
Abstract
Diffuse axonal injury is a frequent pathologic sequel of head trauma, which, despite its devastating consequences for the patients, remains to be fully elucidated. Here we studied the release of interleukin-6 (IL-6) into CSF and serum, as well as the expression of IL-6 messenger ribonucleic acid (mRNA) and protein in a weight drop model of axonal injury in the rat. The IL-6 activity was elevated in CSF within 1 hour and peaked between 2 and 4 hours, reaching maximal values of 82,108 pg/mL, and returned to control values after 24 hours. In serum, the levels of IL-6 remained below increased CSF levels and did not exceed 393 pg/mL. In situ hybridization demonstrated augmented IL-6 mRNA expression in several regions including cortical pyramidal cells, neurons in thalamic nuclei, and macrophages in the basal subarachnoid spaces. A weak constitutive expression of IL-6 protein was shown by immunohistochemical study in control brain. After injury, IL-6 increased at 1 hour and remained elevated through the first 24 hours, returning to normal afterward. Most cells producing IL-6 were cortical, thalamic, and hippocampal neurons as confirmed by staining for the neuronal marker NeuN. These results extend our previous studies showing IL-6 production in the cerebrospinal fluid of patients with severe head trauma and demonstrate that neurons are the main source of IL-6 after experimental axonal injury.
Traumatic brain injury (TBI) initiates a complex series of cellular and molecular events. The inflammatory response and the process of tissue repair in the brain are regulated by cytokines, which also promote the secretion of neurotrophic factors (Kossmann et al., 1996, Morganti-Kossmann et al., 1997). However, if released in high amounts and for extended periods, cytokines may contribute to neurodegeneration and secondary brain insult (Morganti-Kossmann et al., 1992; Rothwell and Relton, 1993).
One of the mediators involved in the inflammatory response occurring in the injured brain is interleukin-6 (IL-6). Interleukin-6 is a multifunctional cytokine that originally was described as an inducer of B-cell differentiation and as a hepatocyte stimulating factor (for review see van Snick, 1990). Interleukin-6 is produced by various cell types on challenge, such as astrocytes, microglia, and neurons. Several functions of neural cells can be modulated by IL-6, including the differentiation of PC12 cells, increased neuronal survival, and induction of nerve growth factor (NGF) synthesis (Satoh et al., 1988; Frei et al., 1989; Selmaj et al., 1990; Benveniste et al., 1990; Hama et al., 1991; Schöbitz et al., 1993; Benveniste, 1995; Ringheim et al., 1995; Von Coelln et al., 1995; Kossmann et al., 1996).
Several studies have shown increased synthesis of IL-6 in a variety of disorders of the CNS, including brain injury (for reviews see Morganti-Kossmann et al., 1992; Benveniste, 1995; Morganti-Kossmann and Kossmann, 1995). Studies performed in patients with severe TBI demonstrated an augmented release of IL-6 into the cerebrospinal fluid (CSF; McClain et al., 1991; Kossmann et al., 1995; Kossmann et al., 1996). Experimental models of focal brain damage have shown an upregulation of IL-6 activity within the injured cortex (Woodroofe et al., 1991; Yan et al., 1992; Taupin et al., 1993; Shohami et al., 1994), as well as its release into the CSF (Woodroofe et al., 1991). However, few data are available on the inflammatory response occurring after axonal injury, which is of high clinical relevance because of its presence in most fatal head injuries and because of its association with high morbidity and mortality (Povlishock, 1992; Gentleman et al., 1995).
Previously, we have shown that high concentrations of IL-6 remain detectable in human CSF up to 3 weeks after severe brain injury. Produced intrathecally, IL-6, once reaching the periphery through a damaged blood-brain barrier, may induce an acute phase response, a frequently observed reaction in head trauma patients (Kossmann et al., 1995). In addition, the upregulation of IL-6 after TBI was found to be associated with the release of NGF into CSF, and human CSF containing IL-6 had the ability to induce NGF production by cultured astrocytes (Kossmann et al., 1996).
Based on these findings, an experimental model of axonal injury was established in the rat with the objective of extending the results obtained in humans. This model resembles injury patterns frequently found in patients after severe head trauma, whereas alternative models of brain injury produce focal contusions but minimal axonal injury (Foda and Marmarou, 1994; Marmarou et al., 1994; Povlishock et al., 1997). In our model system, the release of IL-6 was monitored in rat CSF and serum for up to 2 weeks, and IL-6 expression at messenger ribonucleic acid (mRNA) and protein level was analyzed by in situ hybridization and immunohistochemical study, respectively, to characterize the kinetics and the cell types synthesizing IL-6.
MATERIALS AND METHODS
Animal model of axonal injury
Fifty-nine male Sprague Dawley Ivanovas rats (Biologisches Zentrallabor, Zuerich, Switzerland) weighing 372 ±57 g (mean ± SD) were used in this study. Under volatile anesthesia (O2/N2O/halothane) (Halothan, Abbott AG, Cham, Switzerland), the skull was exposed, and a round metal disk was fixed in the midline with dental acrylic (De Trey Zinc, De Trey Dentsply, Konstanz, Germany). Traumatic brain injury was induced by dropping a weight of 250 g on the disk from a height of 2 m through a vertical tube, as described by Marmarou et al. (1994). Because a central respiratory depression is mainly responsible for mortality in this model, the animals were mechanically ventilated. For trauma delivery, rats were briefly (about 15 seconds) disconnected from the respirator. Thereafter, they were immediately ventilated with 100% O2 until recovering to sufficient and spontaneous respiration. According to this protocol, no episodes of apnea were observed during the experiments. The disk was removed, the wound sutured, and the animals were returned to their cages with food and water ad libitum. Rats were killed with a lethal dose of fentanyl/droperidol (Innovar-Vet Injection, Pitman-Moore, Inc., Mundelein, IL, U.S.A.) at different time points (1, 2, 4, 8, 16 hours, and 1, 2, 4, 7, 14 days) after trauma. For each time point, five rats were used, with the exception of the group corresponding to the 4-hour time point (n = 4). Two animals died immediately and two within 2 hours after the traumatic impact, and were excluded from the study. For control, three sham-operated animals were treated in the same fashion, except for the delivery of the trauma, and were killed at 2 hours and 2 days after surgery. Blood was taken from the right heart ventricle and collected in glass tubes on ice. The CSF was obtained by puncture of the cisterna magna after exposure of the dura between the occiput and the C1 vertebra. About 50 µL (15 to 100 µL) were collected from each rat. Serum and CSF were immediately centrifuged at 170 g for 10 minutes at 4°C, and supernatants frozen at −70°C until analyzed. The brains were removed immediately after taking CSF, divided along the midline, shock frozen on dry ice, and kept at −70°C. The animal protocol was approved by the Kantonales Veterinaramt Zürich (no. 138/93), and procedures and care were conducted in conformity with national and international laws and policies.
