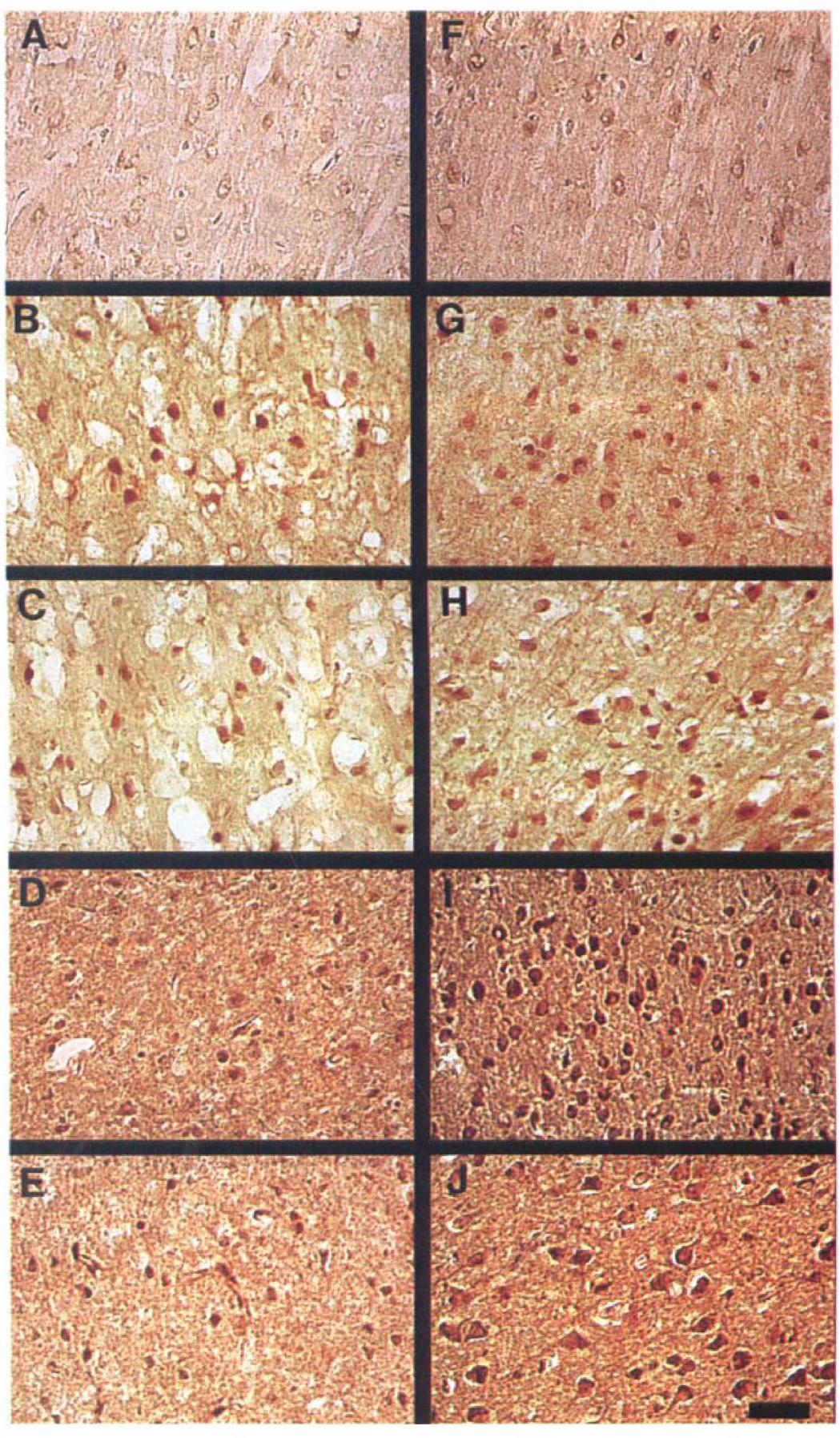
Representive photomicrographs showing immunoreactivity with anti-IL-6 antibody in the ischemic core
There were two types of IL-6–immunoreactive cells. One type was pyramid-shaped neurons, and their immunoreactivity was prominent in the cytoplasm. Double staining showed that they stained simultaneously with anti-MAP2 antibody (Figs. 1B and 1C). Most of the IL-6–immunoreactive cells in the contralateral cerebral cortex were pyramidal neurons that persisted throughout the entire observation period, and they increased and peaked at 24 hours of reperfusion (Fig. 1E). Hippocampal neurons (CA1, CA2, CA3, and dentate gyrus) also expressed IL-6 protein bilaterally from 12 to 24 hours of reperfusion onward (Fig. 1F).
