
Abstract
Inhibitors of apoptosis and of excitotoxic cell death reduce brain damage after transient and permanent middle cerebral artery occlusion. We compared the neuroprotective effects of two caspase family inhibitors with the N-methyl-
Keywords
Several laboratories provide evidence for cell death by an apoptotic mechanism in animal models of cerebral ischemia (Li et al., 1995a,1995b,1995c; MacManus et al., 1995a,1995b; Charriaut-Marlangue et al., 1996). Apoptosis is a cell suicide program under active cell control. The interleukin-1β converting enzyme (ICE) family caspases, implicated in apoptotic cell death (Yuan and Horvitz, 1990; Ellis et al., 1991), are the human homologues of the nematode Caenorhabditis elegans CED-3. Eleven members of this family have been identified, which share a common QACXG consensus sequence (Alnemri et al., 1996). Caspase 1 (ICE) cleaves a pro-interleukin-1β 31-kilodalton (kd) protein to generate mature 17.5-kd interleukin-1β, which is involved in inflammatory reactions (Dinarello, 1994), apoptosis (Friedlander et al., 1996), as well as focal and global cerebral ischemia (Saito et al., 1996; Liu et al., 1993). Caspase 3 (CPP32), another important family member implicated in apoptotic cell death, cleaves poly(ADP-ribose)polymerase and DNA-dependent protein kinase delta, among other substrates (Tewari et al., 1995; Nicholson et al., 1995).
Reports from our laboratory show that peptide inhibitors of the caspase family (N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone [z-VAD.FMK] and N-benzyloxycarbonyl-Asp-Glu-Val-Asp-fluoromethyl ketone [z-DEVD.FMK]) are neuroprotective after focal reversible cerebral ischemia in mice and rats (Hara et al., 1997a). Both drugs reduced tissue injury and associated neurologic deficits after 2-hour middle cerebral artery occlusion (MCAO) when assessed at 24 and 72 hours (Hara et al., 1997a). Interleukin-1β formation, a product of ICE cleavage, was significantly blocked by z-VAD.FMK, a rather unselective caspase inhibitor, but not by the putative caspase 3 inhibitor z-DEVD.FMK. Smaller infarcts also were observed in transgenic mice expressing a dominant negative mutation of ICE (Friedlander et al., 1997; Hara et al., 1997b). Cleavage of pro-interleukin-1β is important to the development of ischemic damage (Relton and Rothwell, 1992; Yamasaki et al., 1995) as is a cascade that may involve activation of caspase 3. We speculated that an important mechanism by which z-VAD and z-DEVD decrease infarct size relates to blockade of ischemia-induced apoptosis and in this report, we tested the effects of peptide methylketones in a murine model of brief (30 minutes) transient focal cerebral ischemia.
Mild injury augments apoptotic cell death in in vitro and in vivo models of cell injury, including brain ischemia. In the rat, infarct development is delayed until 3 days after 30 minutes of distal middle cerebral artery occlusion whereas tissue damage develops much earlier (within 24 hours) after 90 minutes or 2 hours of reversible MCAO. In this rat model, terminal deoxynucleotidyl transferase—mediated dUTP nick-end labeling (TUNEL)–positive cells and laddered DNA are present in the periinfarct zone, suggesting a role for apoptotic cell death (Du et al., 1996).
In the current study, we developed a model of mild ischemia in mice showing delayed infarct development and delayed appearance of several apoptosis markers (TUNEL staining, DNA laddering). We then evaluated the efficacy and the treatment window of two caspase inhibitors. We determined whether inhibition of caspase family members by the use of peptide methylketones decreases DNA laddering in ischemic tissue as measured by densitometry on agarose gels. Delayed therapeutic intervention—possibly directed against apoptotic death—may become an important therapeutic strategy to reduce mild ischemic insults (Du et al., 1996).
METHODS
Drugs
Both z-VAD.FMK and z-DEVD.FMK were obtained from Enzyme Systems Products (Dublin, CA, U.S.A.). The compounds were dissolved in 0.3% dimethylsulfoxide (DMSO; MC/B, Norwood, OH, U.S.A., prepared with 0.1 M phosphate-buffered saline [PBS] pH 7.4). Cycloheximide (Chex) was purchased from Sigma (St. Louis, MO, U.S.A.) and dissolved in PBS. We obtained (+)-MK-801 hydrogen maleate (MK-801) from Research Biochemicals International (Natick, MA, U.S.A.) and dissolved it in PBS.
Physiology
Regional cerebral blood flow (rCBF) was measured by Laser-Doppler flowmetry (PF2B, Perimed, Stockholm, Sweden) along with arterial blood pressure and heart rate as described (Hara et al., 1996, 1997a,
b
). Arterial blood samples (50 μL) were analyzed for pH, oxygen (Pa
Ischemia model
Adult male 129/SV mice (18 to 20 g, Taconic farms, Germantown, NY, U.S.A.) were anesthetized with 1.5% halothane for induction and maintained on 1.0% halothane in 70% N2O and 30% O2 using a Fluotec 3 vaporizer (Colonial Medical, Amherst, NH, U.S.A.). Ischemia was induced with a 8.0 nylon monofilament coated with silicone resin/hardener mixture (Xantopren and Elastomer Activator, Bayer Dental, Osaka, Japan), as described previously (Hara et al., 1996). The filament was introduced into the left internal carotid artery up to the anterior cerebral artery. For filament withdrawal, the animals were briefly reanesthetized. The MCAO procedures were performed by two investigators (M. E. examined the effects of z-VAD.FMK and MK-801 experiments, whereas M. S. -S. tested z-DEVD.FMK).
Treatment protocol
Both z-VAD.FMK (120 ng) and z-DEVD.FMK (160 ng or 480 ng) were injected intracerebroventricularly (2 μL; bregma −0.9 mm lateral, −0.1 mm posterior, −3.1 mm deep) either 10 minutes before ischemia or 6, 12, or 18 hours after reperfusion. Control animals were injected with 2 μL of 0.3% DMSO. Cycloheximide (10 mg/kg) was administered intraperitoneally 1 day before ischemia. MK-801 (3 mg/kg) was administered intraperitoneally 10 minutes before ischemia or 3 or 6 hours after reperfusion. Control animals were injected intraperitoneally with a corresponding volume of PBS. Since hypothermia is a well known consequence of MK-801 treatment, animals were kept in an incubator (ThermoCare System, Incline Village, NV, U.S.A.) at 30 to 31°C for 6 hours after treatment to maintain a body temperature of 37°C. Core temperatures were measured using a thermometer (BAT-12, Physitemp, Clifton, NJ, U.S.A.) after 3 and 6 hours. Control mice (36.8° ± 0.2°C at 3 hours and 36.7° ± 0.2°C at 6 hours) did not differ from MK-801–injected animals (36.7° ± 0.1°C at 3 hours and 36.7° ± 0.2°C at 6 hours).