Immunohistochemistry
Cryosections of rat brains were cut at a thickness of 14 µm and stained using the Vectastain ABC kit (Vector Laboratories, Inc., Burlingame, CA, U.S.A.). Sections were dried for 1 hour and then fixed in acetone and rehydrated with decreasing ethanol concentrations (100%, 75%, 50%). After blocking with 2% horse serum and 2% rat serum in phosphate-buffered saline (PBS) for 30 minutes at room temperature, sections were incubated overnight at 4°C with an antibody against neurofilament 70 and 200 kd to visualize axonal changes (1:40; Bioscience Products AG, Emmenbrticke, Switzerland), NeuN as a marker for neurons (1:100; Chemicon, Temecula, CA), glial fibrillary acidic protein (GFAP) for astrocytes (1:10; Boehringer Mannheim GmbH, Mannheim, Germany), and complement receptor type 3 (OX-42) for microglia/macrophages (1:100; Serotec Ltd, Oxford, UK). For negative control, the first antibody was omitted and replaced by blocking buffer. Sections then were washed 3 × 5 minutes in PBS, incubated for 10 minutes in 1% peroxide in PBS to exhaust endogenous peroxidase, and finally incubated with biotinylated horse anti-mouse antibody and avidin-peroxidase (5 mg/mL) each for 1 hour at room temperature. Positive cells were revealed by developing with 3,3-diaminobenzidine (Sigma Chemie, Buchs, Switzerland) in Tris buffer. Additionally, some sections were stained with hematoxylin and eosin.
For IL-6 staining, sections were air dried for 1 hour, fixed in acetone at room temperature for 5 minutes, and blocked with 4% milk powder and 2% normal horse serum in PBS for 1 hour. After washes in PBS, a goat polyclonal anti-IL-6 antibody (R-19) (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) at 1:1000 dilution (0.2 mg/mL) was incubated overnight at 4°C. After washing three times in PBS, biotinylated rabbit anti-goat antibody (Vector Laboratories) was diluted 1:400 in 2% milk powder in PBS and incubated for 1 hour at room temperature. The subsequent staining steps did not differ from the procedure described above. To demonstrate specificity, antibody solution was incubated with increasing concentrations of recombinant IL-6 (0.01 to 2 mg/mL) overnight at 4°C before application to the brain sections for immunohistochemical study. For these experiments, three TBI animals per time point (1 hour up to 2 weeks), two sham-operated (2 and 48 hours, respectively), and untreated animals were analyzed.
Interleukin-6 bioassay
The IL-6 activity in serum and CSF samples was determined using 7TD1 cells (kindly provided by Dr. K. Frei, University Hospital Zuerich, Switzerland), an IL-6-dependent murine hybridoma cell line (Van Damme et al., 1987) based on a colorimetric assay (Mosmann, 1983). Samples were distributed in a 1:3 dilution series in 50 µL volumes in 96-well plates. Fifty microliters of 7TD1-cells (104/mL) were added to each well in Dulbecco's Modified Eagle Medium containing 10% fetal calf serum and incubated for 4 days at 37°C in a humidified carbon dioxide incubator. 3-[4,5-Dimethylthiazol-2-y1]-2,5-diphenyltetrazoliumbromide (MTT; Sigma) at the concentration of 10 mg/mL in PBS was added to the cultures (10 µL per well) for 3 hours at 37°C, and the cells were lysed with 100 µL 10% sodium dodecyl sulfate (Sigma). The absorbance was determined at 550 nm using a microplate reader (MR5000, Dynatec Laboratories, Inc., Chantilly, Va, U.S.A.). Measurements were performed in triplicate and concentrations calculated against a standard curve (1 to 1000 pg/mL) using mouse rIL-6 (Genzyme Diagnostics, Cambridge, MA, U.S.A.). The detection limit of this assay was 7 pg/mL. The IL-6 activity was specifically inhibited by adding anti-IL-6 antibody to the samples provided by the “EU Concerted Action: Cytokine in the Brain” (Dr S. Poole, National Institute for Biological Standards and Control, U.K.).
In situ hybridization
For the detection of IL-6 mRNA on rat brain, in situ hybridization was performed using a digoxigenin (DIG)-labeled cRNA probe after a modified protocol (Schaeren-Wiemers and Gerfin-Moser, 1993). Briefly, rat IL-6 cDNA (ATCC, Rockville, MD, U.S.A.) was isolated by alkaline lysis (Plasmid Maxi Kit, Qiagen, Inc., Chatsworth, CA, U.S.A.). Template cDNA was linearized, and sense and anti-sense cRNA runoff transcripts were labeled with DIG (DIG RNA Labeling Kit, Boehringer Mannheim GmbH). Transcripts of the sense-strand were routinely used as controls. For better tissue penetration, the labeled probes were alkali-hydrolyzed to an average length of 0.3 kb. Furthermore, the nucleotide concentrations were checked spectrophotometrically (using GeneQuant II, Pharmacia Biotech, Uppsala, Sweden); the length of hydrolyzed nucleotides was checked by agarose gel electrophoresis; labeling efficiency was tested on dot blots, as well as by blotting the gels, on a nylon membrane followed by immunodetection of the probes with anti-DIG-AP antibodies. This revealed sense and anti-sense probes of almost identical length and labeling intensity.
Rat brains were cut coronally at 3, 5, and 7 mm rostral from interaural line according to Paxinos and Watson (1986); three animals were analyzed per time point after injury (1, 2, 4, 8, 16, and 24 hours), and two sham-operated animals 2 hours after surgery. Cryosections (14 µm) were mounted on glass slides (Superfrost*/Plus, E. Merck AG, Dietikon, Switzerland), air dried for 1 hour, and fixed 20 minutes in 4% paraformaldehyde in PBS. Sections were washed 3 × 5 minutes in PBS, acetylated 10 minutes (1.2% triethanol-amine, 0.5% acetic anhydride in dH2O), permeabilized 20 minutes (0.1% Triton X-100 in PBS), and treated 7 minutes with 1 mg/mL proteinase-K in TE buffer (100 mmol/L Tris, 50 mmol/L ethylenediamine tetraacetic acid, pH 8.0) at room temperature. After each step, slides were washed 3 × 5 minutes in PBS.
Prehybridization was performed for 4 hours at 61°C with hybridization buffer (50% formamide, 5× standard saline citrate (SSC), 5× Denhardt's reagent, 1 mg/mL yeast tRNA, 0.5 mg/mL herring sperm DNA) and followed by an overnight hybridization with the same solution containing 250 ng/mL DIG-cRNA. The slides were washed at 61°C in 5× SSC for 5 minutes and in 0.2× SSC for 1 hour. Sections then were equilibrated to room temperature in 0.2× SSC and washed in maleic acid buffer (100 mmol/L maleic acid, 150 mmol/L NaCl, pH 7.5). After blocking (Blocking Reagent, Boehringer Mannheim) the sections for 1 hour, DIG-labeled hybrids were detected immunologically by adding anti-DIG-alkaline-phosphatase (1:250; Boehringer Mannheim) for 1 hour. Slides were rinsed twice with maleic acid buffer and equilibrated in detection buffer (100 mmol/L Tris, 100 mmol/L NaCl, 50 mmol/L MgCl2, pH 9.5). The color reaction was developed in the dark using a detection buffer containing 175 mg/mL 5-bromo-4-chloro-3-indolyl-phosphate, 500 mg/mL levamisole, and 340 mg/mL nitro blue tetrazolium chloride (Sigma). The alkaline phosphatase reaction was terminated by adding TE buffer when the desired intensity was reached and sections were mounted in Kaiser's glycerol gelatin (Merck).
