
Abstract
Interleukin-1β (IL-1β) is expressed after cerebral ischemia and blocking its action reduces subsequent ischemic brain injury. However, the mechanisms by which IL-1β affects ischemic brain are not understood. To investigate the role of IL-1β in regulation of tumor necrosis factor-alpha (TNF-α) and intercellular adhesion molecule-1 (ICAM-1) during focal cerebral ischemia, the authors studied mutant mice deficient in the IL-1 converting enzyme (ICE) gene (ICE knockout [KO] mice). Ninety-four adult male ICE KO and wild-type mice underwent 3, 6, 12, and 24 hours of permanent middle cerebral artery occlusion using the suture method. Expression of TNF-α and ICAM-1 protein in ischemic brain was examined using immunohistochemistry and Western blot analysis. Neither ICE KO nor wild-type mice had significant differences in CBF and body temperature measurements during the ischemic procedure. TNF-α expression increased in the ipsilateral hemisphere after 3 hours of occlusion, peaked at 12 hours and decreased at 24 hours of ischemia in both ICE KO and wild-type mice. ICAM-1 immunohistochemistry showed that the number of ICAM-1–positive vessels in the ischemic hemisphere was reduced in ICE KO mice (P < .05). Western blot analysis showed that ICAM-1 protein expression was significantly attenuated in the ipsilateral hemisphere in the ICE KO mice, which paralleled the immunohistochemistry results, The authors' results indicate that TNF-α expression is increased in both ICE KO and wild-type mice suggesting that TNF-α expression is not related to or upregulated by IL-1β. ICAM-1 expression is significantly reduced in the ICE KO mice suggesting that IL-1β plays an important role in the upregulation of adhesion molecules during focal cerebral ischemia.
Keywords
Research on experimentally induced cerebral ischemia has revealed the importance of immune or inflammatory molecules in the pathogenesis of cerebral ischemia. Interleukin-1β (IL-1β), a proinflammatory cytokine, is markedly increased during global and focal cerebral ischemia. Exogenous application of IL-1β exacerbates ischemic brain injury (Feuerstein et al., 1997; Loddick and Rothwell, 1996; Minami et al., 1992; Yamasaki et al., 1995; Yamasaki et al., 1992), while administration or overexpression of IL-1 receptor antagonists greatly reduces ischemic brain injury in rats and mice (Betz et al., 1995; Garcia et al., 1995; Relton et al., 1996; Yang et al., 1997). IL-1β converting enzyme (ICE) cleaves proIL-1β to IL-1β, the biological active form. The observations that mice expressing a mutant ICE inhibiting protein or having the ICE gene knocked out, have reduced brain damage, further supports a crucial role for IL-1β during cerebral ischemia (Hara et al., 1997a; Schielke et al., 1998).
Similarly, tumor necrosis factor-alpha (TNF-α) expression is markedly increased after permanent and temporary cerebral ischemia (Botchkina et al., 1997; Pantoni et al., 1998). The action of TNF-α may worsen brain injury after ischemia (Buttini et al., 1996; Liu et al., 1994; Yang et al., 1998) because administration of a TNF-α antagonist or soluble TNF-α receptor greatly reduces ischemic brain injury in rats and mice (Nawashiro et al., 1997; Shohami et al., 1996). In contrast, Bruce et al. (1996) reported that ischemic brain injury was increased in mice genetically deficient in the TNF receptor, suggesting that TNF-α may have a neuroprotective function. However, TNF-α may play a primary role in “setting the stage” for inflammatory and immune reactions elicited by cerebral ischemia (Feuerstein et al., 1994).
Although IL-1β and TNF-α are implicated as mediators of ischemic brain injury in experimental models and in humans, the relationship between the release of IL-1β and TNF-α during cerebral ischemia is still unclear. Whether the actions of these two cytokines are independent or whether the action of one cytokine is mediated by the other has yet to be determined.