Infarct measurement
For survival longer than 24 hours, infarct size was measured from hematoxylin-eosin (H&E)–stained sections, whereas triphenyltetrazolium chloride (TTC; Sigma, St. Louis, MO, U.S.A.) stained 2-mm coronal sections were used to assess injury after shorter periods of occlusion. For H&E-stained sections, the brains were first immediately frozen in 2-methylbutane on dry ice and then sectioned. For TTC staining, animals were decapitated, and the brains were divided into five coronal 2-mm sections using a mouse brain matrix (RBM-2000C, Activational Systems, MI, U.S.A.) and stained with 2% 2,3,5-TTC. Infarction volume was quantitated in TTC-stained sections or in H&E-stained cryostat section (12 μm) with an image analysis system (M4, Imaging Research, St. Catherines, Ontario, Canada) and calculated by summing the volumes of each section determined directly (Huang et al., 1994) or indirectly using the following formula: contralateral hemisphere (in cubic millimeters) minus undamaged ipsilateral hemisphere (in cubic millimeters) (Swanson et al., 1990). Differences between “direct” and “indirect” volumes are likely to be accounted for by brain swelling.
Neurologic deficits
Mice were tested for neurologic deficits and scored as described by Bederson and others (1986) with the following minor modifications (Hara et al., 1996): 0, no observable neurologic deficit (normal); 1, failure to extent right forepaw (mild); 2, circling to the contralateral side (moderate); 3, loss of walking or righting reflex (severe). The rater was naive to the treatment protocol and to the group's identity. Assessments were made at 30 minutes after onset of ischemia and 24 and 72 hours (if necessary) after reperfusion.
DNA analysis
Samples were obtained at different time points after 30 minutes of transient MCAO. Ischemic striatal tissue was taken from the third 2-mm section along with homologous tissue from contralateral side after the brain was cut coronally with a brain matrix. For quantitation of DNA damage, a terminal transferase—dependent [32P]ddATP end-labeling method was used (Tilly and Hsueh, 1993) with minor modifications, as described previously (Hara et al., 1997b). Three micrograms of DNA were used in the labeling procedure together with 35 ng of a 100-base pair (bp) DNA fragment as an internal standard. Electrophoresis was performed on the DNA on a 2.0% agarose gel (agarose 3:1, Amresco, Solon, OH, U.S.A.), autoradiographed together with a [32P] standard, and analyzed with the M4 image analysis system. DNA less than 10 kb was used as an index of total DNA fragmentation. To measure oligonucleosomal damage more specifically, laddered DNA less than 1000 bp was measured by summing the areas of each peak (areas under the curve) minus baseline densitometry readings. The method of quantitation was validated using an artificial “smear-ladder” system in which smeared DNA was extracted from a decapitated mouse brain and incubated at 37°C for 48 hours (MacManus et al., 1995b). In a total amount of 1 μg of DNA, 0.02, 0.04, 0.08, and 0.12 μg of a commercially available 200-bp ladder (Invitrogen, Carlsbad, CA, U.S.A.) were added to a constant amount of smeared DNA (Fig. 1). In another experiment, increasing amounts of smeared DNA (0.3, 0.5, 0.7, 0.9 μg) were added to a constant amount of ladder (0.1 μg). The measurements for “total DNA fragmentation” and “DNA laddering” were related linearly to the amount of total (smeared) and laddered DNA (error < 10%).
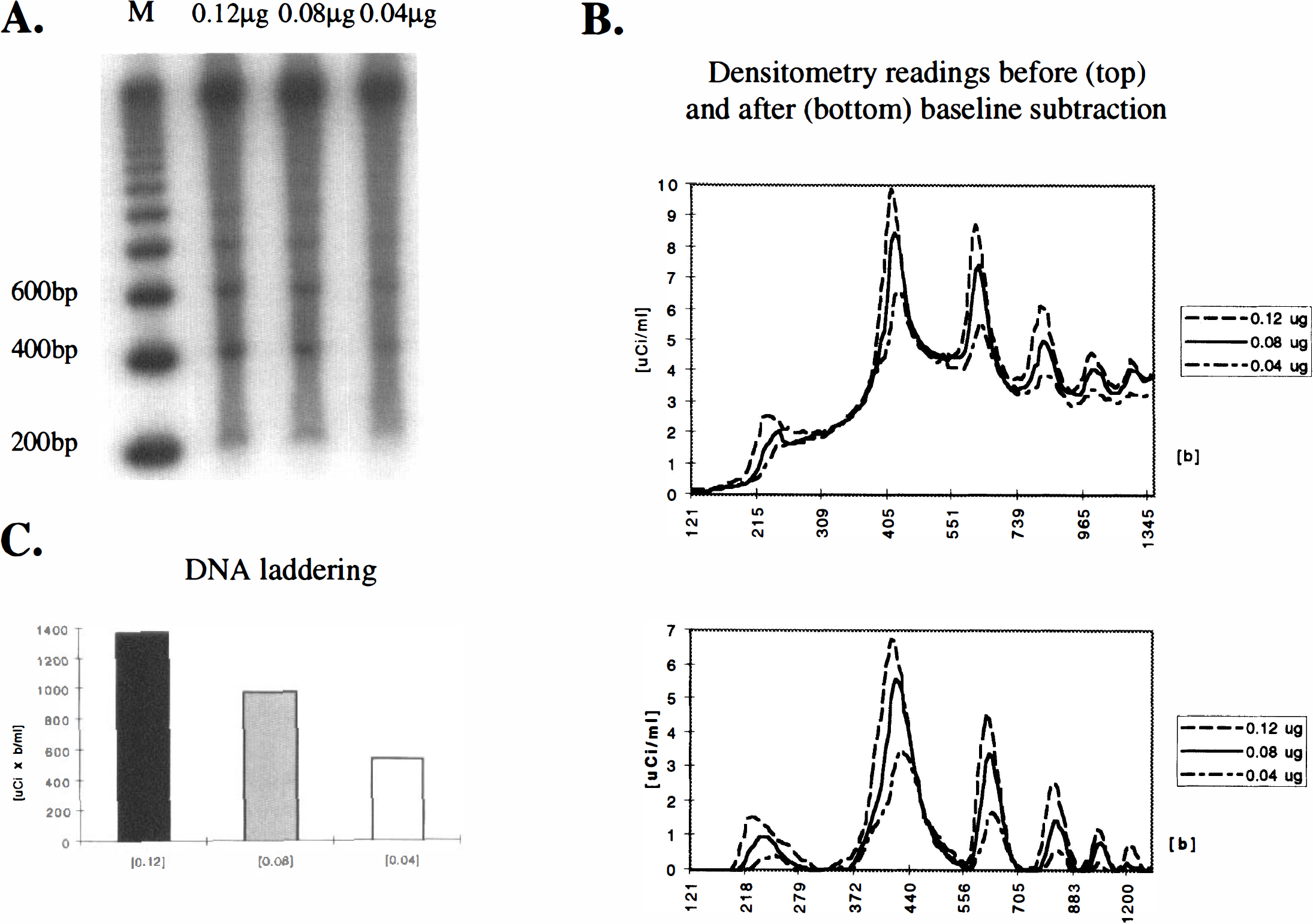
Quantitative evaluation of “laddered” and “smeared” DNA by agarose gel electrophoresis. To validate the method, decreasing amounts of laddered DNA (0.12 μg, 0.08 μg, 0.04 μg) were added to constant amounts of smeared brain DNA (MacManus et al, 1995b), end-labeled with [32P]ddATP, separated by agarose gels (
Terminal deoxynucleotidyl transferase—mediated dUTP nick-end labeling
The TUNEL staining was carried out according to the method of Gavrieli and others (1992) with minor modifications according to Wood and associates (1993). Terminal deoxynucleotidyl transferase and biotinylated dUTP were obtained from Boehringer Mannheim (Mannheim, Germany). The biotinylated dUTP was visualized by the avidin—biotin method with 3′–3′ diaminobenzidine as chromogene. The reaction was intensified with 0.04% nickel chloride. For negative controls, either terminal deoxynucleotidyl transferase or biotinylated dUTP was omitted. For positive controls, the sections were treated with DNase I.
