
Abstract
In the brain, the expression of the pleiotropic cytokine interleukin-6 (IL-6) is enhanced in various chronic or acute central nervous system disorders. However, the significance of IL-6 production in such neuropathologic states remains controversial. The present study investigated the role of IL-6 after cerebral ischemia. First, the authors showed that focal cerebral ischemia in rats early up-regulated the expression of IL-6 mRNA, without affecting the transcription of its receptors (IL-6Rα: and gp130). Similarly, the striatal injection of N-methyl-
Keywords
Increasing evidence suggests that, in addition to apoptotic and excitotoxic pathways (Choi, 1996), the immune response to tissue damage may also significantly influence the neuronal outcome after ischemic brain injury. For example, the striatal injection of the proinflammatory cytokine interleukin-1β (IL-1β) worsens ischemic brain lesions (Stroemer and Rothwell, 1998), whereas the intracerebral injection of the immunosuppresive cytokine transforming growth factor-β (TGF-β) is neuroprotective in experimental cerebral ischemia (Prehn et al., 1993). Although the involvement of immune/inflammatory cytokines, such as IL-1β, TNF-α or TGF-β, in the ischemic neuronal death has been extensively studied (Loddick et al., 1997; Feuerstein et al., 1998; DeGraba, 1998; Buisson et al., 1998), the role of IL-6 remains to be elucidated.
First identified as a B-cell stimulating factor (Hirano et al., 1986), IL-6 belongs to a subfamily of related hematopoietic cytokines, including leukemia inhibitory factor, ciliary neurotrophic factor, oncostatin-M, cardiotrophin-1 and IL-11. To exert its biological effects, IL-6 first binds to either the soluble or the membrane-associated receptor IL-6Rα. This complex then recruits the signal transducer gp130, a transmembrane glycoprotein common to the IL-6 related cytokines, as an obligatory component for signal transduction. The subsequent dimerization of gp130 activates the Jak/STAT and MAPK pathways, allowing the transcriptional modulation of target genes (Akira, 1997; Heinrich et al., 1998; Hirano, 1998).
The expression of IL-6 in the brain increases in numerous neurological disorders, such as Alzheimer's disease, Parkinson's disease, meningitis, trauma, and stroke (Gruol and Nelson, 1997; Benveniste, 1998). IL-6 has been reported to be up-regulated after focal permanent ischemia in rats (Wang et al., 1995) and transient global ischemia in gerbils (Saito et al., 1996). However, conflicting data have been reported with regard to the potential role of IL-6 production during both acute and chronic insults to the brain. The intracerebral injection of IL-6 in rats reduces the infarcted volume after focal permanent cerebral ischemia (Loddick et al., 1998) and protects striatal cholinergic neurons against N-methyl-
The aim of the present investigation was to assess the possible involvement of IL-6 in cerebral ischemia. First, the expression of IL-6 and its receptors after permanent or transient focal cerebral ischemia in rats was examined. Then, the expression of IL-6 and its signalling receptors after the exposure of brain tissue to NMDA both in vivo and in vitro was investigated. Finally, the authors characterized the effect of IL-6 against the neuronal death induced by either excitotoxic or apoptotic paradigms in murine cortical cultures.
MATERIALS AND METHODS
Polymerase chain reaction and reverse transcriptase system kits were purchased from Promega (Charbonnières, France). Dulbecco's Eagle's minimal essential medium, Poly-D-lysine, cytosine β-D-arabinoside (AraC), horse serum, fetal calf serum, Tween 20, ionomycin (A23187), staurosporine, Extravidin, MAP2 and glial fibrillary acidic protein (GFAP) antibodies and the synthetic IL-6 competitive inhibitor were obtained from Sigma Chemical Co. (L'Isle D'Abeau, France). NMDA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA), 6-cyano-7-nitroquinoxaline-2,3-dione, kainate and (+)5-methyl-10,11-dihydro-5H-dibenzo(a,d)cyclohepten-5,10-imine maleate (MK-801) were from Tocris (Bristol, U.K.). Laminin was from Life Technologies (Cergy Pontoise, France). Mouse recombinant IL-6 (specific activity: 105 U/μg) was purchased from R&D Systems (Oxford, U.K.). Antibodies raised against IL-6Rα and gp130 were from Santa Cruz (Heidelberg, Germany).
In vivo procedures
All experiments were performed on male Sprague-Dawley rats weighing 280 to 320g (CERJ, France). Anesthesia, induced by halothane 4% and maintained at 1–1.5%, was administrated through a vaporizer in a gas mixture of 30% O2:70% N2O.
Middle cerebral artery occlusion
All of the rats used for ischemia procedures were intubated and a femoral artery was cannulated for continuous arterial pressure monitoring and gas analysis. Body temperature was maintained at 37.5°C ± 0.2°C with a heating pad. Focal cerebral ischemia was induced by intraluminal middle cerebral artery occlusion (MCAO) (Longa et al., 1989). Briefly, a midline neck incision was made and the right common carotid artery was exposed. After coagulation of its branches, the external carotid artery was severed distally. A nylon thread (0.18mm in diameter) having a distal cylinder (3mm long and 0.38mm in diameter) of thermofusible glue (3M:3764; Radiospares composantes, Paris, France) was inserted in the lumen of the external carotid artery and pushed into the internal carotid artery up to the origin of MCA. In the case of transient focal ischemia, the nylon thread was removed and cut 90 minutes later, so as to restore blood flow in the MCA. Rats were killed 3, 6, 24 and 72 hours after MCAO, and cortices were isolated for reverse transcription polymerase chain reaction (RT-PCR) analysis.
Excitotoxic lesions
Rats received a unilateral injection (3 μL) into the left striatum (coordinates 0.2 mm posterior, 3 mm lateral, 5.5 mm ventral to the bregma; Paxinos and Watson, 1986) with either vehicle (phosphate-buffered saline [PBS], pH 7.4) or NMDA (75 nmol in PBS, pH 7.4). Five minutes after insertion of the needle, the solution was injected over the next 6 minutes using a Hamilton syringe pump (Carnegie, Stockholm, Sweden) at a rate of 0.5 μL/min. The needle was removed 5 minutes later. Rats were killed after 1, 6 and 24 hours and the striata were collected for RT-PCR analysis.
Cell culture
Mixed cortical neuron and glial cell cultures were prepared from fetal OF1 mice at 15 to 16 days gestation (Rose et al., 1993). After removing the meninges, isolated cerebral cortices were mechanically dissociated in media stock (MS, minimal essential medium with 25 mmol/L glucose). The cell suspension was adjusted with MS supplemented with 2 mmol/L glutamine, 5% fetal calf serum, and 5% horse serum, before plating on a preformed confluent layer of astrocytes. The latter (containing no neurons) was derived from 1- to 3-day-old mice following the same procedure, with the exception that the culture medium was MS supplemented with 2 mmol/L glutamine, 10% fetal calf serum and 10% horse serum.
Near pure neuronal cultures containing <5% glial cells were obtained as described above, but were plated on a poly-D-lysine and laminin layer.
All cultures were kept at 37°C in a humidified 5% CO2 atmosphere. Non– neuronal cells proliferation was halted by adding 10 μmol/L AraC after 3 days in vitro for near pure neuronal cultures and 7 days in vitro for mixed cultures. Three days before the experiments, culture media were exchanged for MS +10 μmol/L glycine.