For the identification of astrocytes, a monoclonal antibody against GFAP (Boehringer Mannheim) was used. Antibody was added simultaneously with the anti-DIG antibody after the hybridization step and followed by an incubation with a secondary antibody fluorescein-conjugated rabbit anti-mouse (Dako, Diagnostics AG, Zug, Switzerland).
Statistical analysis
The IL-6 values were log-transformed for statistics to achieve normal distribution for paired t test. Calculations were performed using commercially available software (StatView 4.0, Abacus Concepts, Inc., Berkeley, CA, U.S.A.).
RESULTS
Axonal injury induces the release of interleukin-6 into cerebrospinal fluid and serum
Axonal injury in this model was confirmed by immunohistochemical examination with an antibody against neurofilament. Widespread axonal damage was found in the white matter, and Fig. 1B shows typical axonal swelling and retraction balls within the medial geniculate body compared to sham-operated animals (Fig. 1A). Furthermore, acidophilic cortical neurons with nuclear pyknosis and lost nucleoli (“pink shrunken neurons”) representing neuronal death were visible after hematoxylin-and-eosin staining (Fig. 1C). These findings are in agreement with previous reports demonstrating neuronal somatic and axonal changes in this model (Foda and Marmarou 1994; Povlishock et al., 1997).
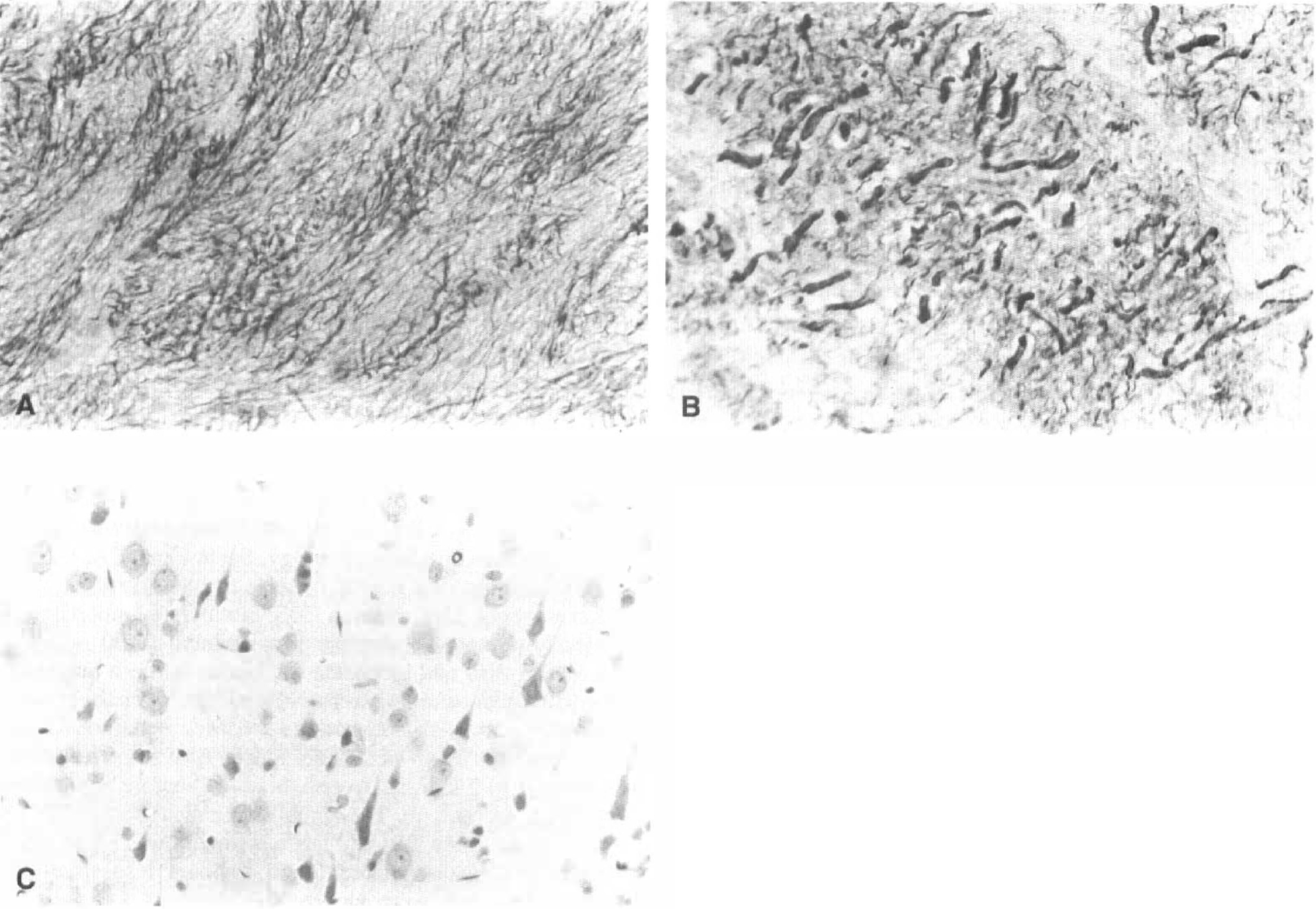
The model of closed head injury causes axonal injury. Immunohistochemical study for neurofilament 70 and 200 kd was performed on 14-µm cryosections, as described in Materials and Methods.
Significant release of IL-6 was detected in CSF and serum of animals with TBI compared with the sham-operated animals (Fig. 2). The IL-6 activity was increased in the CSF 1 hour after injury, reaching a maximum of 762.5 pg/mL (geometric mean 135 pg/mL) and peaked between 2 and 4 hours, reaching a maximal concentration of 48,720 and 82,108 pg/mL (geometric means: 11,365 and 12,771 pg/mL), respectively. Thereafter, IL-6 returned to control values as measured in sham-operated animals. A peak of IL-6 also was observed in serum after 4 hours but at a much lower concentration (maximum 393 pg/mL, geometric mean 278 pg/mL) compared with CSF. The concentrations of IL-6 in CSF were significantly higher compared with those in serum between 2 and 8 hours after trauma (2 hours, P = 0.0002; 4 hours, P = 0.027; 8 hours, P = 0.01; paired t test; Fig. 2). Comparably low detectable IL-6 levels were found in serum of both trauma and sham-operated rats, which was attributed to the surgical procedure.