The other type was round, activated microglia with few processes, and their immunoreactivity was observed in the cytoplasm, the nuclei, or both. Double immunostaining demonstrated that the round-type microglia that stained with GSAI-B4 were immunoreactive for IL-6 (Figs. 3A through 3C).
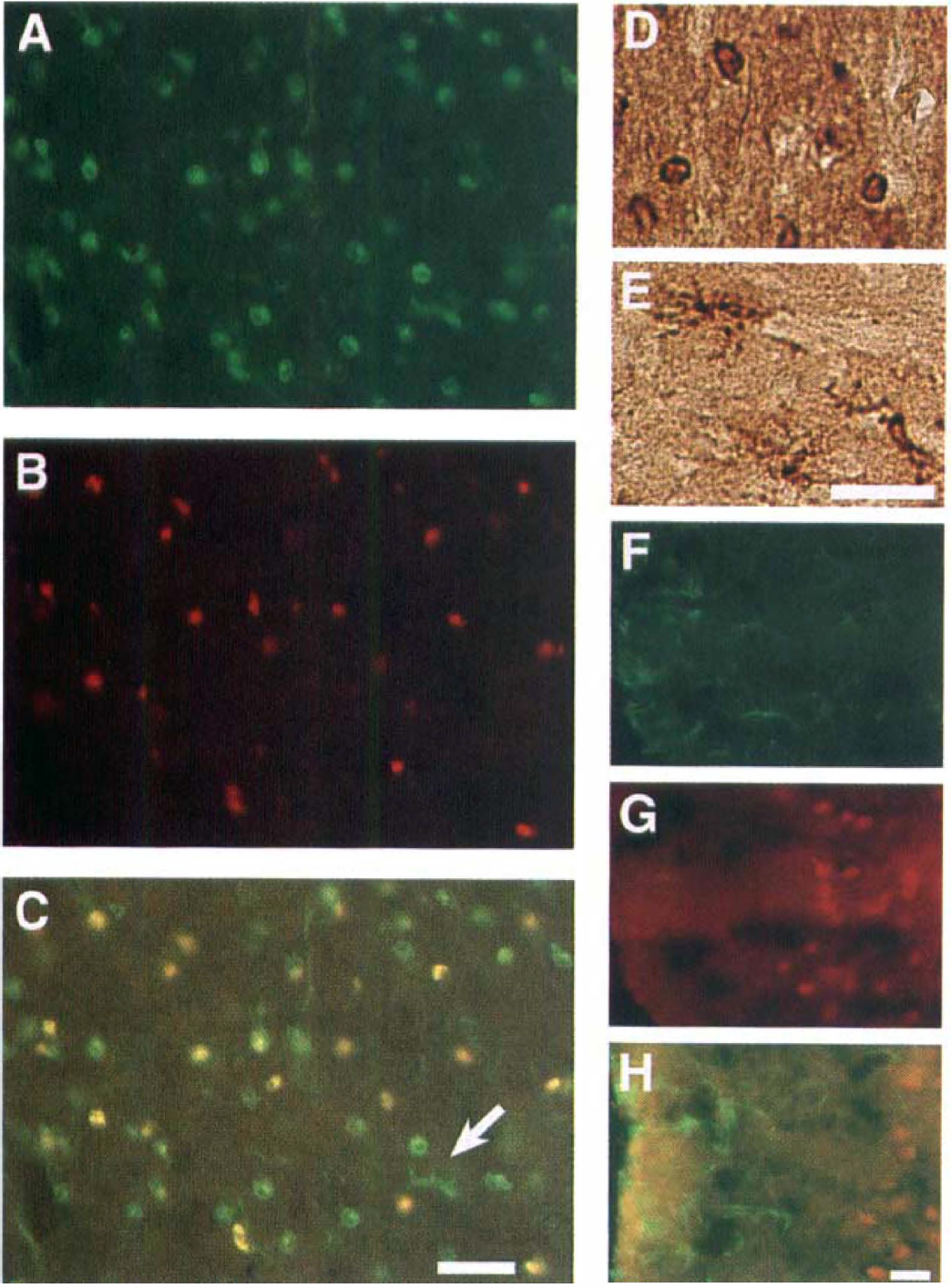
(A–C) Double immunohistochemical staining for GSAI-B4 and IL-6 in the ischemic core after 24 hours of reperfusion after 1.5 hours of MCAO.