Semiquantitative reverse transcription polymerase chain reaction
Total RNAs were isolated from rats cerebral cortices and striata or murine cell cultures through the use of the RNaxel kit (Eurobio, Les Ulis, France), or RNeasy kit (Qiagen, Courtaboeuf, France), respectively. One μg of total RNAs was reverse transcribed into cDNA using poly(dT) oligonucleotides that allow a specific targeting of poly(A+)mRNA. For semiquantitative experiments with β-actin as a housekeeping gene, an aliquot of the cDNA library (1 μL) was amplified by PCR with specific sets of oligonucleotides (Table 1). Amplification conditions using a Thermolyne thermocycler (Amplitron II; Barnstead, Dubuque, IA, U.S.A.) were the following: 95°C-30 seconds, 55°C-30 seconds, and 72°C-1 minute for β-actin, IL-6 and gp130; and 95°C-30 seconds, 58°C-30 seconds, and 72°C-1 minute for IL-6Rα. The number of cycles was chosen corresponding to 50% of the saturation curve. Amplified products were separated by agarose gel electrophoresis and visualized by ethidium bromide staining. Analysis was based on visual inspection of the agarose gel electrophoresis.
PCR oligonucleotides used for 1L-6, IL-6Rα, gp130, and β-actin

PCR product sizes are expressed in base pairs (bps). For each set of primers, upper and lower sequences respectively stand for sense and antisense probes. PCR, polymerase chain reaction.
Immunocytochemistry
Murine cortical cultures were fixed with 4% paraformaldehyde and incubated overnight with the primary antibody raised against either glial fibrillary acidic protein, microtubul associated protein-2 (GFAP, 1:5000 and MAP2, 1:200 in PBS plus 1% bovine serum albumin plus 0.1% Tween 20) or IL-6 receptors (IL-6Rα and gp130, 1:200 in PBS plus 1% bovine serum albumin). Cells were then washed and incubated for 1 hour with the appropriate secondary biotin-conjugated antibody. Antibody-antigen complexes were amplified with avidin (Vectastain ABC kit; Vector Laboratories, Burlingame, California) and revealed by H2O2-peroxydase reaction.
SDS-PAGE and Western Blotting
Murine cultured cortical neurons and astrocytes were lysed in Tris-NaCl-Triton buffer and centrifuged for 5 minutes (2500 g) to obtain whole cell extracts. Then, sodium dodecyl sulfate-polyacrylamide gel electrophoresis was performed before immobilization on a polyvinyl difluoride membrane. Western blotting were exposed for 1 hour to the primary antibodies (1:1500), 1 hour to the appropriate secondary biotin-conjugated antibody (1:1000), and for 1 hour to Extravidin (1:2000) before revelation using a chemiluminescence kit (NEN, Paris, France).
In vitro toxicity experiments
Excitotoxicity
Slowly triggered excitotoxicity was induced at 37°C in mixed neuron-glia or pure neuronal cortical cultures (DIV 14) by a 24-hour exposure to 12.5 μmol/L NMDA, 10 μmol/L AMPA or 50 μmol/L kainate in MS supplemented with glycine (Choi, 1992). MK-801 (10 μmol/L) was always added concurrently with AMPA or kainate to block secondary activation of NMDA receptors. Excitotoxins were applied alone or concurrently with mouse recombinant IL-6 (mrIL-6).
Neuronal death was estimated by examining the cultures under phase-contrast microscopy and quantitated by measuring lactate dehydrogenase (LDH) release from damaged cells into the bathing medium one day after the onset of exposure to the appropriate excitotoxin (Koh and Choi, 1987). The LDH level corresponding to complete neuronal death (without glial death) was determined in parallel dishes exposed to 200 μmol/L NMDA. Background LDH levels were determined in sister cultures subjected to sham wash and subtracted from experimental values to yield the signal specific for experimentally-induced injury.
Apoptosis
Serum deprivation was initiated by transferring near pure neuronal cultures (DIV 7) for 24 hours into growth medium lacking serum (Martin et al., 1988) in the presence or absence of mrIL-6. Secondary NMDA receptor activation was blocked by the addition of MK-801 to the bathing medium. Neuronal death was assessed by phase-contrast cell counting after staining with 0.4% trypan blue dye. The percentage of neuronal death was estimated as the ratio of trypan blue positive neurons after serum deprivation to the total neurons labeled with the same dye after paraformaldehyde permeation.
Staurosporine exposure was performed at 37°C for 24 hours by transferring mixed cortical cell cultures in MS supplemented with glycine, containing 200 nmol/L of staurosporine (Koh et al., 1995) with or without mrIL-6. MK-801 was added concurrently to staurosporine to block secondary NMDA receptor activation. Neuronal death was assessed by the measurement of LDH release.
Statistical analysis
Results are expressed as mean ± SD. Statistical analysis consisted in one-way variance analysis, followed by Bonferronni-Dunn's test.
RESULTS
Specificity of the PCR products
Oligonucleotide probes were designed from the published cDNA sequence of IL-6 (Chiu et al., 1988), IL-6Rα (Sugita et al., 1990) and gp130 (Saito et al., 1992). Each set of oligonucleotides used for semiquantitative RT-PCR studies gave the PCR product of the expected size (638 bps for IL-6, 320 bps for IL-6Rα and 405 bps for gpl30), as illustrated in Fig. 1. The specificity of the PCR products was confirmed by restriction digest controls. IL-6 and IL-6Rα PCR products were digested using AluI endonuclease, whereas the gp130 PCR product was submitted to EcoRI digestion. Fig. 1 shows that restriction enzymes generated the expected fragments of 365, 166, and 107 bps for IL-6 (Fig. 1A); 155 and 111 bps for IL-6Rα (two expected fragments of 46 and 8 bps were not visualized because of their small size) (Fig. 1B); 215 and 190 bps for gpl30 (Fig. 1C).
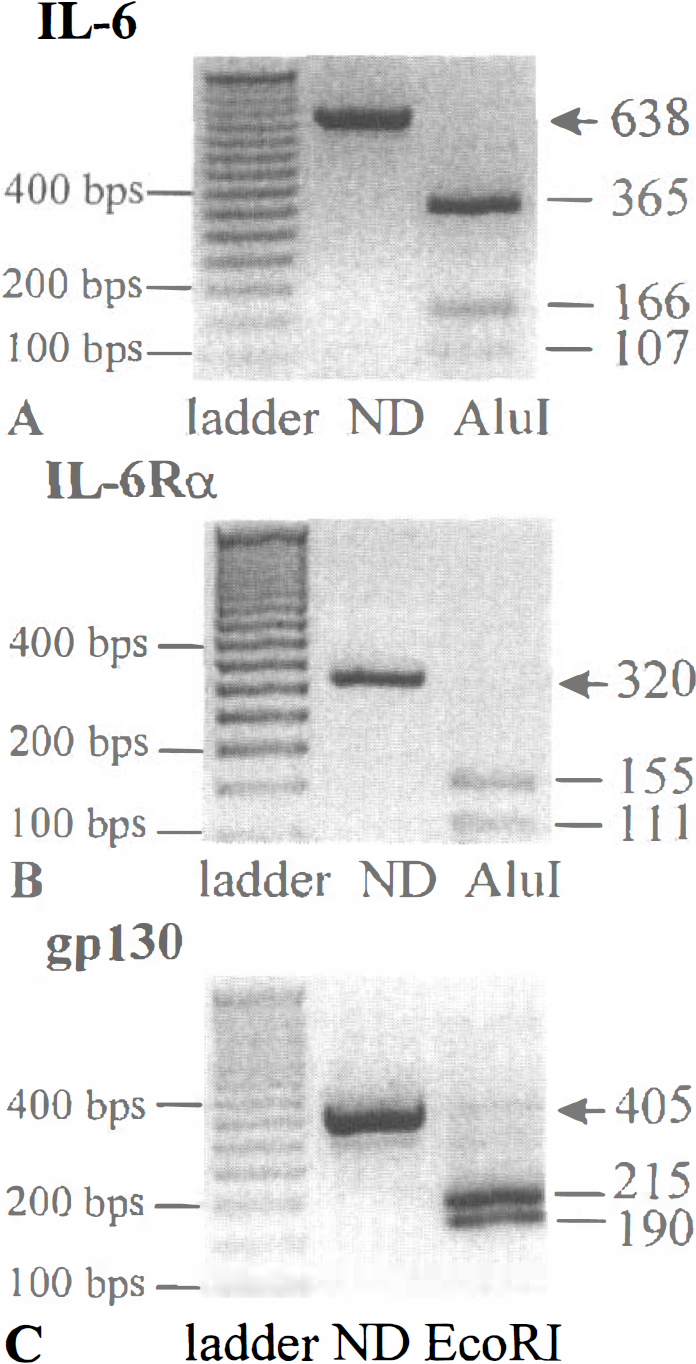
Specificity of the polymerase chain reaction (PCR) products. Total RNAs were extracted from murine cerebral cortex and reverse transcribed using a poly(dT) primer. PCR amplification was then performed using specific sets of primers (Table 1). Agarose gel electrophoresis of the IL-6 PCR product digested or not digested by Alul
As predicted by the high degree of homology between the rat and the mouse cDNA sequences encoding for IL-6 and its receptors, similar results were obtained with cDNAs derived from rat cerebral cortex (data not shown).