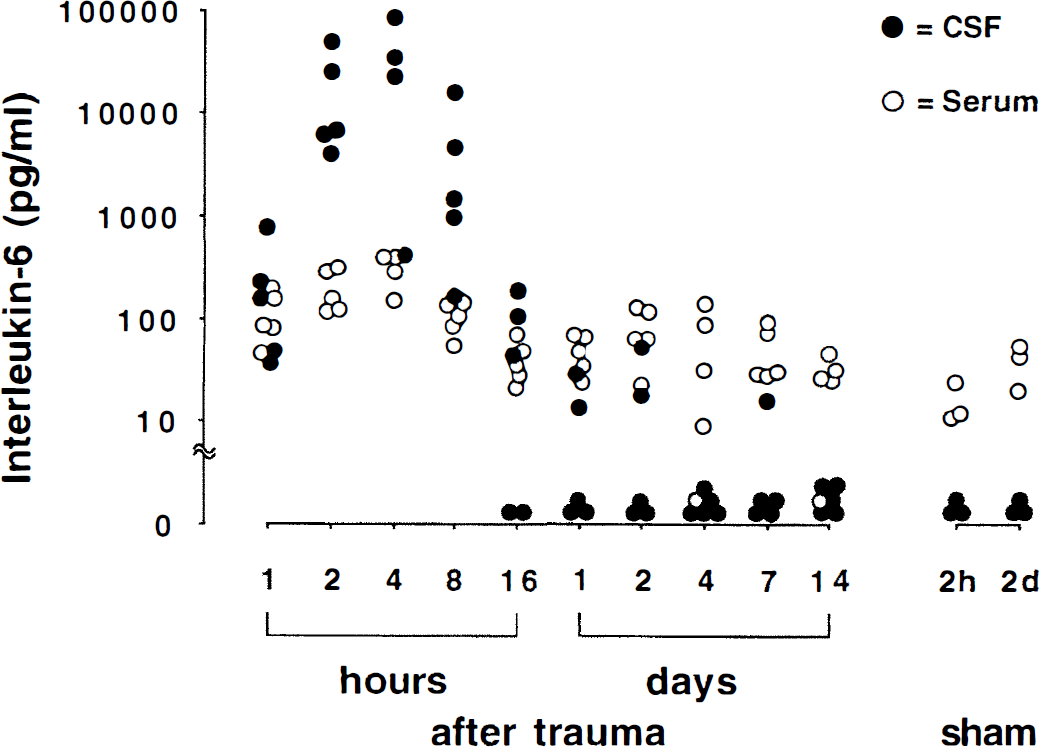
Release of interleukin-6 (IL-6) in CSF and serum of rats after brain injury. Concentrations of IL-6 in CSF and serum were determined at different time points (1 hour to 14 days) using the 7TD1 cell line, as described in Materials and Methods. Animals showed an increase of IL-6 in the CSF within 1 hour after trauma and significantly higher concentrations in the CSF than in serum between 2 and 8 hours (2 hours, P = 0.0002; 4 hours, P = 0.027; 8 hours, P = 0.01; paired t test). The closed circles represent the CSF-derived IL-6, and the open ones the serum derived IL-6 activities of individual animals. The IL-6 was no longer detectable in CSF after 24 hours and also was not found in sham-operated animals at 2 hours and 2 days after injury.
Localization of interleukin-6 mRNA according to in situ hybridization
Rat brains were analyzed by in situ hybridization in different coronal planes (about 3, 5, and 7 mm rostral from the interaural line according to Paxinos and Watson [1986]) for localization of IL-6 mRNA expression after axonal injury. These regions were chosen because they are located just below the area covered by the metal disk and thus below the center of the impact between lambda and bregma. Expression of IL-6 mRNA was observed in three regions: (1) thalamus, (2) basal subarachnoid space, and (3) parietal and basal cortex.
Thalamus. Different thalamic nuclei showed an expression of IL-6 mRNA. The cells were located in the midline (intermediodorsal, mediodorsal, paraventricular, reuniens, rhomboid, and xiphoid thalamic nuclei) and more caudally in lateral parts (ventrolateral, lateral posterior, and posterior thalamic nuclei) (Figs. 3A through C, and 4). Expression of IL-6 mRNA also was found in the medial geniculate body. The distribution pattern of IL-6 mRNA-expressing cells localized in different anatomical nucleus areas implies that these cells are neurons. This also is corroborated by the fact that (1) OX-42-positive microglia and macrophages stained on consecutive sections show different morphologic features and distribution, and (2) sections double-stained for GFAP and IL-6 mRNA or protein lacked overlapping signals (data not shown).
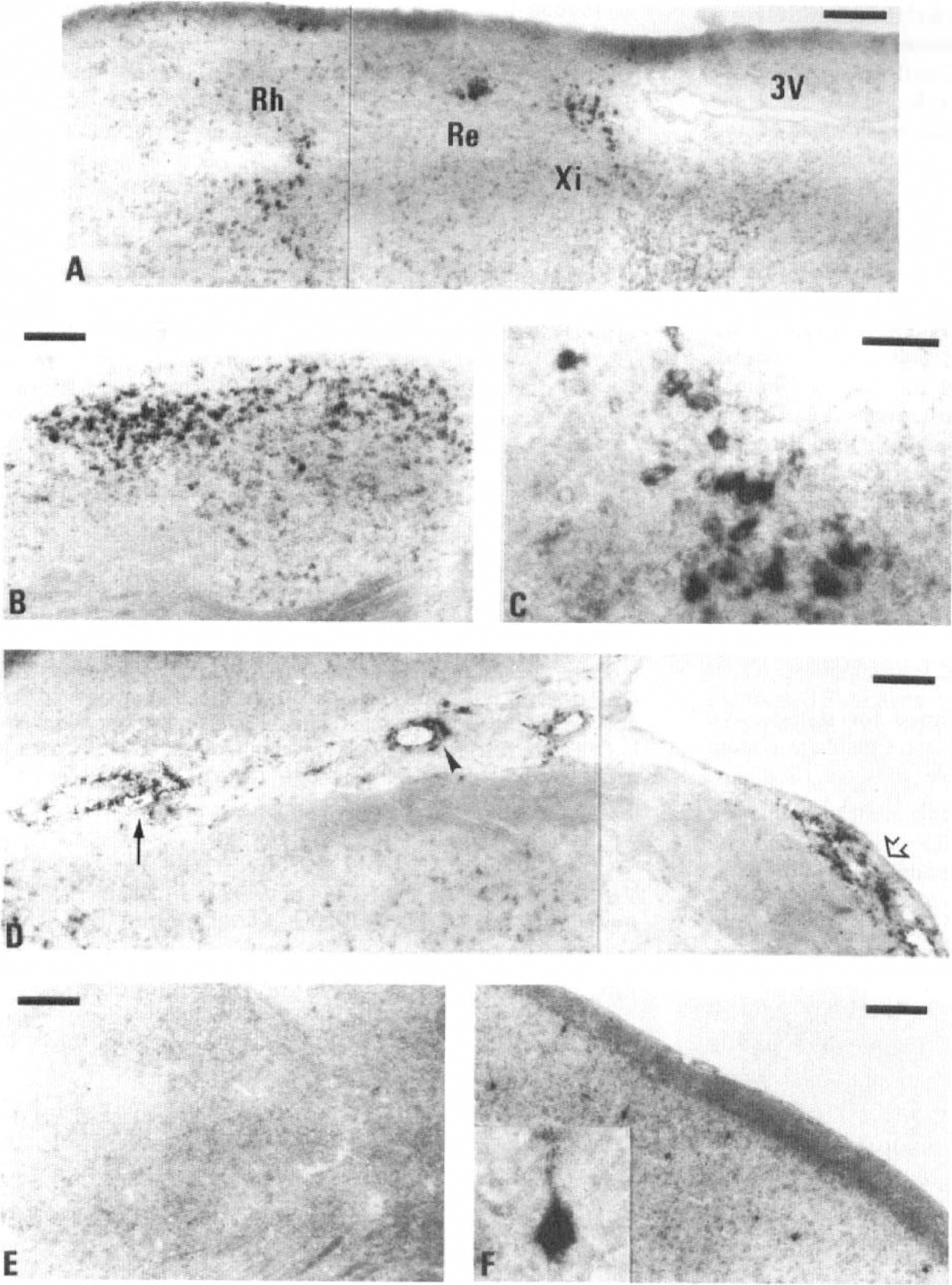
In situ hybridization for IL-6 in rat brain after brain injury. The brains of three rats were analyzed for IL-6 mRNA expression at various time points (1, 2, 4, 8, 16, and 24 hours) after axonal injury, and also the brains of two sham-operated animals at 2 hours after surgery. For each brain, 14-µm thick coronal cryosections at 3, 5, and 7 mm rostral from the interaural line were used. In situ hybridization using a digoxigenin-labeled cRNA probe was performed as described in Material and Methods.