Based on these histologic observations, the IL-6–immunoreactive cells in the ischemic core at 3.5 hours of reperfusion were confirmed to be neurons and activated microglia (Fig. 2B). Round-type reactive microglia were observed from 3.5 hours of reperfusion onward (Fig. 3D). The cresyl violet staining and immunohistochemical study with anti-MAP2 antibody showed that several neurons with apparent ischemic changes were seen in the ischemic core from 12 hours of reperfusion onward. Some IL-6–immunoreactive neurons were shrunken and beaded at 24 hours of reperfusion, but they were no longer visible at 24 to 48 hours of reperfusion (Figs. 2D and 2E). Because there were fewer neurons, the proportion of IL-6–immunoreactive microglia in the ischemic core was relatively high at 24 to 48 hours of reperfusion (Figs. 2D and 2E).
In the periischemia region, the IL-6–immunoreactive cells from 3.5 to 48 hours of reperfusion were neurons and the round, reactive microglia (Figs. 20 through 21). Staining with OSAI-B4 detected highly ramified microglia with large, thick processes (Fig. 3E), but they did not express IL-6 (Fig. 3C).
Some ED1-positive small, round cells appeared at 24 hours of reperfusion and gradually increased. These cells were observed mainly in the ischemic core, showing clearly different distribution from that of IL-6 immunoreactivity. Astrocytes labeled with anti-GFAP antibody were increased near the surface of the ischemic cortex from 24 hours of reperfusion onward (Fig. 3F). Double-staining immunohistochemical examination revealed that the IL-6-positive cells were not co-localized with the GFAP-staining cells (Figs. 3F through 3H), indicating that the IL-6–immunoreactive glial cells were intrinsic reactive microglia, not astrocytes. Morphologically, only the round microglia, and not the highly ramified microglia, expressed IL-6 protein.
Immunoreactivity for IL-6 was not detected when the anti-IL-6 antibody was preincubated with recombinant mouse IL-6 (Fig. 10) or when the primary antibody was omitted.
Figure 4 summarizes the number of IL-6-immunoreactive cells in three different regions: the ischemic core, the periischemia region, and the contralateral cerebral cortex. In the sham group, the number of immunopositive cells was similar in each region. After 3.5 hours of reperfusion, the number of immunopositive cells was markedly increased in all regions examined, and the increases in the periischemia region were marked and peaked at 24 hours of reperfusion. The number of IL-6–positive cells remained significantly higher in each region compared with the sham group at 48 hours of reperfusion. Table 1 summarizes the proportion of IL-6–immunoreactive neurons and microglia.
Proportion of IL-6 immunoreactive cells

IL, interleukin; N, neurons; M, microglia; N > M, neurons are more predominant than microglia; N = M, neurons and microglia almost equally exist; N < M, microglia are more predominant than neurons.