For cell counts, sections were counterstained lightly with H&E. Following a systematically random sampling scheme and based on stereologic techniques (Gundersen, 1992; West, 1993; Gómez-Isla et al., 1996), five coronal sections (12 μm) were selected, taken at equally spaced intervals (2 mm) through the full rostrocaudal extent of the brain. The boundaries of the area occupied by TUNEL-positive cells within each section were marked using the Bioquant Image analysis system (Nashville, TN, U.S.A.). The entire volume of the lesion occupied by TUNEL-positive cells was calculated according to the principle of Cavalieri (Cavalieri, 1966). Within each section with TUNEL-positive cells, a systematically random sampling scheme was applied to count cells. The total number of TUNEL-positive cells in each brain was estimated by using ~80 optical dissectors and a 40 × objective. Each optical dissector was a 100 × 100 μm sampling box with extended exclusion edges. The appropriateness of the sampling scheme chosen was evaluated by calculating the precision of the estimates in each animal, expressed as the coefficient of error (West and Gundersen, 1990). In all cases, the coefficient of error was less than 0.10, suggesting that a minimal amount of variance in the counts can be attributed to the technique. The calculation of total number of TUNEL-positive cells was performed by multiplying the volume density of TUNEL-positive cells by the total volume occupied by TUNEL-positive staining.
Glial fibrillary acid protein immunohistochemistry/TUNEL double staining and CD-45 (leukocyte common antigen) immunohistochemistry
Glial fibrillary acid protein (GFAP)/TUNEL double staining was performed to determine if glial cells show TUNEL staining. The TUNEL staining protocol was performed as described earlier. Subsequently, sections were washed and blocked with 10% normal goat serum in PBS. GFAP double labeling was performed with a rabbit GFAP polyclonal antibody (1:500, Dako Corporation, Carpintiera, CA, U.S.A.) using a three-stage avidin—biotin method and biotinylated goat anti-rabbit IgG as secondary antibody. The reaction product was visualized with 3-amino-9-ethylcarbazole reagent (Elite PK 6101 Kit, Vector Laboratories, Burlingame, CA, U.S.A.). For controls, single GFAP immunostaining was performed in adjacent sections.
To identify inflammatory cells, CD-45 (leukocyte common antigen) immunohistochemical study was performed on adjacent sections with a purified rat anti-mouse antibody (1:500, Clone 30F11.1, Pharmingen, San Diego, CA, U.S.A.) and a three-stage avidin—biotin method with a biotinylated rabbit anti-rat IgG (BA 4001, Vector Laboratories) as secondary antibody.
Statistical analysis
Data are presented as mean ± SD. Statistical comparisons were made by one-way analysis of variance and followed by Dunnett test (rCBF) or Tukey test (physiology). For neurologic deficits, Mann-Whitney rank sum test was applied for two groups, and Kruskal-Wallis one-way analysis of variance on ranks followed by Dunn test for three or more groups. For unpaired data (infarction volume and area, DNA laddering, cell counts) two-tailed Student's t test was applied. Analysis was made using the software SigmaStat (Jandel Corporation, San Rafael, CA, U.S.A.) or. Excel (Microsoft, Redmond, WA, U.S.A.). P < 0.05 was considered statistically significant.
RESULTS
Mouse model of mild focal cerebral ischemia
Physiologic parameters before, during, and after ischemia were within the normal range and did not differ between groups. Mean arterial blood pressure remained stable at 110 ± 12 mm Hg, and heart rate at 480 ± 70 beats per minute. Blood gases were stable at pH 7.4 ± 0.1, Pa
No evidence for injury was detected 24 or 72 hours (TTC staining) after 10 minutes of MCAO in pilot experiments (n = 5 each). Twenty-four hours after 30 minutes of MCAO, ischemic changes were not detected by TTC (n = 5) or grossly by H&E (n = 5), although infrequent striatal cells showed light microscopic changes consistent with ischemia.
At 72 hours of reperfusion after 30 minutes of MCAO, clear evidence for infarction was found. Infarct size was 32.7 ± 8.3 mm3 (n = 17) after vehicle (DMSO) was injected intracerebroventricularly 6 hours after reperfusion. Vehicle injection did not alter infarct size, since noninjected animals showed the same injury volume (31.4 ± 19.2 mm3, n = 12). The lesion was located predominantly in the striatum where cells were severely pyknotic. Cortical injury, by comparison, was minor. Lesion volume did not increase significantly by 7 days (33.5 ± 21.1 mm3, n = 8, no intracerebroventricular injection), and brain swelling did not contribute significantly to infarct size, as assessed by an indirect method for measurement of infarct size. Cycloheximide (Chex) pretreatment (10 mg/kg, n = 1) reduced infarct size (10.5 ± 9.0 mm3 versus 31.4 ± 19.2 mm3, P < 0.05) at 72 hours, suggesting the importance of protein synthesis inhibition.
Inhibitors of the caspase family reduce infarct size
Table 1 contains the data for experiments using caspase inhibitors and MK-801. Infarct size in vehicle-treated controls were smaller in the z-DEVD.FMK experiments (22.6 to 26.5 mm3) versus the other two vehicle groups (27.1 to 38.1 mm3), probably because the surgical procedure for z-DEVD.FMK was performed by a different investigator (see Methods).