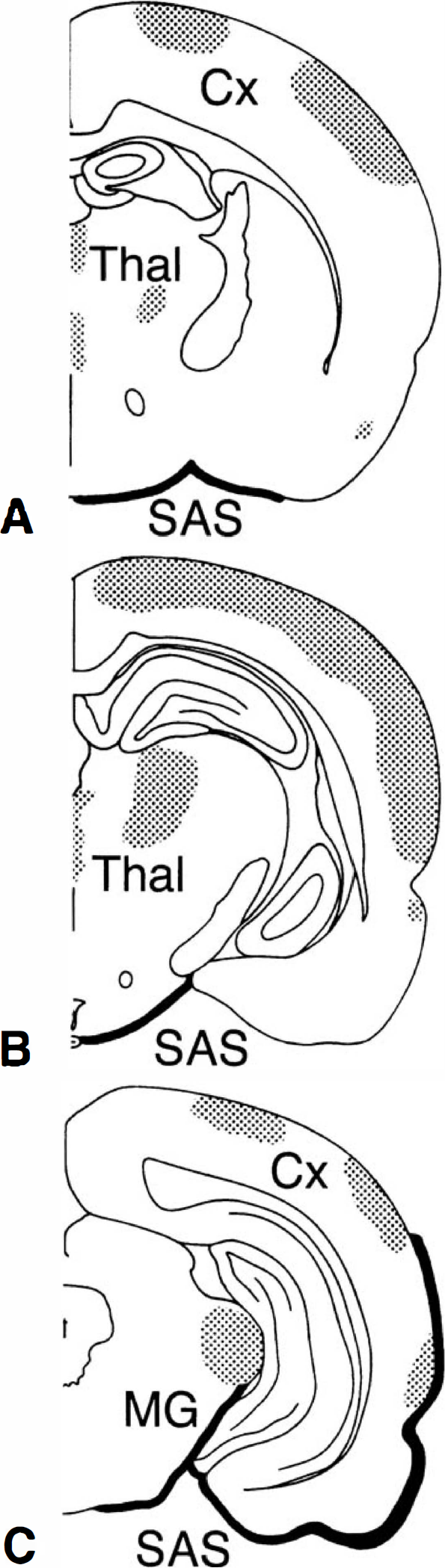
The IL-6 mRNA distribution in various brain regions revealed by in situ hybridization. Schematic drawing summarizes the greatest extent of IL-6 mRNA expression found in coronal sections at 7, 5, and 3 mm rostral from the interaural line (designated as A, B, and C, respectively) from 1 hour up to 24 hours after axonal injury. Increased IL-6 mRNA was seen within the subarachnoid space (SAS) in the more caudal sections (black), whereas in the parenchyma (shaded) the staining is mainly localized below the center of the traumatic impact in the cortex (Cx), as well as in thalamic nuclei (Thal) and in the medial geniculate body (MG). The IL-6 mRNA expression was found symmetrically in both hemispheres.
Basal subarachnoid space. Round or elliptically shaped cells, occasionally with small ramifications, were found intravascularly and perivascularly in the basal subarachnoid space (Fig. 3D), extending rarely into the superficial cortex. These cells were most readily seen in the more caudal sections (interaural 3 mm). Especially in the basal subarachnoid space and on the ventral side of the brain stem, petechial bleedings were seen macroscopically in a few animals. Furthermore, this is underlined by the presence of many cells in this area expressing major histocompatibility complex class II and OX-42 on serial sections (Csuka, et al., 1997). However, the cells found within and by the vascular lumen were instead considered to be adherent monocytes, endothelial cells, or pericytes, also known to synthesize IL-6 (Terebuh et al., 1992; Fabry et al., 1993).
Parietal and basal cortex. Positively stained pyramid-shaped neurons were scattered throughout the parietal and lateral cortex between the rhinal fissure and the interhemispheric fissure in various layers (Figs. 3F and 4). The highest density of these cells was localized below the center of the impact at 5 mm interaurally.
In sham-operated animals, no cells stained positively for IL-6 mRNA. Sense probe showed no specific signal in either trauma and sham-operated animals (Fig. 3E). A schematic diagram summarizes the distribution of IL-6 mRNA on coronal brain sections at 3, 5, and 7 mm rostral from the interaural line (Figs. 4A through C, respectively). Data shown represent the greatest extent of expression, as observed in all animals killed between 1 and 24 hours after injury. Expression of IL-6 mRNA was found symmetrically in both hemispheres.
Immunohistochemistry for interleukin-6
To investigate whether IL-6 mRNA expression was followed by a protein synthesis in brain parenchyma, immunohistochemical study using a polyclonal anti-IL-6 antibody was established (Fig. 5). Control brain showed a low constitutive expression of IL-6 in the isocortex layers II through IV, in the hippocampus, in thalamic nuclei, and in parts of the basal ganglia. A dramatic enhancement of IL-6 staining was visualized as early as 1 hour after injury throughout the first 24 hours and declined to baseline afterward. Immunoreactivity of IL-6 was localized intracellularly and paralleled IL-6 mRNA expression with regard to location and time. However, whereas IL-6 mRNA was restricted to single cells or to cell clusters, IL-6 protein production was demonstrated to be widespread in most brain regions. A strong signal appeared within 1 hour after injury in cortical cells, often pyramid-shaped, in the layers II through VI through the piriform cortex (Fig. 5A). From 48 hours on, the staining intensity became weaker, and also, less cells showed immunoreactivity for IL-6 located in these layers. At 1 and 2 weeks after trauma, IL-6 protein expression was comparable with that found in sham-operated or untreated animals. Within the hippocampus, a stronger staining was observed in the CA1 and CA3 regions compared with the CA2, whereas the dentate gyrus showed only a weak immunoreactivity (Fig. 5D). A more diffuse but unequivocal positive staining also was present in other regions. The IL-6 protein was upregulated in parts of the basal ganglia, like caudate-putamen and globus pallidus (Fig. 5B), in septal nuclei, in the thalamus (Fig. 5C), and also in the magnocellular preoptic nucleus, a component of the hypothalamic region. Although not as many cells were stained in these regions as seen in the cortex, the immunoreactivity remained clearly visible up to 1 week after trauma. In all brains, white matter and fiber tracts did not show IL-6 immunoreactivity. In some animals, the plexus epithelium also showed IL-6 production, whereas ependymal cells and the endothelium remained unstained (Fig. 5E). The latter staining pattern was found between 2 hours and 2 weeks after injury, and there was no correlation with concentrations of IL-6 in the CSF.