Transcriptional expression of IL-6 and its receptors after permanent or transient MCAO in rats
Focal cerebral ischemia was induced by transient (90 minutes) or permanent intraluminal occlusion of the middle cerebral artery. In both cases, MCAO induced, after 24 hours a severe lesion affecting both the striatum and the cortex, and representing approximately 40% (≃ 200 mm3) of the total volume of the hemisphere.
Total RNAs from ipsi- and contra-lateral cortices were harvested at the indicated times after permanent or transient MCAO. RNA samples (1 μg) were then reverse transcribed with poly-dT oligonucleotides. An aliquot of the cDNA libraries (1 μl) was amplified by PCR with specific sets of oligonucleotides for β-actin, IL-6, IL-6Rα or gp130. The expression pattern of IL-6 and its related receptors in a model of permanent cerebral ischemia was then studied. As shown in Fig. 2A, all PCR reactions showed the same level of the chosen housekeeping gene (β-actin). The IL-6 cDNA product was not detected in ipsi- and contra-lateral cortices obtained 6 hours after the intervention in sham rats. However, in the injured cortex the expression of IL-6 mRNA enhanced at 3 hours, peaked at 24 hours, and returned to baseline levels after 3 days. In contrast to IL-6, the expression of IL-6Rα and gp130 receptors mRNAs remained unchanged. The expression of IL-6 mRNA in a model of 90 minutes transient cerebral ischemia displayed a pattern slightly delayed when compared to the model of permanent cerebral ischemia (Fig. 2B).
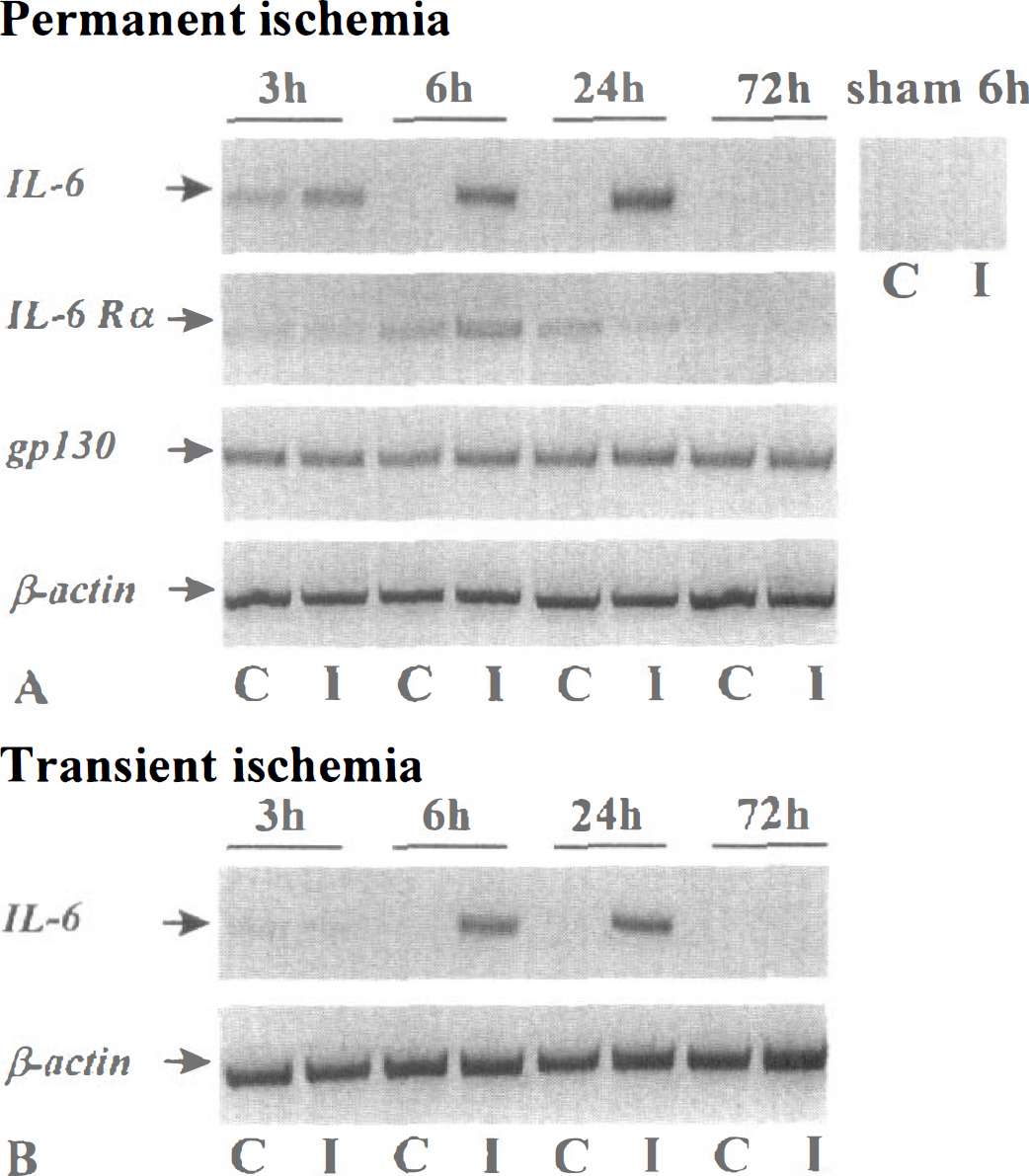
Transcriptional expression of IL-6 and its receptors after permanent or transient middle cerebral artery (MCAO) occlusion in rats.
After both transient and permanent focal ischemia, a transient increase in IL-6 mRNA levels was observed in the contralateral cortex. As previously suggested, this increase could result from the physical deformation of the contralateral hemisphere induced by the swelling in the injured ischemic side (Loddick et al., 1998).
Overexpression of IL-6 mRNA after the intrastriatal injection of NMDA
Histologic analysis revealed that, whereas the striatal injection of vehicle (PBS) alone did not induce any significant lesion, rats injected with NMDA (75 nmol/L in PBS) exhibited a lesion volume of 27.0 ± 2.5 mm3 (mean ± SD, n = 8) (Ruocco et al., 1999).
Total RNAs (1 μg) harvested from the control and injected rat striata were submitted to reverse transcription using poly-dT oligonucleotides and PCR was performed for IL-6, IL-6Rα, gp130, and β-actin mRNAs, as described above. All PCR reactions showed the same level of expression of the chosen housekeeping gene (β-actin) (Fig. 3).