Effects of the nonselective caspase inhibitor z-VAD.FMK, the putative caspase 3 inhibitor z-DEVD.FMK, and the NMDA-antagonist MK-801 on infarct volume after 30 minutes of MCAO
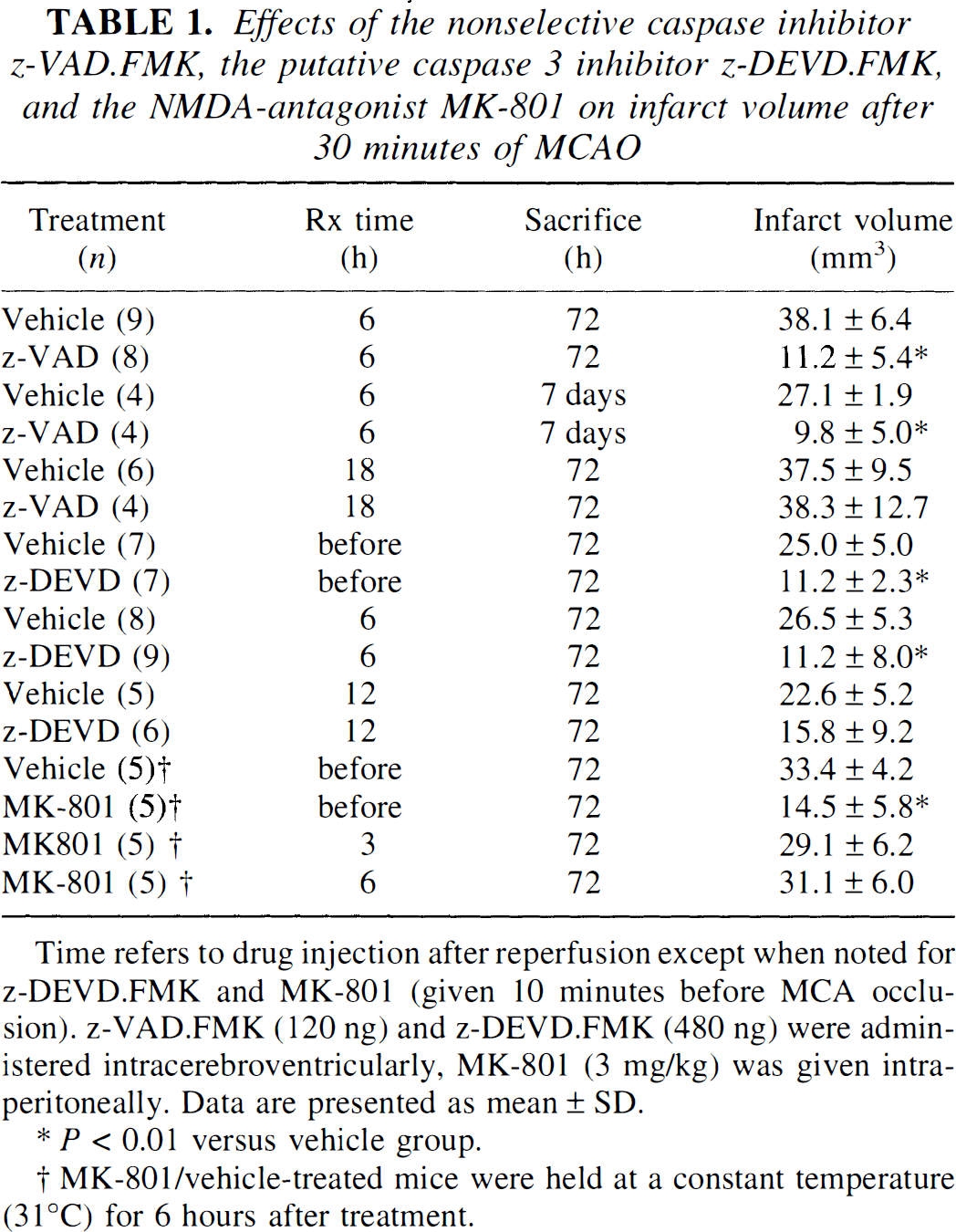
Time refers to drug injection after reperfusion except when noted for z-DEVD.FMK and MK-801 (given 10 minutes before MCA occlusion). z-VAD.FMK (120 ng) and z-DEVD.FMK (480 ng) were administered intracerebroventricularly, MK-801 (3 mg/kg) was given intraperitoneally. Data are presented as mean ± SD.
P < 0.01 versus vehicle group.
MK-801/vehicle-treated mice were held at a constant temperature (31°C) for 6 hours after treatment.
A dose of z-VAD.FMK (120 ng intracerebroventricularly) given 6 hours after reperfusion decreased infarct size (Table 1) and neurologic deficits at 72 hours (1.0 ± 0.9 versus 2.4 ± 0.9, P < 0.05). The protective effects sustained for at least 7 days (Table 1). When given at 18 hours after reperfusion, however, the drug was not effective in reducing gross infarct size (Table 1); neurologic deficits did show some improvements in this group, however (1.2 ± 0.8 versus 2.4 ± 0.9).
A dose of z-DEVD.FMK (480 ng given intracerebroventricularly 10 minutes before ischemia) significantly reduced infarction volume at 72 hours (Table 1). A lower dose (160 ng) did not reach statistical significance (18.0 ± 3.3 mm3 versus 24.7 ± 5.0 mm3, n = 7). Infarct size and neurologic deficits (0.4 ± 0.5 versus 1.7 ± 0.5, P < 0.05) were reduced when 480 ng z-DEVD.FMK was administered at 6 hours after reperfusion (Table 1). Infarct volumes, although decreased, did not reach statistical significance when z-DEVD was given 12 hours after reperfusion (Table 1).
The rCBF during ischemia and after reperfusion did not differ between groups (Table 2 and Table 4). There were also no significant group differences in physiologic parameters after z-VAD.FMK (Table 3) or z-DEVD.FMK treatment (Table 4).
rCBF during and after 30 minutes of filamentous middle cerebral artery occlusion in z-VAD.FMK and vehicle (0.3% DMSO) groups

z-VAD.FMK (120 ng) or vehicle were administered intracerebroventricularly in a volume of 2 μL 6 hours after reperfusion following 30 minutes of MCAO. rCBF was measured by Laser-Doppler-flowmetry (expressed as % of baseline). Data represent mean ± SD (n = 5–6). There are no significant differences between groups.