Intercellular adhesion molecule-1 (ICAM-1) has been identified as a ligand for leukocyte adhesion molecules (Lindsberg et al., 1996), including LFA-1, Mac-1, and p150, 95. There is increasing experimental evidence in vivo that the β2 integrins on leukocytes may be important in the pathogenesis of inflammatory diseases (Chopp and Zhang, 1996). The reaction of ICAM-1 on the endothelial cells is thought to facilitate lymphocyte migration and inflammatory cell infiltration (Adams and Shaw, 1994). Numerous reports suggest that ICAM-1 is upregulated after various brain disorders, including cerebral ischemia, and inhibition of ICAM-1 expression can reduce ischemic brain injury (Connolly et al., 1996a; Handa et al., 1995; lander et al., 1995). Recent studies show that infarct volume is significantly reduced in ICAM-1 knock out mice after cerebral ischemia and reperfusion (Soriano et al., 1996; Soriano et al., 1998). Because IL-1β and TNF-α increase adhesion molecule expression in the endothelial cell cultures (Fabry et al., 1992; McCarron et al., 1994), expression of these cytokines in ischemic brain may be linked to tissue injury through ICAM-1 expression.
This study was designed to identify (1) the time course of the expression of TNF-α after middle cerebral artery occlusion (MCAO) in mice deficient in the ICE gene (ICE KO) and wild-type (WT) mice; (2) the relationship between IL-1β deficiency and TNF-α expression after ischemic insult in these mice; and (3) the possible role of IL-1β and TNF-α in the regulation of ICAM-1 during cerebral ischemia.
MATERIALS AND METHODS
Animal preparation
Procedures for the use of laboratory animals have been approved by the institutional animal care and use committee. The ICE KO mice were created and characterized by the BASF Corporation, Worcester, MA (Li et al., 1995). These mice contain a null mutation in the ICE gene (ICE-/-) generated by homologous recombination in embryonic stem cells. The wild-type (WT, ICE+/+) mice were generated from the same chimeric founder, and interbred among themselves. Both strains have a mixed background of both SV129 and C57BL/6-derived genes.
Experimental groups
Ninety-four adult mice weighing 30 to 35 gm were used for three experiments: (1) ICE KO (n = 16) and WT (n = 16) mice underwent 3, 6, 12, and 24 hours of permanent MCAO (n = 4 at each time point). Two mice at each time point underwent sham surgery. After the animals were killed by decapitation, the brains were removed and prepared for TNF-α immunohistochemistry. (2) Thirty-two animals underwent a similar procedure with the brains being processed for ICAM-1 immunostaining. (3) ICE KO (n = 11) and WT (n = 11) mice underwent 3, 6, or 24 hours of MCAO or sham surgery, and were used to measure ICAM-1 protein using immunoprecipitation and Western blot analysis.
Middle cerebral artery occlusion
Adult ICE KO and WT mice were anesthetized with 4% isoflurane in 70%N2O/30%O2 and maintained at 1.5% isoflurane delivered through a face mask. The body temperature was maintained with a heated blanket using a feedback control system (73A, Yellow Springs, OH). The middle cerebral artery was permanently occluded for 3 to 24 hours in all groups of mice. Middle cerebral artery occlusion was performed as described in our previous studies (Yang et al., 1994). Briefly, the left common carotid artery was exposed through a midline incision. The internal carotid artery was isolated and its branch, the pterygopalatine artery, was ligated close to its origin. A 2-cm length of 5-0 rounded tip nylon suture was gently advanced from the external carotid artery through the common carotid artery and then up to the internal carotid artery for a distance of 11.0 ± 0.5 mm.
CBF measurement
Surface blood flow was measured using a laser-Doppler flowmetry monitor (Model BPM2 System, Vasamedics Inc., St. Paul, MN, U.S.A.) equipped with a 0.7-mm diameter probe (P-433, Vasamedics). Blood flow was measured 5 minutes before and 15 minutes after MCAO at three points: (1) the ischemic core area: 3.5 mm lateral to the sagittal suture and 1 mm posterior to the coronal suture on the ipsilateral hemisphere; (2) the control area: 3.5 mm lateral to the sagittal suture on the contralateral hemisphere; and (3) the perifocal area: 1 mm lateral to the sagittal suture on the ipsilateral hemisphere and 1 mm posterior to the coronal suture. Blood flow values are expressed as a percentage of baseline values.