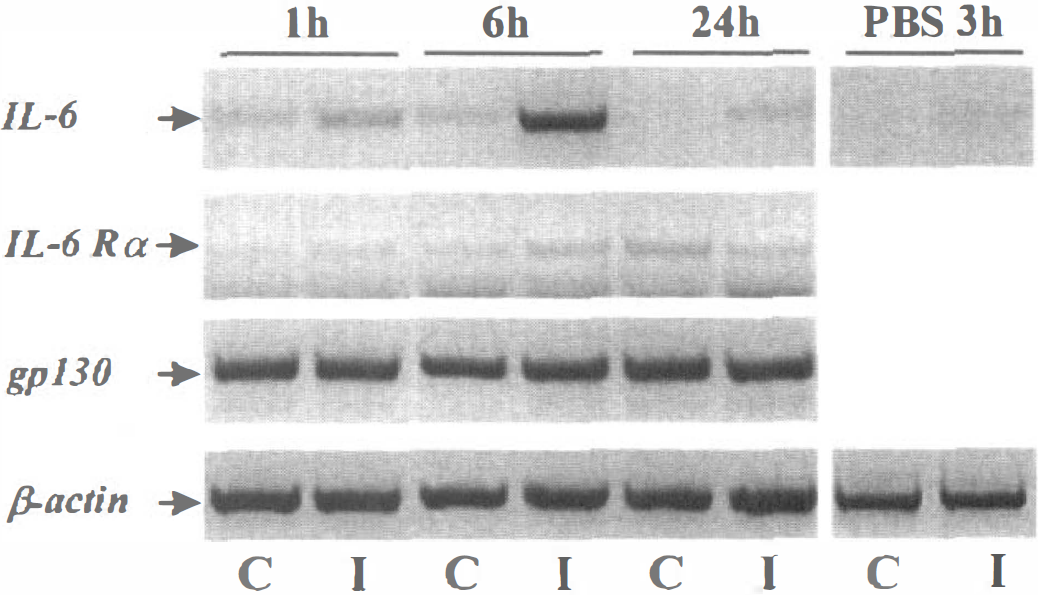
Overexpression of IL-6 mRNA after the intrastriatal injection of N-methyl-
Figure 3 shows that, in contrast to the transcription of both IL-6 related receptors (IL-6Rα and gp130), the expression of IL-6 mRNA after striatal injection of NMDA displayed a marked overexpression after 6 hours, that returned to control levels after 24 hours. As observed after focal ischemia (Fig. 2), a minor increase in IL-6 mRNA was detected in the contralateral striatum at 1 and 6 hours. After 3 hours, the injection of the vehicule (PBS) into the striatum induced only minor modifications in IL-6 mRNA levels.
NMDA treatment induces the overexpression of IL-6 mRNA in cultured cortical neurons
As both ischemic and excitotoxic insults to the brain induce an early up-regulation of IL-6 expression in vivo, the question arose whether the activation of ionotropic glutamatergic receptors in cultured cortical neurons may also induce an increase of this cytokine. By using semiquantitative RT-PCR, the authors studied the transcriptional expression of IL-6 in cultured neurons after exposure to AMPA or NMDA. Near pure neuronal cultures (DIV 14), obtained from the cortex of mice were exposed for a short period (less than 3 hours) to high concentrations (50 μmol/L) of glutamatergic agonists (the AMPA exposure was performed in the presence of 10 μmol/L MK-801 to prevent secondary NMDA receptor activation). These applications induced an acute swelling of neuronal bodies but did not alter the membrane permeability, as estimated by the LDH release into the bathing medium (data not shown). As shown in Fig. 4A, AMPA exposure did not obviously modify the transcriptional activity of IL-6 and its related receptors (IL-6Rα and gp130) in cultured cortical neurons. The expression of IL-6Rα and gp130 mRNA was equally unchanged after NMDA exposure. However, NMDA treatment enhanced the transcription of IL-6 in a time-dependent manner. This neuronal overexpression of IL-6 was specifically induced by NMDA, because it was completely blocked in the presence of the NMDA antagonist, MK-801 (Fig. 4B). After 3 hours, exposure to MK-801 (10 μmol/L) had no effect on IL-6 transcription as compared to control cultures (data not shown). The authors thus postulated that this induction of IL-6 mRNA could result from the calcium influx mediated through the activation of NMDA receptors. The authors tested this hypothesis by exposing near pure neuronal cultures to the calcium ionophore, ionomycin (5 μmol/L in the presence of 10 μmol/L MK-801 to prevent excessive NMDA receptor activation). The addition of ionomycin induced an up-regulation of IL-6 mRNA over the same interval of time as seen after the application of NMDA (Fig. 5).
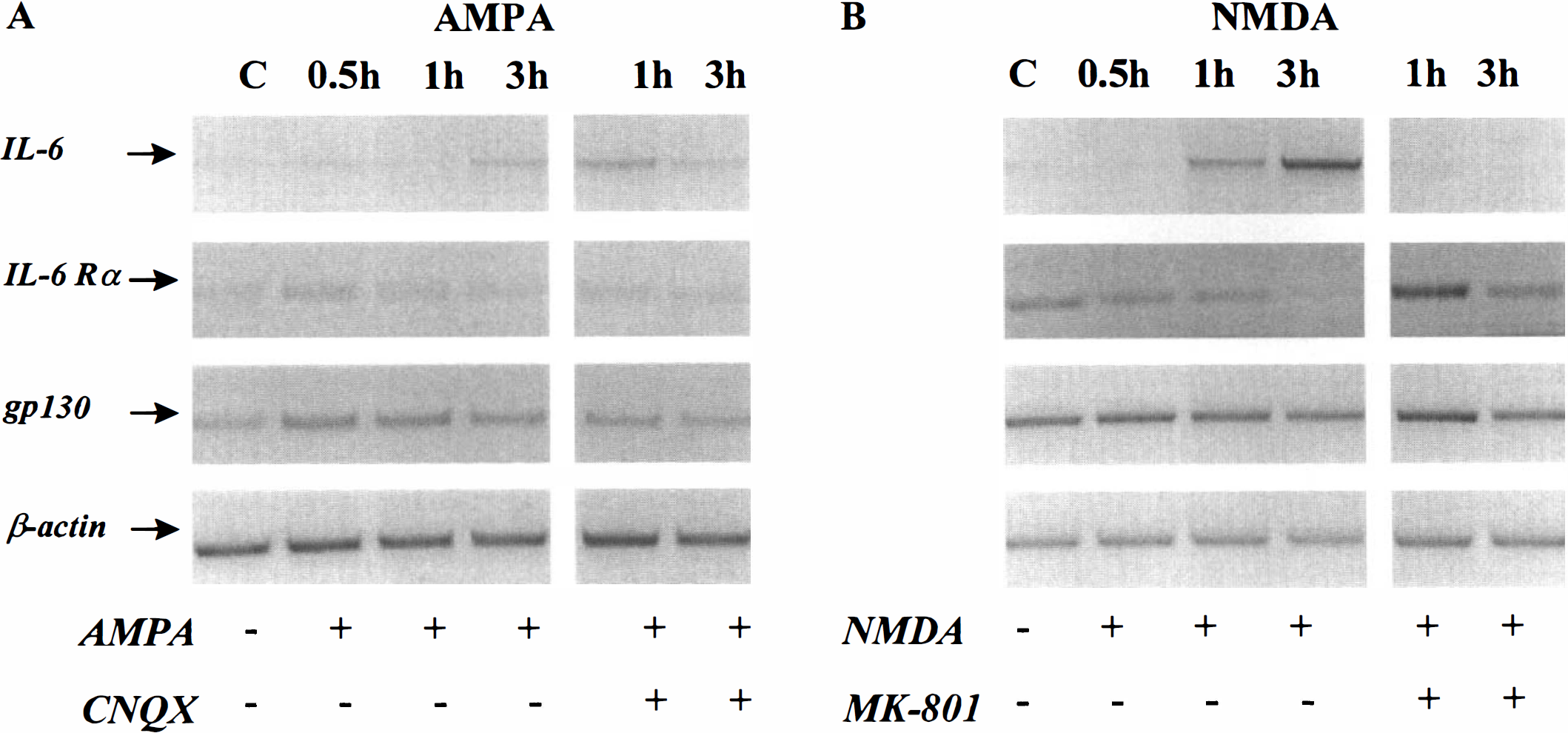
The application of N-methyl-
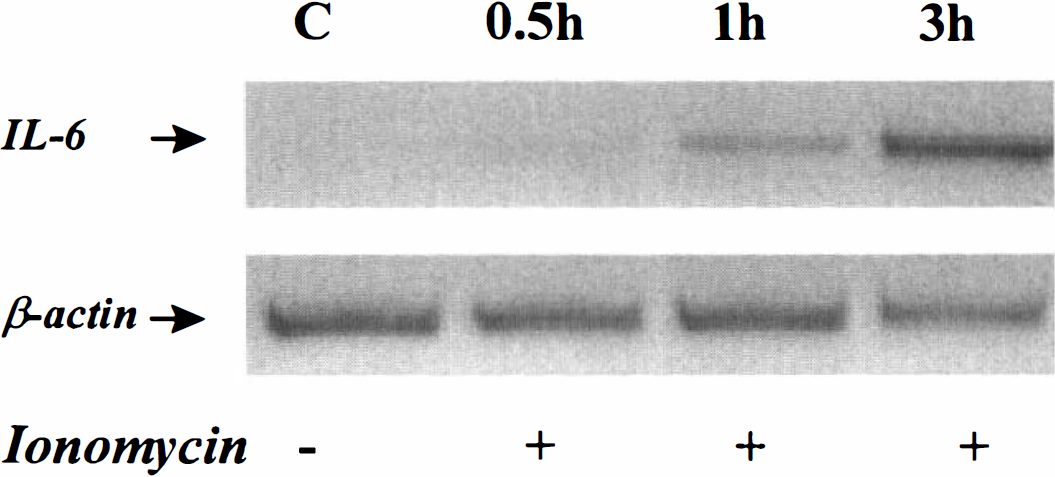
Ionomycin treatment induces an overexpression of IL-6 mRNA in cultured cortical neurons. Pure cortical neuron cultures were incubated for the indicated times with ionomycin (5 μmol/L) and MK-801 (10 μmol/L). Total RNAs were then isolated and submitted to reverse transcription polymerase chain reaction for IL-6 and B-actin mRNAs. Presented electrophoresis is representative of three independent experiments.