Physiologic variables 10 minutes before and until 1 hour after injections of z-VAD.FMK or vehicle (0.3% DMSO)
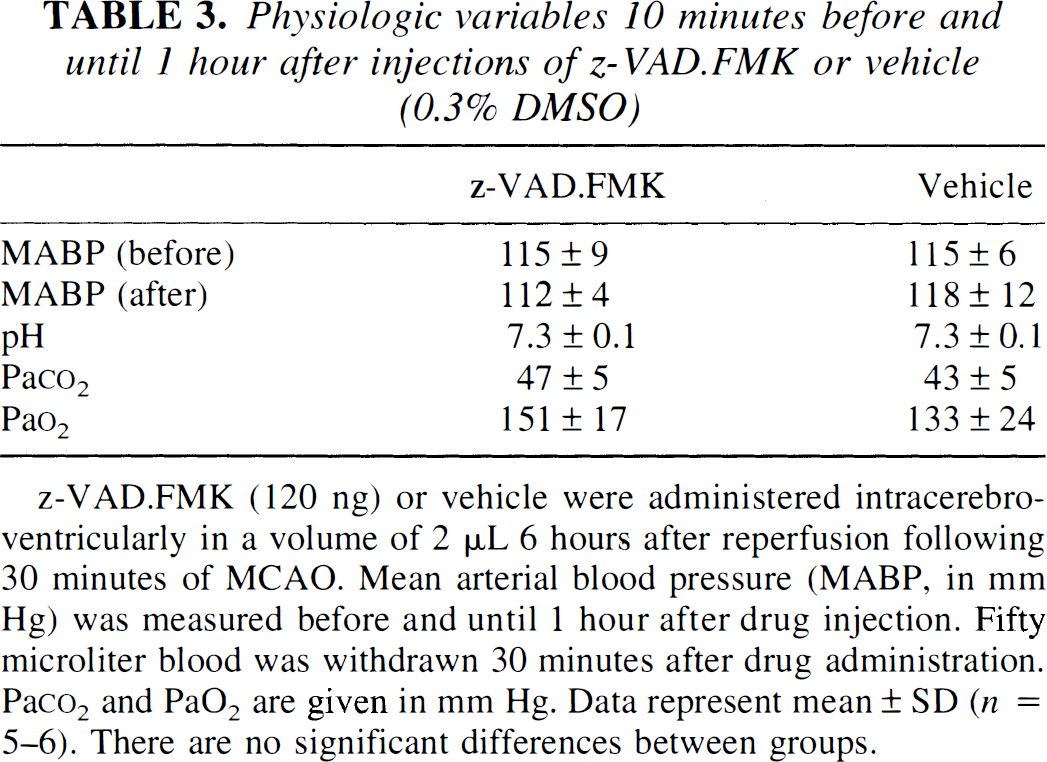
z-VAD.FMK (120 ng) or vehicle were administered intracerebroventricularly in a volume of 2 μL 6 hours after reperfusion following 30 minutes of MCAO. Mean arterial blood pressure (MABP, in mm Hg) was measured before and until 1 hour after drug injection. Fifty microliter blood was withdrawn 30 minutes after drug administration. Pa
Physiologic parameters before, during and after 30 minutes of ischemia (129/SV mice) after z-DEVD.FMK or vehicle (0.3% DMSO) pretreatment
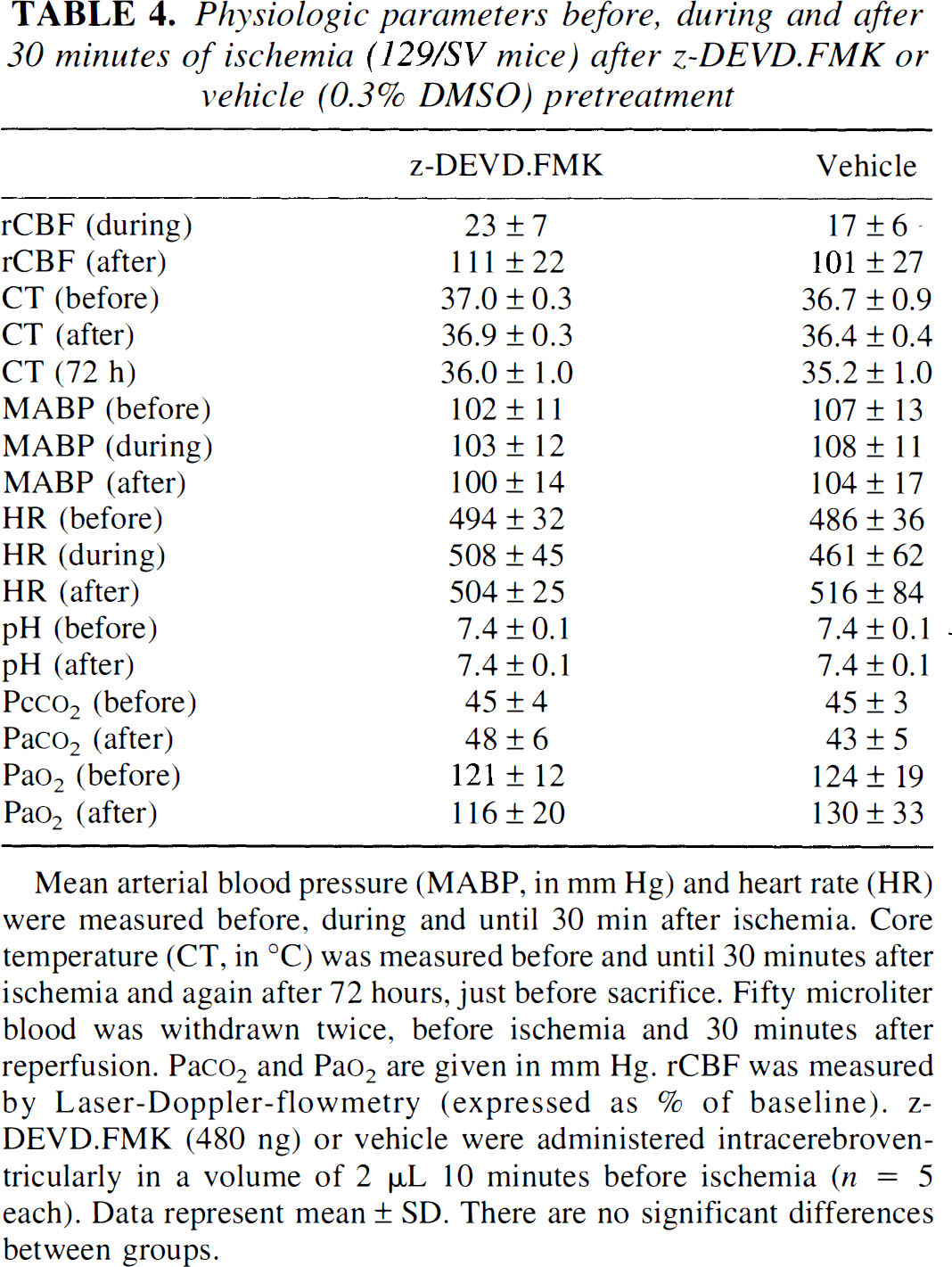
Mean arterial blood pressure (MABP, in mm Hg) and heart rate (HR) were measured before, during and until 30 min after ischemia. Core temperature (CT, in °C) was measured before and until 30 minutes after ischemia and again after 72 hours, just before sacrifice. Fifty microliter blood was withdrawn twice, before ischemia and 30 minutes after reperfusion. Pa
Treatment with MK-801 significantly reduced infarct size if given before MCAO but not when given 3 or 6 hours after reperfusion (Table 2 and Table 3). Neurologic deficits tended to be lower after pretreatment but did not differ from controls when MK-801 was given at 3 or 6 hours after reperfusion (0.4 ± 0.6 [pretreatment], 1.0 ± 0.8 [3 hours], 1.2 ± 0.8 [6 hours] versus 1.0 ± 0.7 [control], n = 4 to 5 per group).
DNA fragmentation
The TUNEL-positive cells, present in low numbers throughout the striatum at 24 hours, profoundly increased at 72 hours. The TUNEL-positive cells were identified morphologically as neurons, many of them with nuclear condensation and apopoptic bodies. Almost no TUNEL-positive cells contained GFAP double staining (Fig. 2, n = 3 for each time point). At 72 hours, low numbers of CD-45–positive inflammatory cells were found within the vasculature; however, only single cells were located within ischemic brain tissue (not shown).