TNF-α and ICAM-1 immunohistochemistry
The mice were decapitated 3, 6, 12, and 24 hours after permanent MCAO, and the brains were removed and frozen immediately in 2-methyl-butane solution (Mallinckrodt) at −40°C. Coronal cryostat sections (20-μm thickness) were cut and mounted on precleaned Probe-on-slides (Fisher Scientific, Pittsburgh, PA). After drying, the sections were fixed with Cornoy fixative solution (60% methanol, 30% chloroform, and 10% acetate) for 20 minutes. Nonspecific binding was blocked by treatment with 10% normal goat serum for 30 minutes at room temperature. The sections were incubated in a 1:100 dilution of poly clonal anti murine TNF-α primary antibody (Genzyme, Cambridge, MA) overnight at 4°C. A negative control was performed using the same concentration of normal rat immunoglobulin G instead of primary antibody. After treatment with 1% H2O2 in 30%/70% methanol/phosphate-buffered saline (PBS) solution, the sections were incubated with a biotinylated goat antirabbit secondary antibody in 1:200 dilution for 90 minutes at room temperature, followed by an ABC process (ABC-Elite Kit, Vector Lab., San Francisco, CA, U.S.A.). Finally, the sections were treated with stable DAB (Research Genetics, Huntsville, AL, U.S.A.) as a peroxidase substrate and counter-stained with hematoxylin. The same procedures were used for ICAM-1 immunohistochemistry except for the use of 10% normal rabbit serum as a blocker of nonspecific binding, 1:300 dilution of rat monoclonal antimouse CDS4 as a primary antibody (Caltag, San Francisco, CA, U.S.A.), and biotinylated rabbit antirat antibody (Vector Lab) as a secondary antibody.
The number of deep brown—stained TNF-α–positive cells and ICAM-1–positive vessels from each mouse at each time point were counted in the three coronal sections located at +0.86, +0.38, −0.10 mm from Bregma. The immunopositive stained cells in the contralateral and the ipsilateral hemisphere were counted separately. The counting was performed manually to exclude the infiltrating cells occasionally expressing TNF-α and/or rCAM-1.
ICAM-1 immunoprecipitation and Western blot analysis
Western blot analysis was performed to verify the reaction specificity of the ICAM-1 antibody referred to above against mouse brain tissues. The mice were killed after 3, 6, and 24 hours of MCAO. Brains were immediately removed and divided into contralateral cortex, contralateral basal ganglia, ipsilateral cortex, and ipsilateral basal ganglia. The tissue samples were homogenized and lysed in a buffer containing ISO mmol/L NaCl, 50 mmol/L Tris-HCl, 