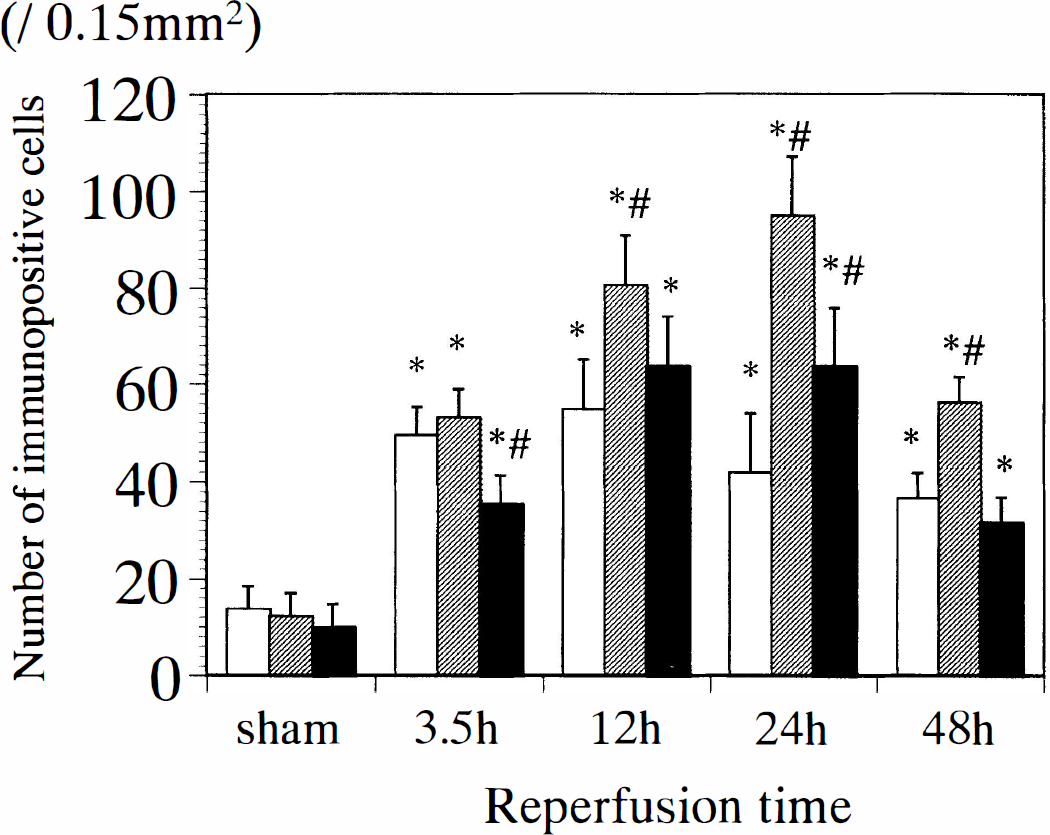
Temporal profiles showing the number of IL-6–immunoreactive cells in three different regions of the cerebral cortex. The ischemic core (blank columns) was located in the parietal cortex area 2 (Paxinos and Watson, 1986) and the periischemia region (shaded columns) in the hindlimb area of the cortex (Paxinos and Watson, 1986). The contralateral cortex corresponds to the parietal cortex area 2 (solid columns) on the nonsurgery side of the MCAO group. *P < 0.01 versus sham group, #P < 0.01 versus the ischemic core.
DISCUSSION
The following summarizes the major findings of the current study: (1) there was no clear expression of IL-6 protein in the brain under the control conditions; (2) IL-6 protein was clearly expressed in the neurons and the round-type reactive microglia during the reperfusion period after focal cerebral ischemia; (3) IL-6 expression was predominantly upregulated in the periischemia region of the cerebral cortex, peaking at 24 hours of reperfusion; and (4) IL-6 immunoreactivity also was observed in the contralateral cerebral cortex and bilateral hippocampi, which were not perfused by the occluded MCA.
Interleukin-6 is a glycoprotein with a molecular mass in the 21- to 28-kD range, depending on the cell source and mode of preparation (Akira, 1993; Heinrich et al., 1998). Since Western blot analysis in the current study detected a single band at 21 kD in the samples from reperfused rat brain, which was identical with that of recombinant mouse IL-6, the anti-mouse IL-6 antibody used here can be assumed to represent an appropriate marker for the specific detection of IL-6 protein in rat brain. Interleukin-6 is a pleiotrophic cytokine that plays a crucial antiinflammatory role in both local and systemic acute inflammatory responses by controlling the level of proinflammatory cytokines (Xing et al., 1998), and is considered to be one of the important neurotrophic factors. A significant increase in IL-6 bioactivity and IL-6 mRNA has been reported beginning 2 hours after cerebral ischemia in experimental animal models (Wang et al., 1995; Saito et al., 1996; Loddick et al., 1998). Despite these important findings, the cellular localization and temporal profile of IL-6 protein after focal cerebral ischemia have not been fully elucidated.