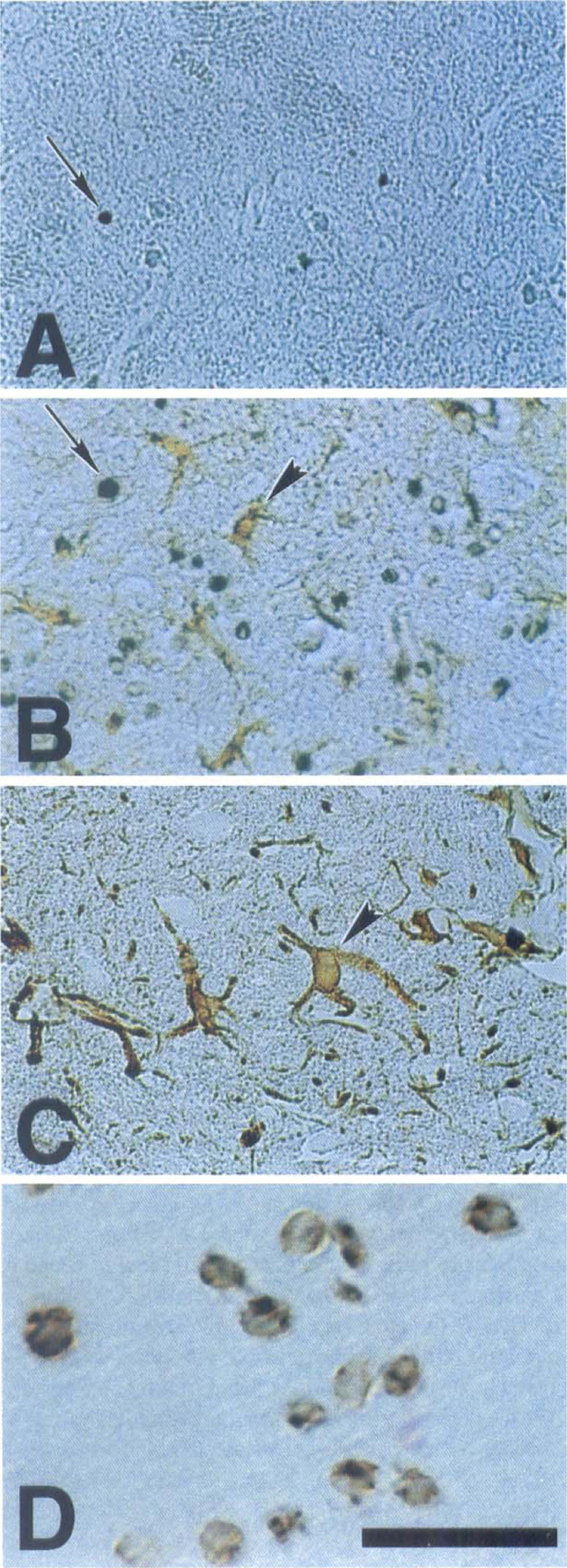
Time-dependent changes in the appearance of terminal deoxynucleotidyl transferase—mediated dUTP nick-end labeling (TUNEL)–positive cells and glial fibrillary acid protein (GFAP) staining in striatum after reperfusion following 30 minutes of middle cerebral artery occlusion (MCAO). Tissue sections (6 μm) obtained at 24 hours (
Laddered DNA, detected first at 24 hours, increased 16-fold at 72 hours and 46-fold at 7 days compared with 24 hours (Fig. 3, n = 3 per group). DNA damage was already present at 6 hours as smeared DNA and increased substantially between 24 and 72 hours (Fig 3). Total DNA damage and laddering were significantly inhibited at 72 hours by Chex pretreatment (Fig. 3 and Fig. 4).
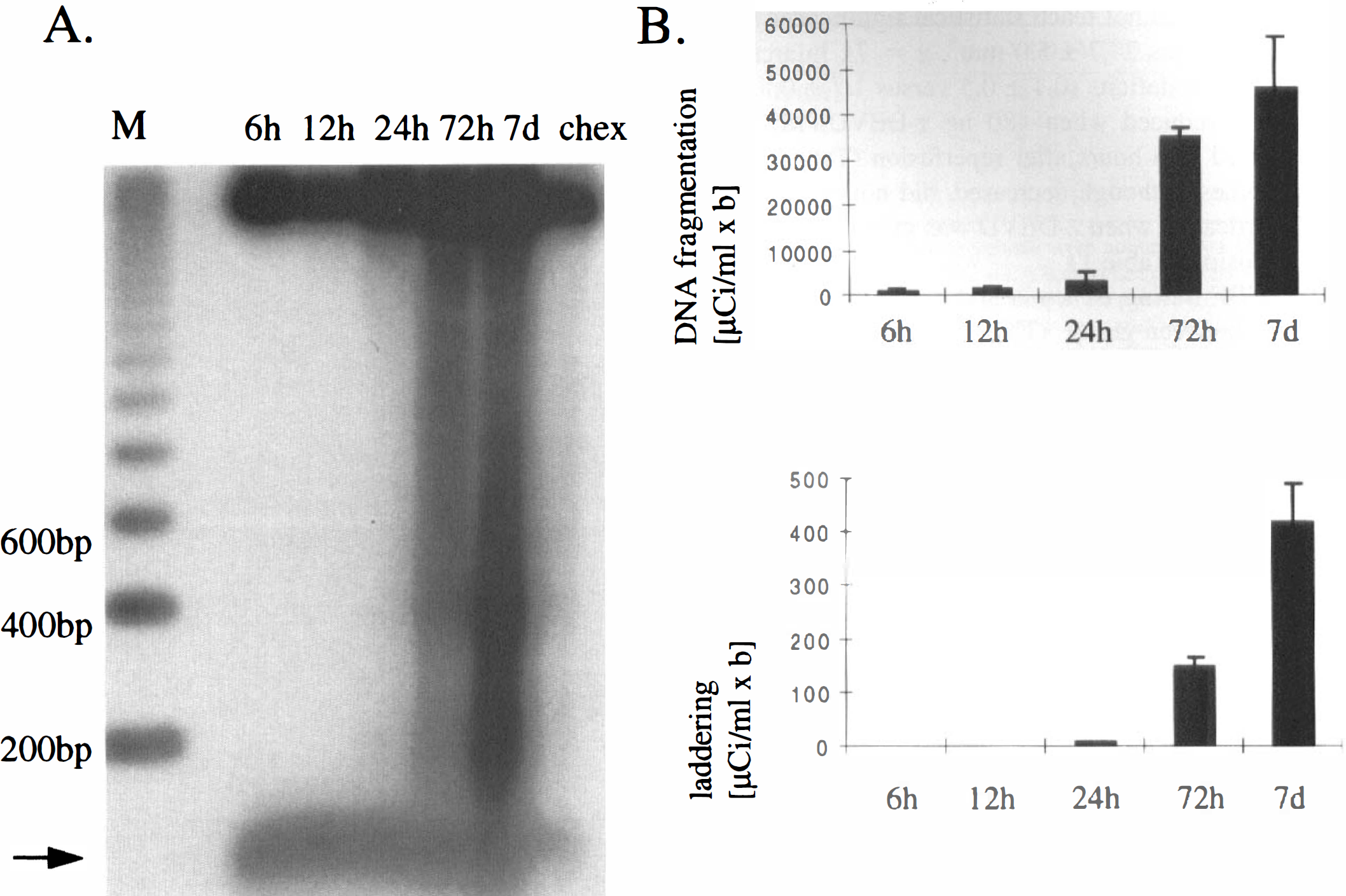
Cycloheximide (Chex, 10 mg/kg) pretreatment inhibited DNA laddering during reperfusion after 30 minutes of MCAO. DNA was isolated from ischemic striatal tissue after various times after reperfusion, end-labeled with [32P]ddATP, electrophoresed on a 2% agarose gel, and autoradiographed. The autoradiogram (
Cycloheximide (Chex) pretreatment (
To test the hypothesis that caspase inhibitors block apoptosis after ischemia, we compared the number of TUNEL-positive cells in z-VAD.FMK—treated animals (120 ng, given at 6 hours, n = 4) versus DMSO (n = 4) at 72 hours. Total number of TUNEL-positive cells was significantly reduced in the treatment group (540,000 ± 258,000 versus 1,390,000 ± 446,000, P < 0.05), and the lesion volume containing TUNEL-positive cells was reduced by 60%. The density of TUNEL-positive cells did not decrease, however (82,700/mm3 ± 6500/mm3 versus 84,900/mm3 ± 9200/mm3).