1% NP-40, 0.5% DOC, 0.1% sodium dodecyl sulfate (SDS), and 100 μg/mL polymethylsulfonylfuoride, as well as 2 μg/mL each of pepstatin, leupstatin, and chymostain. Lysates were cleared by centrifugation at 14,000g for 10 minutes at 4°C. The protein concentration of each sample was determined with a Bio-Rad assay (Bio-Rad Laboratories, Hercules, CA, U.S.A.). ICAM-1 protein was immunoprecipitated by the Harlow method (Harlow and Lane, 1988). (1) Normal goat serum (10 μL) and protein A/G-sepharose-4B beads (20 μL) were applied to the supernatant and mixed for 120 minutes at 4°C. The nonspecific binding protein was discarded by centrifugation at 4000g for 10 minutes at 4°C. (2) Goat polyclonal rCAM-1 antibody (1:100, Santa Cruz Biotechnology Inc., Santa Cruz, CA. U.S.A.) and the total protein extracted from the supernatant was added to the beads. The antigen-antibody-bead complex was rotated overnight at 4°C and washed in 100 mmol/L PBS buffer. The target protein sample (10 μL) was dissociated from the complex by 0.2% SDS solution. Western blot analysis was performed using standard techniques (Sambrook et al., 1989). The sample was boiled at 100°C in SDS gel loading buffer (100 mmol/L Tris-Cl, 4% SDS, 0.2% bromophenol blue and 20% glycerol) for 5 minutes. The total protein sample was loaded onto a 7.5% SDS polyacrylamide gel. After electrophoresis, size-fractionated proteins were electrotransferred to a polyvinylidene difluoride membrane (Bio-Rad Laboratories) in a transfer buffer. The membrane was placed in 5% powdered milk in Tween PBS (100 mmol/L PBS solution with 0.1 % Tween 20) for 60 minutes at room temperature to block nonspecific binding. The transferred polyvinyJidene difluoride membrane was probed with polyclonal goat anti mouse ICAM-1 antibody (Santa Cruz Biotechnology) 1:500 in blocking buffer overnight at 4 DC. After washing in a solution of 0.1 % Tween-20, 0.5% bovine albumin and 1% nonfat dry milk three times for 10 minutes each, the membrane was immunoprobed with peroxidase-conjugated antigoat IgG-HRP (Santa Cruz Biotechnology) at 1:2000. An enhanced chemiluminescence kit (Amersham, Buckinghamshire, England) was applied to detect the target protein ICAM-1. The membrane was wrapped in plastic wrap and exposed to Kodak film (X-omat AR, Eastman Kodak Company, Rochester, NY, U.S.A.). The film was developed according to the instructions of the manufacturer. Immunoreactivity for ICAM-1 on each individual lane of the membrane was quantified by a gel densitometric scanning program using a National Institutes of Health 1.61 image analysis system.