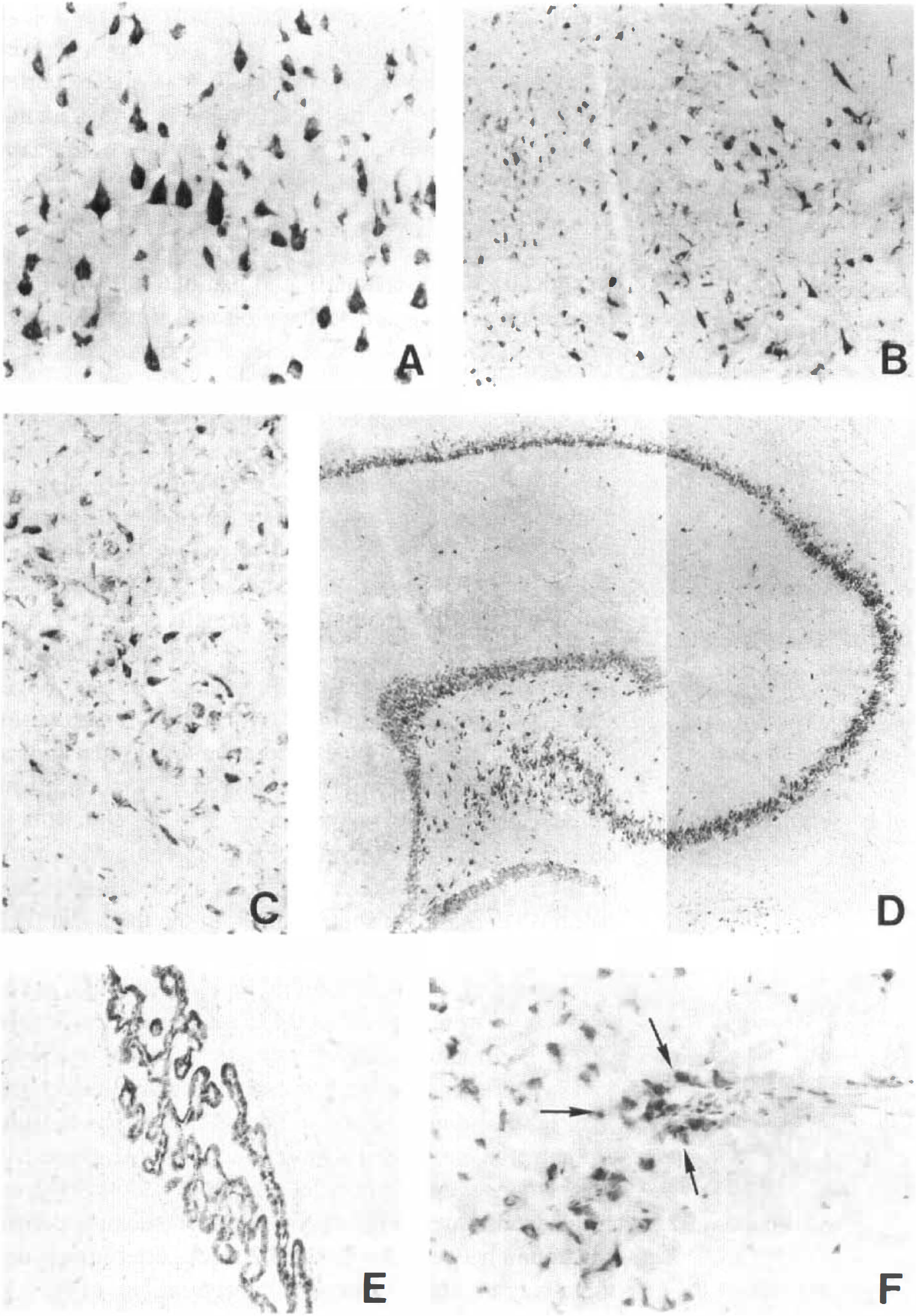
Brain regions displaying enhanced IL-6 protein production. Rat brain cryosections were stained using a goat polyclonal anti-IL-6 antibody as described in Materials and Methods. The IL-6 immunoreactivity is represented in various brain regions within the first 24 hours after trauma: cortex and globus pallidus at 8 hours in
The pattern of IL-6 staining was compared with the patterns obtained on consecutive slides using antibodies against markers for neurons (NeuN), astrocytes (GFAP), and for macrophages/microglia (OX-42) (Fig. 6). This revealed an almost identical cell distribution for IL-6 and NeuN (Fig. 6A and B, respectively), but not for GFAP (Fig. 6C) or OX-42 (Fig. 6D), indicating that, as suggested by the in situ hybridization results, neurons are the main cell type producing IL-6 in this model of axonal injury. Concerning macrophages, only few cells localized at the vicinity of vessels in the basal cortex stained for IL-6 (Fig. 5F). Staining specificity was demonstrated by preabsorbing anti-IL-6 antibody with recombinant IL-6 (1 mg/mL) before application to the brain sections, resulting in a complete abolishment of IL-6 immunoreactivity (Fig. 6F).