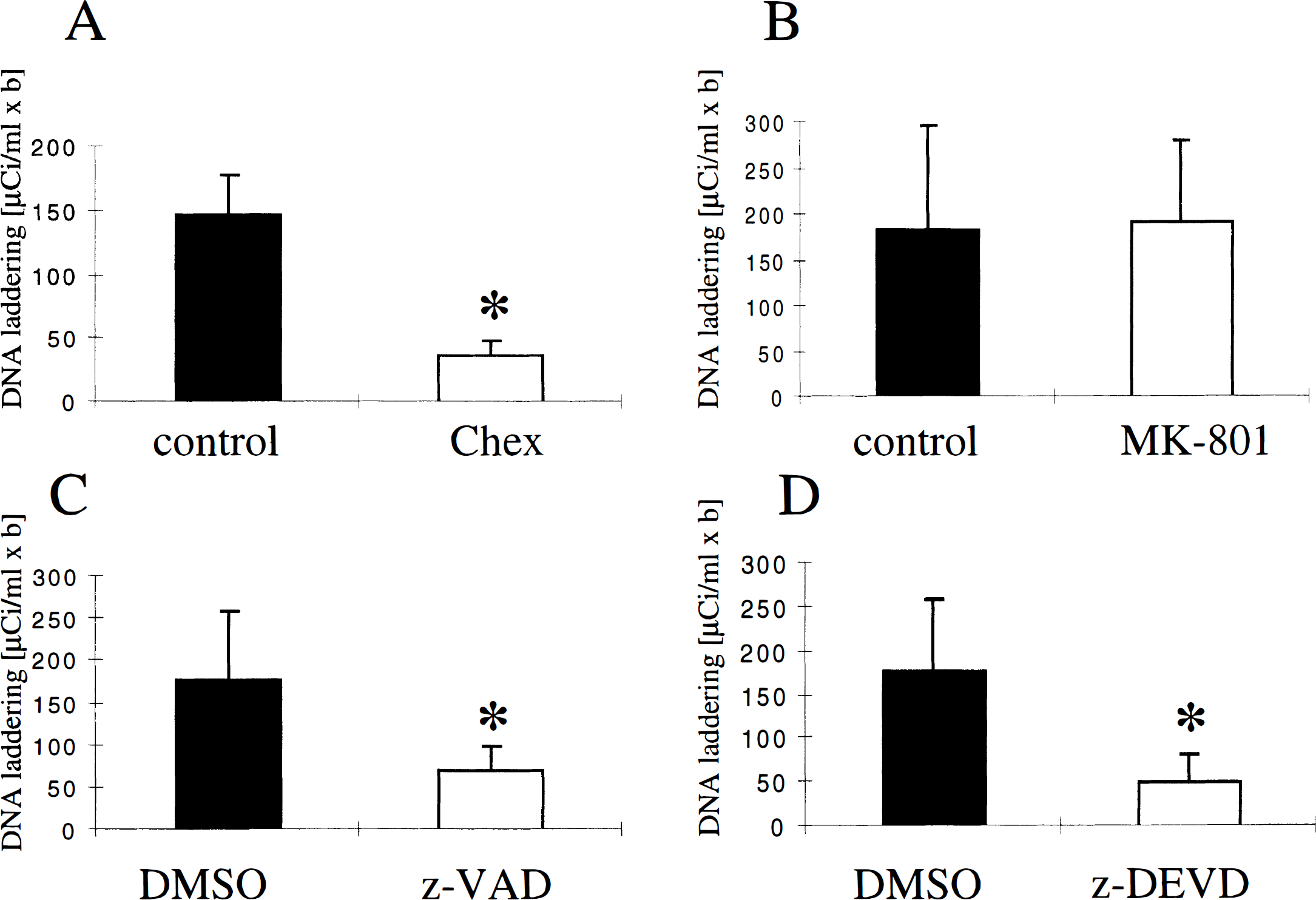
A significant reduction in total DNA fragmentation and DNA laddering within ischemic tissue was found at 72 hours in z-VAD.FMK—treated (n = 4) and z-DEVD.FMK—treated (n = 5) animals (administered 6 hours after reperfusion) compared with controls (n = 9) (Fig. 4 and Fig. 5), whereas MK-801 pretreatment (3 mg/kg, n = 4) did not reduce DNA fragmentation or DNA laddering compared with saline-injected controls (n = 4) (Fig. 4 and Fig. 5).