Expression of IL-6 related receptors in cultured cortical neurons and astrocytes
The cellular expression of IL-6 related receptors in murine cortical cultures was determined at both the mRNA and protein levels. RT-PCR studies revealed that both cultured cortical neurons (DIV 14) and astrocytes exhibited mRNAs for IL-6Rα and gp130 (Fig. 6A). Western blotting analysis performed with an antibody raised against IL-6Rα (80 kDa) or gp130 (130 kDa) revealed the presence of a single band at the expected molecular weight (Fig. 6B); thus demonstrating that neurons and astrocytes in culture express both IL-6 related receptors, as described in the rodent adult brain (Schöbitz et al., 1993; Gadient and Otten, 1994; Watanabe et al., 1996). The expression of IL-6Rα and gp130 in cortical astrocytes and neurons was confirmed by immunocytochemistry (Fig. 6C).
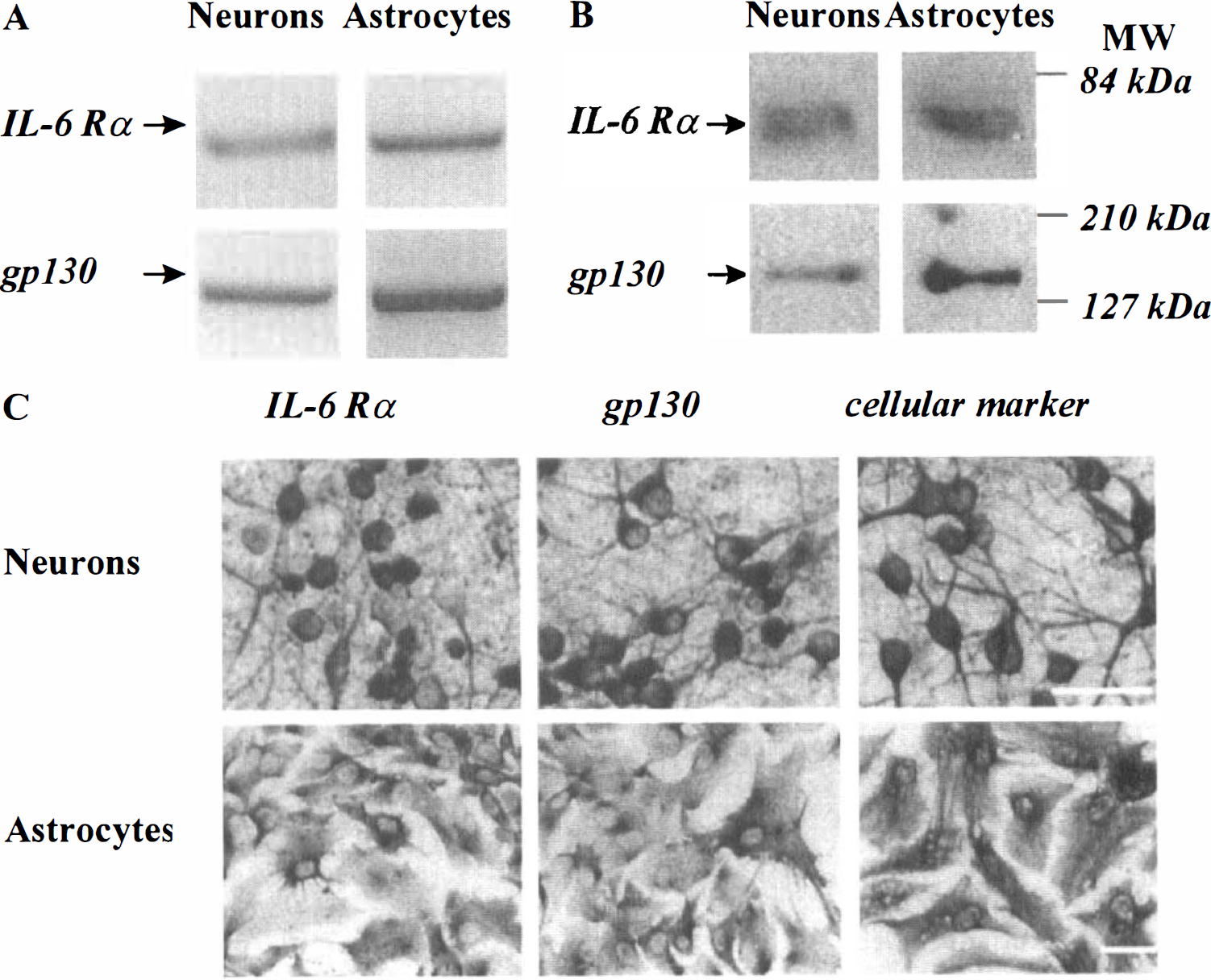
Expression of IL-6 related receptors in cultured cortical neurons and astrocytes. The cellular expression of IL-6Rα and gp130 was studied on murine astrocytic or neuronal cortical cultures at the mRNA
IL-6 is neuroprotective against NMDA-induced neuronal death
To test whether IL-6 might alter neuronal outcome after an ischemic insult, the authors determined the effects of mouse recombinant IL-6 (mrIL-6) on murine cortical cell cultures exposed to apoptotic or necrotic paradigms.
To induce apoptosis, pure cortical neuronal cultures were transferred after 7 days in vitro (DIV 7) to a serum-deficient medium supplemented with MK-801 (Martin et al., 1988), that provokes the degeneration of approximately 50% of neurons over 24 hours. This treatment resulted in three features typical of apoptosis: (1) a gradual shrinkage of the cell body was exhibited by neurons; (2) death was blocked by the addition of cycloheximide (Chx), a protein synthesis inhibitor; and (3) death was accompanied by the appearance of DNA fragmentation (data not shown). The addition of mrIL-6 to the bathing medium (50 ng/ml) failed to modify the extent of neuronal degeneration (Fig. 7) (n = 10). This lack of effect was not due to the absence of IL-6 receptors in 7-day-old cultured neurons, because both IL-6Rα and gp130 (mRNAs and proteins) are expressed in these cultures (data not shown).