According to the current findings, the main localization of IL-6 protein is in neurons. Distribution of IL-6 mRNA has been demonstrated in hippocampal neurons (Schöbitz et al., 1992; 1993). Similarly, we have found that cerebral neurons in both the cerebral cortex and hippocampus express IL-6 protein after transient forebrain ischemia in gerbils (Suzuki et al., 1999). In the current study, double staining for MAP2 (Li et al., 1998) revealed that IL-6 protein was expressed in cerebral and hippocampal neurons. In the ischemic core, some neurons express IL-6 from 3.5 to 24 hours of reperfusion, although clear ischemic neuronal damage was first observed after 12 hours of reperfusion, and most neurons had died and disappeared by 48 hours of reperfusion. Neurons in the periischemia region, on the other hand, expressed IL-6 protein and MAP2 from 3.5 to 48 hours of reperfusion, and were spared ischemic damage. We therefore think that the expression of IL-6 in the neurons observed in the periischemia region may have been crucial to the survival of these neurons. Lemke et al. (1998) found that neurons can produce IL-6 protein under pathologic conditions. Based on all of these findings, we speculate that neurons may be able to produce IL-6 in response to ischemic stress and that IL-6 may act as a neuroprotective mediator in a paracrine manner.
The second source of IL-6 is thought to be microglia. The reaction of microglial cells to ischemia is rapid, and several morphologic types of reactive microglia have been detected with GSAI-B4 (Streit and Kreuzberg, 1987). Although GSAI-B4 also stains cells of the monocyte lineage and endothelial vessels, we consider that GSAI-B4 mainly detected microglia in the current study because of the distribution and morphologic features of the IL-6–positive cells. In the current study, round-type reactive microglia were observed in the ischemic core from 3.5 to 48 hours of reperfusion. By contrast, highly ramified microglia were observed mainly in the periischemia region. Cytotoxic and inflammatory reactions caused by microglial activation have been implicated in the pathogenesis of ischemia/reperfusion brain injury (Kato et al., 1996). In fact, IL-1β and tumor necrosis factor-α expressed by glial cells aggravate ischemic neuronal damage (Barone et al., 1997; Davies et al., 1999). On the other hand, we speculate that activation of IL-6–positive microglia may be associated with neuroprotective mechanisms of brain tissue against cerebral ischemia. Morphologically, only round-type microglia expressed IL-6, and highly ramified microglia did not. The mechanisms underlying the expression of IL-6 in a particular type of microglia currently are unknown.
Whereas culture studies show that astrocytes represent the main source of IL-6 production (Sawada et al., 1992; Saito et al., 1993; Schwanger et al., 1997), the current results demonstrate that the increased activated astrocytes around the surface of the ishemic cortex did not express IL-6. To accurately identify the role of astrocytes in IL-6 production, further experiments with longer reperfusion periods may be required. Although ED1-immunopositive cells may not fully correspond to blood-borne leukocytes (Zhang et al., 1997), the current study reveals that these cells do not express IL-6 protein.
Expression of IL-6 also was enhanced in the contralateral cerebral cortex and bilateral hippocampal neurons, which were not perfused by the occluded MCA. Most of the IL-6–immunoreactive cells in these areas were found to be neurons in the current study, and microglia activation did not contribute to IL-6 expression. In the permanent rat MCAO model, IL-6 bioactivity also is increased on the contralateral side and peaks at 8 hours after ischemia (Loddick et al., 1998). Because cerebral infarction is known to change CBF in remote regions, the contralateral induction may be induced by ischemia in regions outside of the MCA territory. On the other hand, IL-6 production is regulated by some immediate early genes, such as c-fos and zif268. Expression of their mRNA is rapid and precedes the expression of IL-6 mRNA (Wang et al, 1995). The contralateral induction of c-fos and jun-B mRNA has been well observed in focal cerebral ischemia, and this phenomenon has been considered to result mainly from spreading depression (Kinouchi et al., 1993; Hata et al., 1998). Taken together, we suspect that spreading depression also may be involved in the IL-6 expression outside of the MCA territory. Measurement of CBF and recording of cortical electrical activity are planned as the future study.