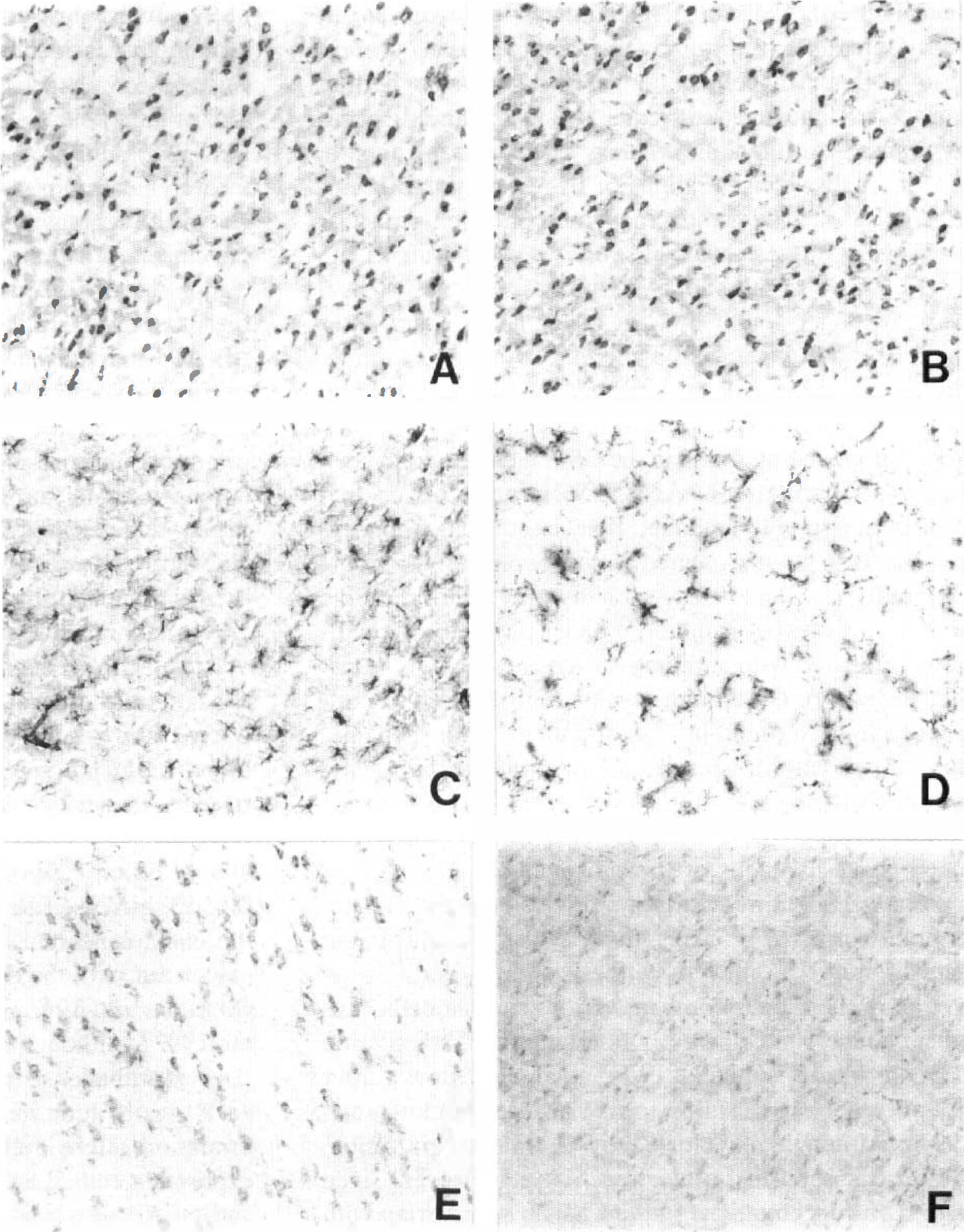
Immunoreactivity of IL-6 resembles neuronal distribution. This panel shows the increased production of IL-6 within the parietal cortex of an animal killed 1 hour after injury. Consecutive sections were stained for IL-6
DISCUSSION
In the current study, we investigated the production of IL-6 in a rat model of axonal injury caused by an acceleration impact to the closed skull (Foda and Marmarou, 1994; Povlishock et al., 1997). This experimental approach was used because diffuse axonal injury is a frequent feature of severe head trauma in humans, leading to dramatic neurologic and functional impairment (Povlishock, 1992; Gentleman et al., 1995). The production of IL-6 in CSF and serum of rats was analyzed to extend to a cellular and molecular level our previous results, which showed the association between release of IL-6 in brain-injured patients and the release of NGF into CSF, as well as with the incidence of the acute phase response (Kossmann et al., 1995, 1996).
In rats, the concentration of IL-6 in CSF increased within 1 hour after trauma and peaked between 2 and 8 hours. This is comparable with findings in various focal injury models such as stab wound (Woodroofe et al., 1991; Yan et al., 1992), fluid percussion (Taupin et al., 1993), and focal contusion (Shohami et al., 1994) where IL-6 levels were maximal between 2 and 5 hours. The similarities seen between the models of focal versus axonal injury indicate that, independent from the technique used to produce trauma, the induction of IL-6 follows a similar time pattern. The intensity of IL-6 immunoreactivity increased already at 1 hour in the brain tissue and persisted for about 24 hours, reflecting the pattern of IL-6 activity demonstrated in rat CSF. This may give an hint regarding the mechanism of transport of this cytokine from the parenchyma to the CSF.
Interestingly, the kinetics of IL-6 in rat CSF were similar to those observed in the CSF of brain-injured patients. However, in the rat maximal induction occurred within 24 hours, whereas in humans the IL-6 peak occurred within the first 3 to 6 days after injury, and the cytokine remained detectable up to 21 days. The prolonged presence of this cytokine in humans may result from multiple focal injuries, similar to models of focal brain damage in which IL-6 was found elevated for a longer time (Woodroofe et al., 1991; Taupin et al., 1993; Shohami et al., 1994). Furthermore, the animals were mechanically ventilated during the experimental procedure, whereas in patients, posttraumatic apnea and hypoxia may aggravate the brain damage. Additionally, it seems that temporal evolution of axonal changes appears much more rapidly in the rat compared with higher mammals (Povlishock, 1992; Povlishock et al., 1997). The hypothesis of intrathecal synthesis of IL-6 after brain injury is supported by the fact that IL-6 concentrations in rat CSF are higher than those in serum, as previously observed in humans (Kossmann et al., 1995). In addition, cerebral synthesis of IL-6 is supported in our experiments by the augmented expression of IL-6 mRNA and the widespread increase of IL-6 protein in various brain regions. How closely IL-6 concentrations in the CSF reflect actual levels of IL-6 in the parenchyma remains to be elucidated. Cells in the subarachnoid space can directly release IL-6 into the fluid compartment, whereas IL-6 produced by cells in the parenchyma has to be first transported into the ventricular or subarachnoid spaces by drainage of interstitial fluid or edema. In this regard, 10-fold higher concentrations of IL-6 were found in mi-crodialysates from the parenchyma compared with cisternal CSF after stab wound injury (Woodroofe et al., 1991). This also might explain the observation that IL-6 was no longer found in CSF at 24 to 48 hours after trauma, despite detectable increased immunostaining. Moreover, since in sham-operated or normal rat brain IL-6 intracellular staining is present at low concentrations without being found in the CSF, it can be concluded that under normal conditions IL-6 is not released into CSF in measurable amounts. If released at low concentrations, IL-6 may be cleared almost completely from the CSF bulk flow, and it may possibly play a different role (e.g., as a “neurotransmitter”) under physiologic conditions compared with an injury situation (as an inflammatory mediator). On the other hand, high IL-6 concentrations have been found in CSF of patients with spontaneous subarachnoid hemorrhage without parenchymal lesions (Mathiesen et al., 1993).
In contrast to the evident increase of IL-6 mRNA in macrophage-like cells of the subarachnoid spaces, IL-6 protein was identified only in a few cells in the vicinity of brain vessels (Fig. 5E). This may suggest a different regulation of IL-6 mRNA synthesis and IL-6 protein secretion in these cells compared with neuronal cells. Another cellular source possibly releasing IL-6 directly into CSF are plexus epithelial cells, as already shown after lipopolysaccharide injection (Vallieres and Rivest, 1997). Although not labeled by in situ hybridization, these cells showed immunoreactivity. However, since high IL-6 concentrations in the CSF do not overlap with the presence of IL-6 immunoreactivity in the plexus epithelium, its contribution to the increased IL-6 activity in CSF remains to be further elucidated.