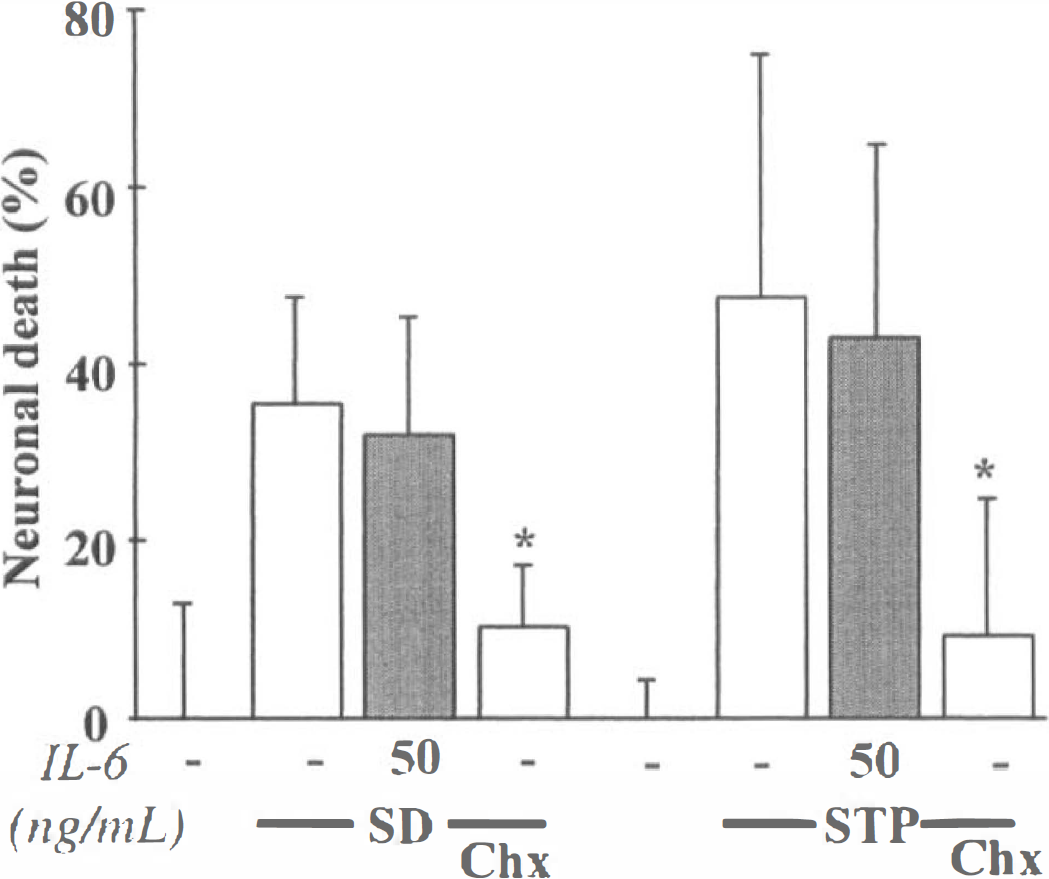
IL-6 does not protect neurons against apoptotic cell death. Neuronal death (%) was assessed after 24 hours by trypan blue dye staining (mean ± standard deviation, n = 10), after serum deprivation (SD) in pure neuronal cultures (DIV 7), or by lactate dehydrogenase release (mean ± standard deviation, n = 14), after exposure of mixed cortical cultures (DIV 14) to staurosporine 200 nmol/L (STP). SD and STP exposure were performed in the presence (shaded bars) or absence (open bars) of mrlL-6 and with or without cycloheximide (Chx) at 1 μg/mL. In both apoptotic paradigms, MK-801 was systematically added to block the secondary activation of N-methyl-
In addition, neuronal apoptosis was also induced by exposing mixed neuron-glia cultures (DIV 14) to staurosporine, a nonspecific protein kinase inhibitor (Koh et al., 1995). Although cycloheximide blocked neuronal death, the addition of mrIL-6 (50 ng/mL) was without effect (Fig. 7) (n = 14).
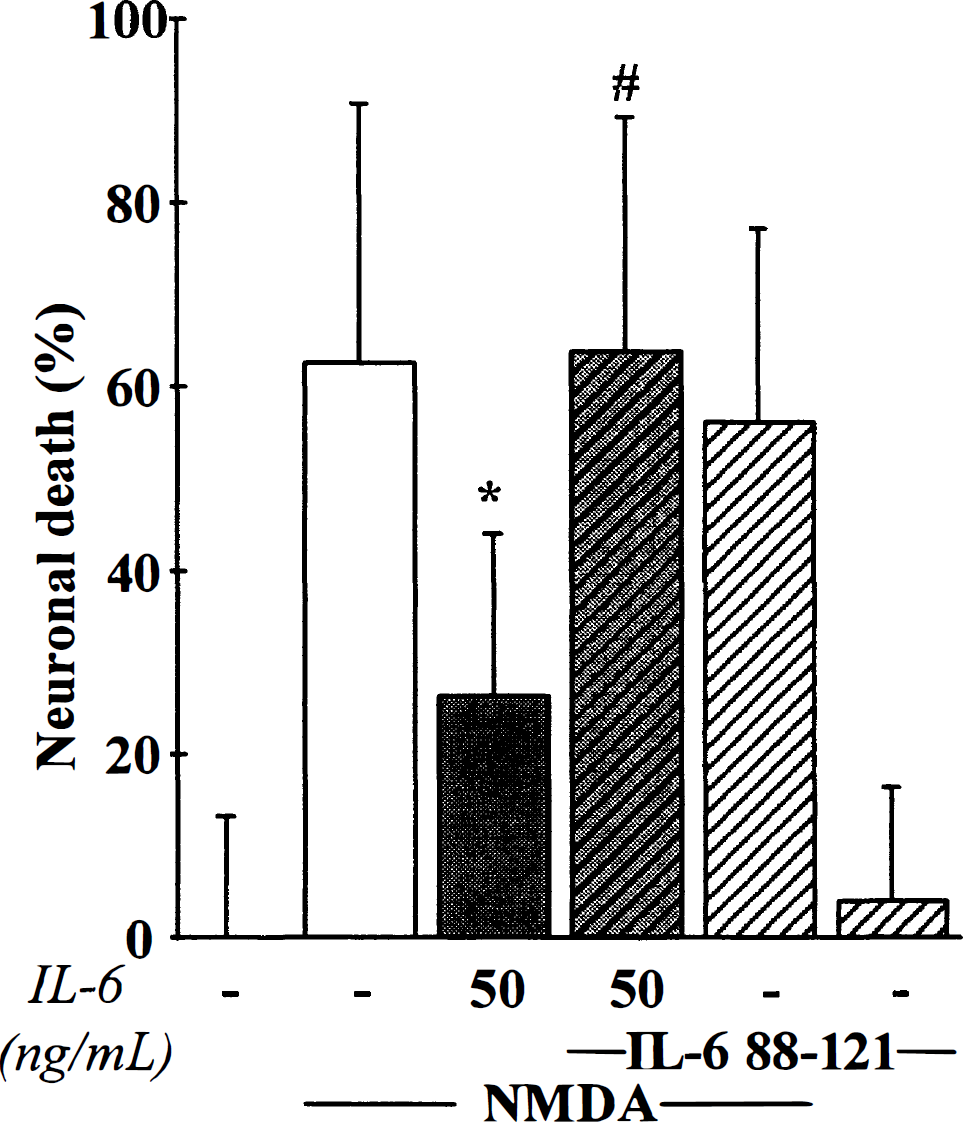
Thereafter, the authors determined the influence of IL-6 on excitotoxic necrosis, a process morphologically distinct from apoptosis and characterized by a prominent and early cell swelling. Necrosis was induced over 24 hours by the following glutamatergic agonists: NMDA (12.5 μmol/L), AMPA (10 μmol/L) and kainate (50 μmol/L) (Choi, 1992). The exposure of mixed cortical neuron-glia cultures (DIV 14) to these agonists produced an acute swelling of neuronal bodies, followed 24 hours later by widespread neuronal degeneration, whereas the glia remained intact. Although mrIL-6 (from 5 to 50 ng/mL) was found ineffective against AMPA- or kainate-induced toxicity (Fig. 8) (n = 12), it significantly reduced NMDA-induced necrotic neuronal death in mixed neuron-glia cultures, as illustrated by the large restoration of MAP-2 immunostaining organization (Fig. 9A). The quantification of the neuroprotective effect of IL-6 revealed that it was dose-dependent (Fig. 9B) (n = 13; P < 0.001). The potential role of astrocytes in the neuroprotective activity of IL-6 was investigated by applying NMDA (12.5 μmol/L) for 24 hours in near pure neuronal cultures (DIV 14). In these cultures, mrIL-6 (50 ng/mL) was able to display a neuroprotective activity against NMDA-induced necrotic death (Fig. 9C) (n = 16; p < 0.0001). To characterize the effect of mrIL-6 on NMDA-induced necrotic neuronal death, the authors tested a polypeptide fragment corresponding to amino acids 88 to 121 of human IL-6 (IL-6 88–121). This peptide has previously been characterized as a competitive inhibitor for IL-6 binding to IL-6Rα (Ekida et al., 1992). Although IL-6 88–121 (2 μg/mL) displayed no toxicity in mixed cortical cultures, it prevented the neuroprotective effect of IL-6 against NMDA toxicity (10 μmol/L), as revealed by the increase in the LDH release in the bathing medium 24 hours after the application of the excitotoxin (Fig. 10) (n = 10; P < 0.0001).