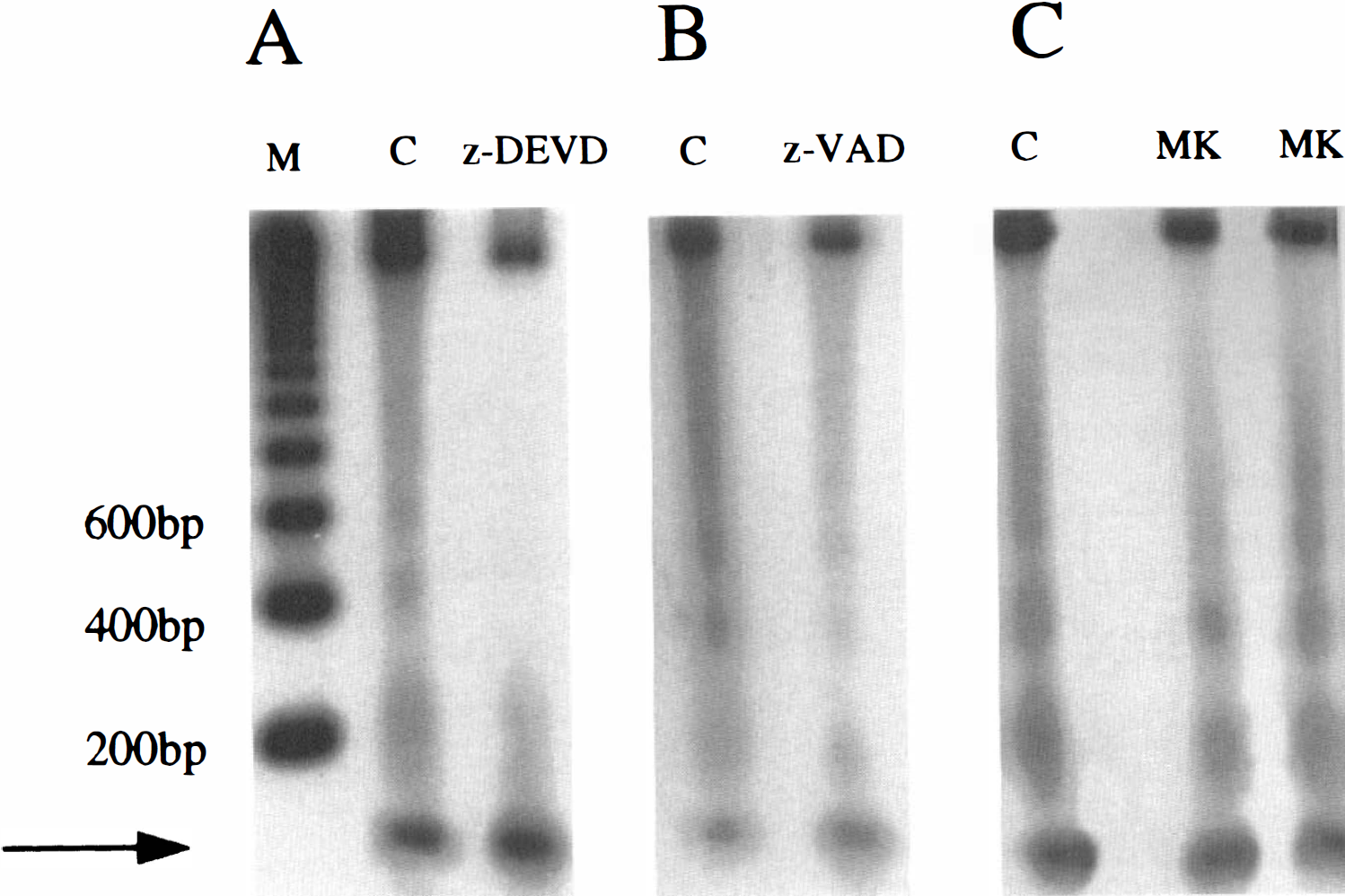
The z-DEVD.FMK (480 ng) (
DISCUSSION
We provide evidence for the importance of apoptosis to the delayed development of tissue injury after mild focal cerebral ischemia (30 minutes of MCAO) in the mouse. In this model, ischemic brain damage became grossly visible at 72 hours and did not expand at 7 days after reperfusion. The TUNEL-positive cells and DNA laddering were observed first 24 hours after reperfusion. Cells staining TUNEL-positive probably were neurons, since they did not stain positive for GFAP or CD-45. In fact, Li and colleagues (1995a, b ) came to a similar conclusion, since they found GFAP co-staining in only a few TUNEL-positive cells (10%) after 2 hours of MCAO in the rat. Treatment with inhibitors of apoptosis such as cycloheximide, z-VAD.FMK, or z-DEVD.FMK reduced ischemic injury after 30 minutes of MCAO, and peptide methylketones were more effective after mild (30 minutes) than after more prolonged ischemia (2 hours of MCAO). In the 30-minute model, z-VAD.FMK and z-DEVD.FMK decreased infarct volume by 70% and 57%, respectively, whereas the degree of protection was 45% and 35%, respectively, after 2-hour MCAO (Hara et al., 1997a; Moskowitz and Ma, 1997). Peptide methylketones also were more successful many hours (6 hours) after 30 minutes of MCAO compared with 0 to 1 hours after more prolonged (2 hours) MCAO (Hara et al., 1997a; Moskowitz and Ma, 1997). Hence, mild focal cerebral ischemia provides a useful experimental tool to assess the contribution of apoptosis to delayed mechanisms of cell death.
DNA laddering may reflect the consequences of a specific cascade promoting tissue injury after ischemia, which is distinct from necrosis. We found that oligonucleosomal damage was a useful indicator of apoptotic cell death and that a newly developed method for quantitation was reliable and reproducible. A linear relation was found between the amount of laddered DNA used in the labeling procedure and the densitometry readings in validation experiments (Fig. 1). Decreases in laddered DNA did not always accompany decreases in infarct size but depended on whether apoptosis was inhibited. For example, MK-801 pretreatment decreased ischemic damage but not DNA laddering (Fig. 4 and Fig. 5). Similar findings were observed after administering 3-aminobenzamide, a widely-used inhibitor of poly(ADP-ribose)polymerase (Endres et al., 1997). On the other hand, inhibitors of apoptotic cell death such as cycloheximide, z-VAD.FMK, or z-DEVD.FMK clearly inhibited oligonucleosomal DNA damage and decreased infarct size (Fig. 3 through Fig. 5; Table 1).
The protective mechanisms of peptide methylketones and MK-801 are different. As observed, MK-801 did not decrease DNA laddering, and its therapeutic window was relatively short in mild ischemia when apoptotic cell death is prominent. We infer that apoptosis probably is not the predominant mechanism of cell death after N-methyl-
The literature supports the notion that MK-801 and caspase inhibitors act through distinct yet complementary and synergistic mechanisms. Choi and colleagues found that NMDA receptor antagonists unmask apoptosis after oxygen—glucose deprivation in neuronal cell cultures in vitro. Caspase inhibitors are protective in culture only after MK-801 pretreatment (Gwag et al., 1995). In vivo, we demonstrated synergistic protective effects after treatment with MK-801 plus caspase inhibitors (Moskowitz and Ma, 1997). A 35% decrease in infarct size was observed in a mouse model of focal ischemia after combining doses and treatment times when neither compound provided protection alone.
Both the mouse and rat model (30 minutes of ischemia) (Du et al, 1996) show delayed infarct development, prominent DNA laddering, and TUNEL-stained cells. Damage in the mouse, however, was primarily within striatum, whereas injury was mostly within cortex in the rat model. Additionally, the lesion in the mouse did not expand after 3 days as it did in the rat. These differences may relate to greater collateral blood flow in the mouse, as well as differences in vascular anatomy or suture placement, with attendant differences in blood flow reduction. Cycloheximide reduced injury in both species, emphasizing the importance of protein synthesis. The ability of cycloheximide to inhibit apoptosis is well documented in ischemia (Linnik et al., 1993; Du et al, 1996). Cycloheximide may at the same time increase bcl-2 expression, which also would reduce tissue injury (Furukawa et al., 1997). In general, cycloheximide suppresses synthesis of all proteins, including those that may afford neuroprotection (e.g., superoxide dismutase, growth factors). Hence, it may be difficult to predict the effects of cycloheximide on tissue injury. In our studies, we demonstrate the relevance of caspases to injury development in a model in which cycloheximide reduces infarct size.
Our data could have important implications for the treatment of stroke or other evolving CNS injuries in humans, or even for procedures that carry a risk of brain damage (e.g., neurosurgical procedures or cardiopulmonary bypass). Caspase inhibitors administered at relatively late time points after mild brain injury or ischemia may protect injured tissue. We conclude that inhibitors of ICE family caspases might become valuable drugs to treat stroke in humans.
Footnotes
Abbreviations used
Acknowledgment
The authors thank Dr. H. Hara for advice.