The use of in situ hybridization allowed us to localize IL-6 mRNA expression to three regions of the injured brain and to different cell types. The cells found in the subarachnoid space are likely migrating peripheral blood leukocytes, since they were seen in the proximity of the vessels. Our data suggest that CR3-expressing activated cells may be recruited in these regions and locally secrete various cytokines, including IL-6, as detected by in situ hybridization. However, the almost identical kinetics observed in the production of IL-6 at protein and at mRNA level imply that preformed IL-6 may be stored in the cytoplasm of these cells and may be promptly released as a result of margination/chemotactic signals. Supportive of this hypothesis are findings showing constitutive IL-6 synthesis and activity in cell lysates of various cell types, including monocytes and neutrophils (Terebuh et al., 1992). Also consistent with our results are data demonstrating that resident cells, such as perivascular and meningeal macrophages, become activated after trauma (Giulian et al., 1989; Gehrmann and Banati, 1995; Aihara et al., 1995), and produce IL-6 (Woodroofe et al., 1991).
Early neutrophil accumulation in brain tissue has been shown in various models of focal injury and along with concurrent production of IL-6 (Woodroofe et al., 1991; Taupin et al., 1993; Clark et al., 1994). Conversely, in our model, macrophages, and not neutrophils, were identified in the subarachnoid space but not within the parenchyma. In accordance with our findings, it was suggested that diffuse neuronal degeneration occurs independently from the recruitment of inflammatory leukocytes, indicating that focal and diffuse injuries to the brain are characterized by distinct mechanisms of cell activation (Soares et al., 1995).
The traumatic impact seems to cause damage to vessels in a contrecoup location, as shown by the presence of subarachnoid hemorrhages within the basal regions (Foda and Marmarou, 1994), thereby facilitating an extravasation of inflammatory blood cells. A damaged blood-brain barrier represents the precondition necessary for the exit of white blood cells to the injury site (Giulian et al., 1989; Aihara et al., 1995; Soares et al., 1995). Blood-brain barrier permeability may be additionally increased by cytokines, including IL-6 itself or the chemokine IL-8, which also has been found to be associated with the dysfunction of this barrier after TBI (Maruo et al., 1992; De Vries et al., 1996; Kossmann et al., 1997). Although astrocytes are capable of producing IL-6 after challenge, they do not seem to contribute to the early posttraumatic IL-6 release, since no GFAP-immunoreactive cells overlapped with IL-6 mRNA-expressing cells. Lack of co-localization of IL-6 mRNA and GFAP also was shown by others after penetrating injury, indicating that an astrocytic response to trauma is unlikely to be responsible for the early IL-6 production (Giulian et al., 1989; Woodroofe et al., 1991; Yan et al., 1992).
The capability of neurons to synthesize IL-6 in vitro was shown only recently (Ringheim et al., 1995). The identification of IL-6 mRNA-positive as well as IL-6-immunoreactive cells with pyramid-like morphologic features similar to the NeuN staining pattern indicates that neurons in vivo release IL-6 after a traumatic challenge. Surprisingly, we did not find constitutive IL-6 mRNA expression in sham-operated animals, which is in contrast to previous reports, suggesting expression of this cytokine in normal rat brain in the hippocampus, nucleus caudatus putamen, striatum, cortex, parts of the hypothalamus, and the optic tract (Yan et al., 1992; Schöbitz et al., 1993; Gadient and Otten, 1994a,b). However, IL-6 immunoreactivity revealed a weak constitutive production in these areas of control brain, thus confirming earlier results at the mRNA level. The discrepancy between our in situ hybridization findings and those reported by others may result from methodology, in this case immunodetection of DIG-labeled probes versus radioactively labeled ones (Yan et al., 1992; Schöbitz et al., 1993), or polymerase chain reaction from tissue extracts (Gadient and Otten, 1994a,b). Therefore, we regard cells exhibiting positive staining as foci of an extremely upregulated transcription/decreased degradation of IL-6 mRNA.
The IL-6 secreted by neurons may function as a neurotrophic factor and as a immune modulator (Wagner, 1996). Interleukin-6 can promote neuronal survival either directly (Hama et al., 1991; Von Coelln et al., 1995) by protecting against glutamate, an excitatory neurotransmitter released in neurotoxic concentrations after TBI (Faden et al., 1989; Baker et al., 1993; Toulmond et al., 1992; Yamada et al., 1994), or indirectly by inducing the synthesis of NGF or other growth-promoting molecules in glial cells (Frei et al., 1989; Giulian, 1993; Kossmann et al., 1996). Interleukin-6 released in the injured brain may trigger astrocyte proliferation and the formation of an astroglial scar (Giulian et al., 1989; Selmaj et al., 1990).
The high IL-6 concentrations in the damaged brain may, on the other hand, activate microglia/macrophages and trigger a series of events ultimately leading to neuronal death and phagocytosis of cellular debris (Giulian et al., 1989, 1993). Interestingly, ceramide, a potent inducer of apoptosis (Hannum and Obeid, 1995; Skowronski et al., 1996), a mechanism of cell death also occurring in neurons as a result of TBI (Rink et al., 1995), was shown to promote IL-6 gene expression and protein release in human astrocytoma cells (Fiebich et al., 1995). Therefore, a lethal condition for neurons may be associated with a prolonged production of IL-6. Overexpression of IL-6 in the brain of transgenic mice has detrimental consequences for the development and physiologic mechanisms of the CNS, leading to a marked neurologic deficit (Campbell et al., 1993). In addition, long-term pretreatment of cultured neurons with IL-6 seems to increase the intracellular calcium response mediated by the N-methyl-D-aspartate receptor, therefore enhancing the neurotoxicity of excitatory amino acids (Qiu et al., 1995). These results taken together suggest that the dual function cytokines may display being either beneficial or deleterious, depending on their concentration and on the duration of their production within the brain in pathologic situations (Morganti-Kossmann et al., 1992; Rothwell and Relton, 1993).
In conclusion, the reported results describe the induction of IL-6 mRNA and protein within a few hours after axonal injury, and the release of IL-6 protein into the CSF. Possible sources for IL-6 synthesis are mainly neuronal cells, invading white blood cells in the subarachnoid space and meningeal macrophages. Depending on the local concentrations and on the cell type activated, IL-6 thus may exert neuroprotective and neurotoxic effects. Similarities with previous findings obtained on humans indicate that this is a suitable model for further studying the complex pathophysiologic mechanisms of axonal injury.
Footnotes
Acknowledgments
The authors thank Ms. E. Ammann at the Division of Research, Department of Surgery, and Ms. R. Frick at the Laboratory of Microsurgery, Department of Neurosurgery, University Hospital Zürich, for their technical assistance. The authors also thank S. Schwyter and N. Wick for the photographic work, and Dr. S. R. Barnum, UAB, for critically reading the manuscript.