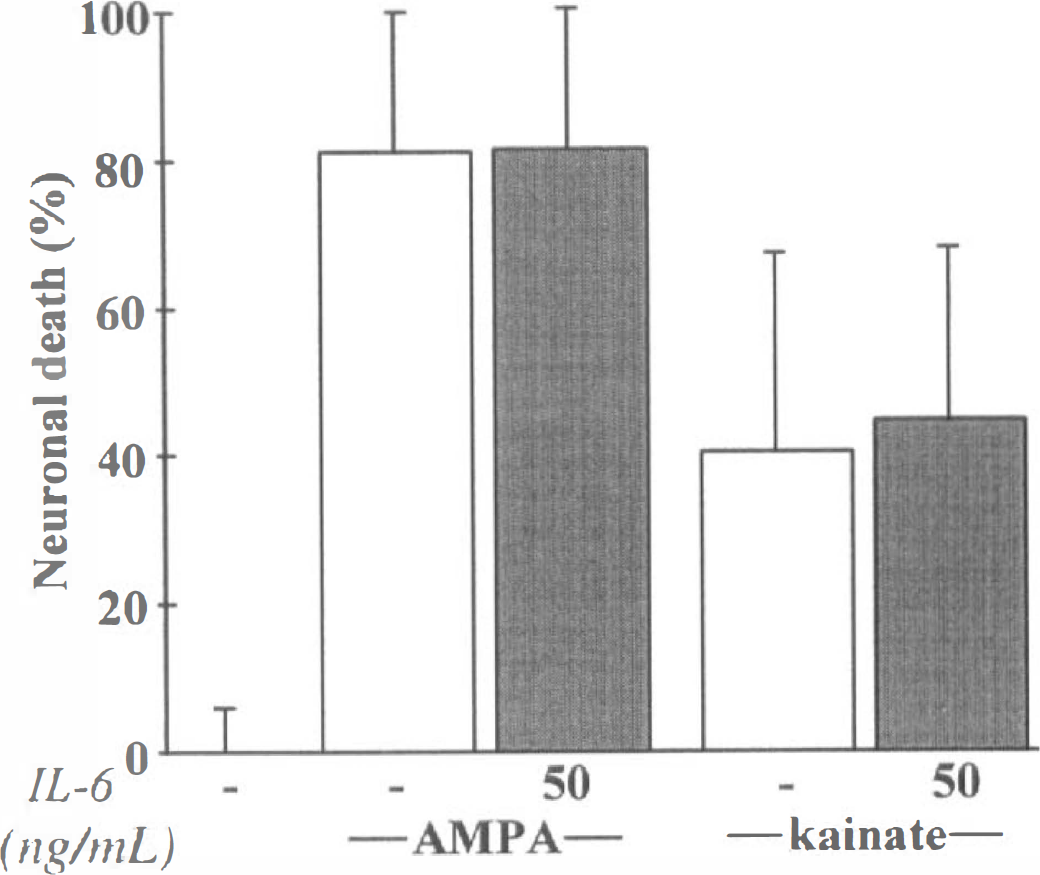
IL-6 fails to modify α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate-(AMPA) or kainate-induced excitoxicity in neurons. Neuronal death (%) was estimated by lactate dehydrogenase release (mean ± SD, n = 12), after a 24-hour exposure to AMPA (10 μmol/L) or kainate (50 μmol/L) in mixed neuron-glia cortical cultures in the presence (shaded bars) or not (open bars) of mrlL-6 (50 ng/mL). Secondary activation of N-methyl-
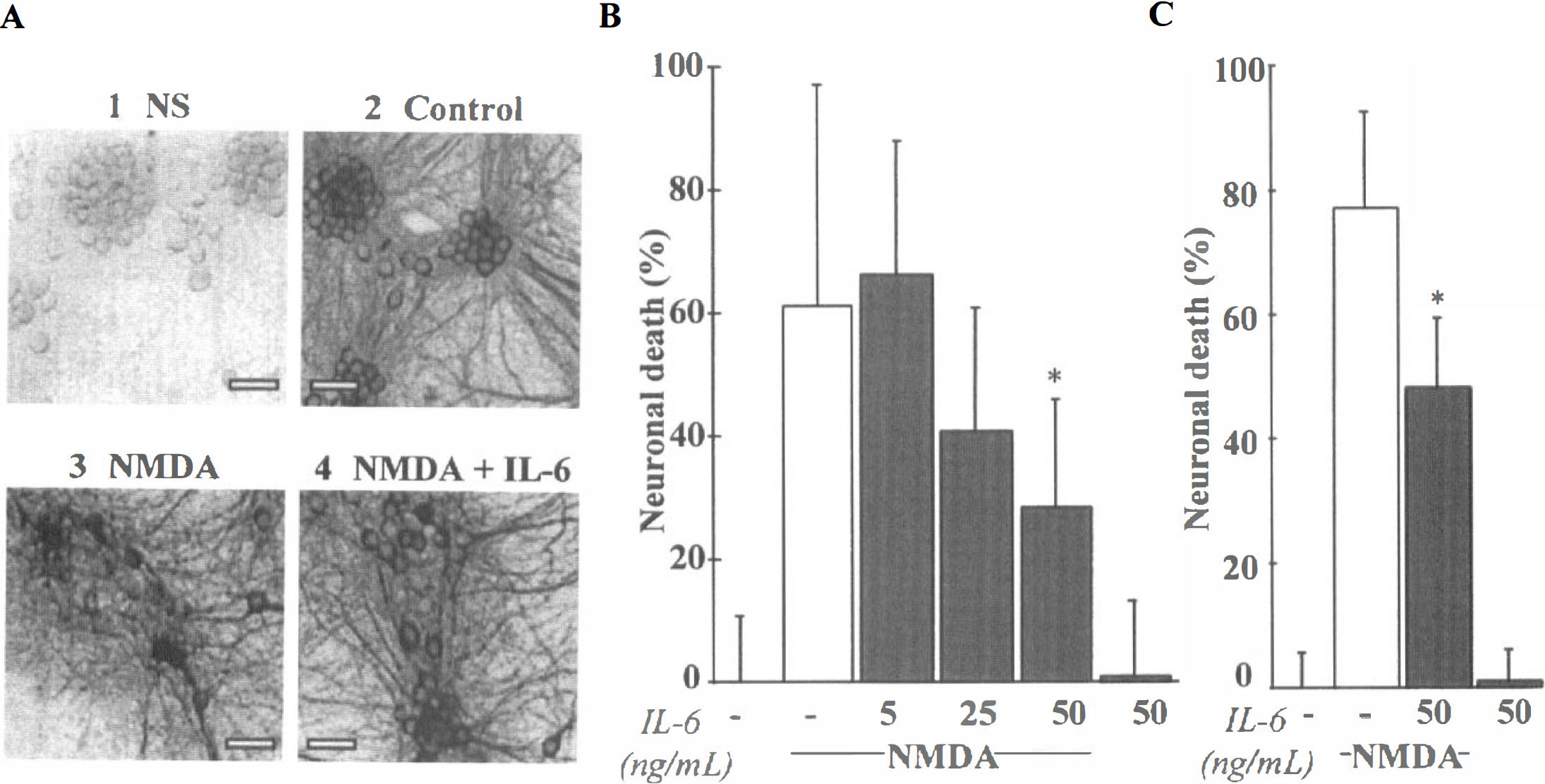
IL-6 is neuroprotective against N-methyl-
IL-6 neuroprotective effect against N-methyl-
DISCUSSION
In the present study, we investigated the implication of IL-6 during cerebral ischemia.
Our observation that the expression of the IL-6 gene is activated early in the injured cortex after focal cerebral ischemia is consistent with previous experimental data (Wang et al., 1995; Hill et al, 1999; Clark et al., 1999). Although we did not address the protein level, the well-documented increase in IL-6 immunoreactivity (Maeda et al., 1994; Saito et al., 1996; Orzylowska et al., 1999) and bioactivity (Loddick et al., 1998) strongly supports the idea that the early occurrence of IL-6 mRNA production (evidenced as early as 3 hours after occlusion) could influence the outcome of ischemic insults. Two major additional findings presented here are the following: (1) the over-activation of NMDA receptors elicits an enhancement of IL-6 transcription in neurons; and 2) IL-6 exerts a neuroprotective effect restricted to NMDA-induced excitotoxic necrosis.
Since the glutamatergic ionotropic receptor-mediated excitotoxic cascade is a major contributory factor for ischemic neuronal death (Choi, 1996), we tested whether the activation of this pathway could reproduce the effect of cerebral ischemia on the expression of IL-6 mRNA. We evidenced that the injection of NMDA resulted in an enhancement of IL-6 transcription in the striatum similar to that provoked at the cortical level by MCAO. These observations suggest that the excitotoxic pathway could initiate the synthesis of IL-6 in the ischemic brain. This hypothesis is strengthened by the induction of IL-6 reported after the striatal injection of another glutamatergic agonist, quinolinic acid (Schiefer et al., 1998). However, because increasing evidence suggests that NMDA may induce both excitotoxic and apoptotic mechanisms (Bonfoco et al., 1995), and to eliminate any effect resulting from tissue lesion elicited by the striatal injection of NMDA, we developed an in vitro model allowing the investigation of the transcriptional modulation(s) mediated during a severe glutamatergic ionotropic receptor-mediated excitotoxic stress. In our conditions, the exposure of pure cultured cortical neurons to high concentrations of glutamatergic agonists, for up to 3 hours, induced the characteristic feature of necrosis (acute swelling of the cell body) without linkage of the neuronal membrane. We showed that cortical neurons are able to overexpress IL-6 mRNA in response to an intense NMDA receptor activation, an effect that is selectively blocked by MK-801. To investigate further the cellular signal(s) responsible for the increased transcription of IL-6, and because NMDA receptor activation is characterized by a raise in the intracellular calcium concentration (MacDermott et al., 1986), we tested the ability of a selective calcium ionophore, ionomycin (A23187) to mimic the activation of IL-6 mRNA expression induced by NMDA. In cultured cortical neurons, ionomycin elicited a time-dependent activation of IL-6 transcription, similar to that resulting from NMDA exposure. Thus, the activation of IL-6 expression in neurons may involve a calcium-dependent pathway. Overall, these data demonstrate that although glial cells have been suggested to be the source of IL-6 in the ischemic brain (Maeda et al., 1994; Schiefer et al., 1998), neurons are a potential parenchymal source of this cytokine. Recent evidence of IL-6 immunoreactive neurons in the ischemic rodent brain (Suzuki et al, 1999a, b ) supports that data. In addition, this is the first demonstration that the calcium ions influx evoked by the over-activation of NMDA receptors could be a critical signal for IL-6 production in the ischemic brain.
When considering the wide variety of central nervous system pathologies that exhibit a locally increased production of IL-6, this cytokine may be viewed as a ubiquitous alarm signal after tissue injury. However, the influence of this IL-6 induction remains controversial (Tarkowski et al., 1995; Campbell, 1998; Qiu et al., 1998; Toulmond et al., 1992; Loddick et al., 1998; Matsuda et al., 1996). To address the role of IL-6 during cerebral ischemia, we used cultured cortical neurons and astrocytes that express both the binding, (IL-6Ra) and the transducing (gp130) IL-6 receptors, and are thus able to respond to IL-6. These cultures were submitted to apoptotic and excitotoxic paradigms, reproducing the major pathways leading to ischemic neuronal death. By using two well characterized apoptotic challenges (serum deprivation and staurosporine exposure), we demonstrated that recombinant IL-6 fails to prevent the neuronal cell death, whereas cycloheximide succeeds. This observation can be related to the lack of protective effect of IL-6 against NO-induced cortical neuronal apoptotic cell death (Toku et al., 1998); but contrasts with the beneficial activity of this cytokine against serum deprivation-mediated PC12 cell death (Umegaki et al., 1996). By using necrotic paradigms, we evidenced that IL-6 does not prevent AMPA/kainate receptor-mediated neuronal death, but dose-dependently protects neurons against NMDA receptor-induced necrosis. We further characterized this selective protective activity of IL-6 by showing that it requires the binding of this cytokine to its receptor, because it is abolished by a competitive inhibitor. Previous data have suggested that IL-6 could support neuronal survival by controlling the astrocytic activation (Fattori et al., 1995; Raivich et al., 1996) or secretion of neurotrophic factors (Kossmann et al., 1996, Marz et al., 1999, Carlson et al., 1999). Here we showed that astrocytes are not necessary for IL-6 neuroprotective activity, as evidenced by the maintenance of IL-6 beneficial effect against NMDA exposure in pure neuronal cultures. The idea that IL-6 directly targets neurons opens new insights into the cellular processes of IL-6-mediated neuroprotection during cerebral ischemia. However, further investigations are required to extend our in vitro findings to the adult brain and to clarify the molecular mechanism(s) that underlies the protective effect of IL-6. In particular, the competitive inhibitor, which prevents the neuroprotective activity of IL-6 in cortical cultures, represents a powerful tool to evaluate the role of IL-6 during cerebral ischemia.
In summary, we demonstrate that during cerebral ischemia and other excitotoxicity-related injuries, neurons could synthesize and release IL-6 in response to the raise in the intracellular calcium concentration mediated through NMDA receptor activation. This induction of IL-6 could serve an endogenous autocrine protective mechanism, directly targeting neurons to prevent NMDA receptor-induced degeneration. These experimental observations, together with the clinical evidence of a cerebral production of IL-6 in stroke patients (Fassbender et al., 1994, Tarkowski et al., 1995), raise the possible relevance of the IL-6 signaling pathway as an early target for the development of new therapeutic strategies.
Footnotes
Acknowledgments
The authors thank A. Ruocco and S. Roussel for technical assistance.