Statistical analysis
Microcomputer-based statistical programs [Statview II (ABACUS, Berkeley, CA, U.S.A.) and Excel (Microsoft Corporation)] were used for statistical analysis. All data are presented as mean ± standard deviation. Parametric data at different time points during MCAO between the ICE KO and the WT mice were compared using analysis of variance followed by the Scheffe's test. A probability value of 0.05% was considered significant.
RESULTS
Physiologic parameters
Changes of body temperature in ICE KO and WT mice before and after MCAO were not significant at 3, 6, 12, and 24 hours of MCAO (P > .05). CBF in the ischemic core area decreased sharply to 16% to 20% and 16% to 22% of baseline 5 minutes after MCAO in the ICE KO and WT mice (Table 1). There were no significant differences between the two groups at the various time points during MCAO (P > .05). The blood flow in the ipsilateral perifocal area decreased to 35% to 51% and 37% to 49% in the ICE KO and the WT mice, respectively. Blood flow in the contralateral hemisphere in both groups remained at 93% to 117% and 101% to 116% of the baseline flow during MCAO (Table 1).
Blood flow changes in the WT and ICE KO mice during middle cerebral artery occlusion
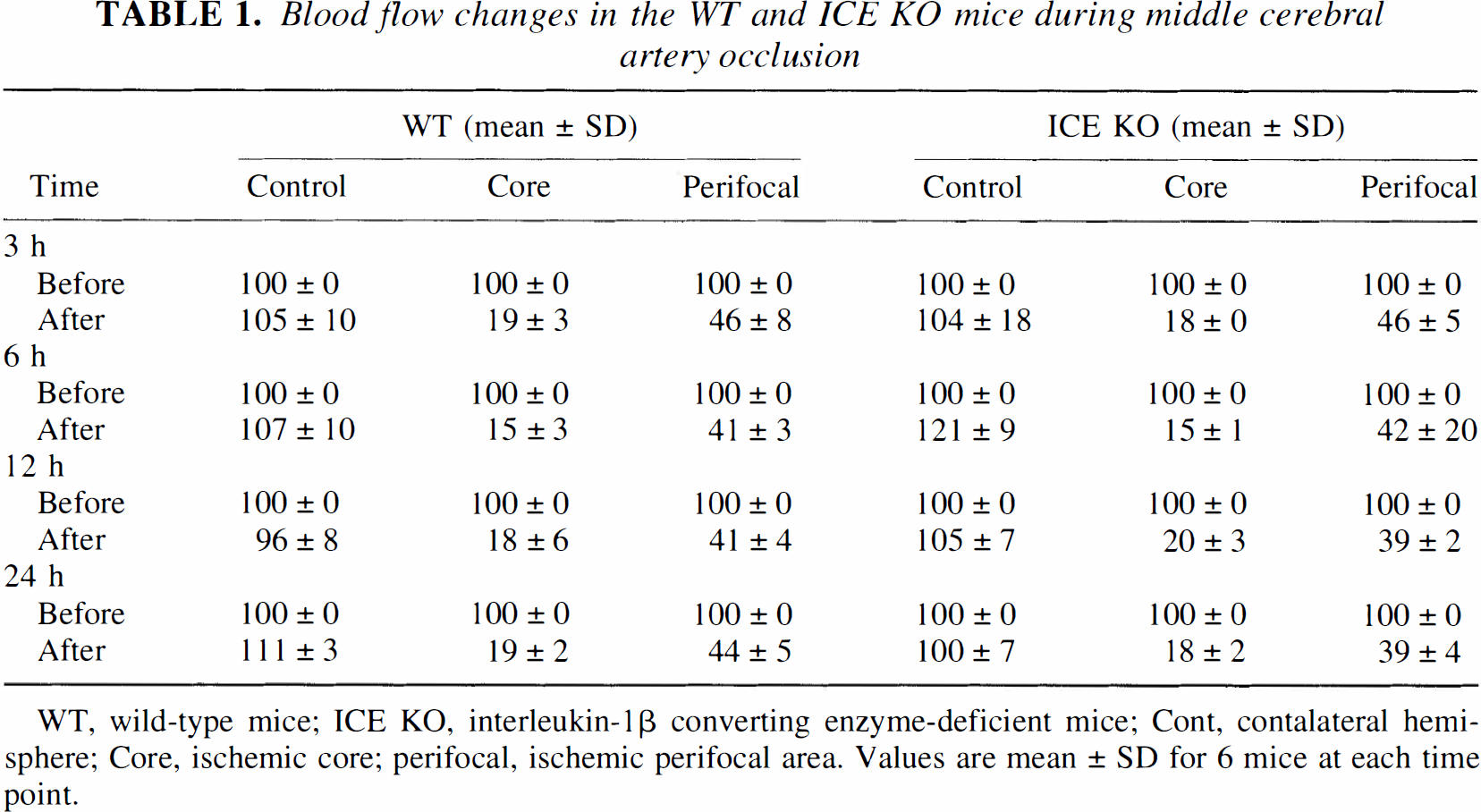
WT, wild-type mice; ICE KO, interleukin-1β converting enzyme-deficient mice; Cont, contalateral hemisphere; Core, ischemic core; perifocal, ischemic perifocal area. Values are mean ± SD for 6 mice at each time point.
TNF-α immunohistochemistry
Little TNF-α immunoreactivity was detected in brain sections from normal- and sham-operated mice. The negative control section (no anti-TNF-α antibody) showed no brown staining. Few TNF-α-positive stained cells were detected in either the sham-operated mice or in the contralateral hemisphere of the experimental animals after MCAO; however, TNF-α immunopositive cells were observed in the ipsilateral hemisphere in the ICE KO and WT groups. The numbers of TNF-α immunopositive cells in the ICE KO and WT groups were not significantly different at any time point (P > .05). TNF-α immunoreactivity developed in the ischemic core area after 3 hours of MCAO, increased at 6 hours of MCAO, peaked at 12 hours of MCAO, and then subsided (Figs. 1 and 2). Most TNF-α–positive cells were distributed in the ipsilateral cortical neuronal layer and displayed neuronal morphology. TNF-α immunoreactivity was also detected in the molecular layer of the hippocampus, the surrounding ventricles, the outer most layer of the core, and the corpus collosum of the ischemic hemisphere.
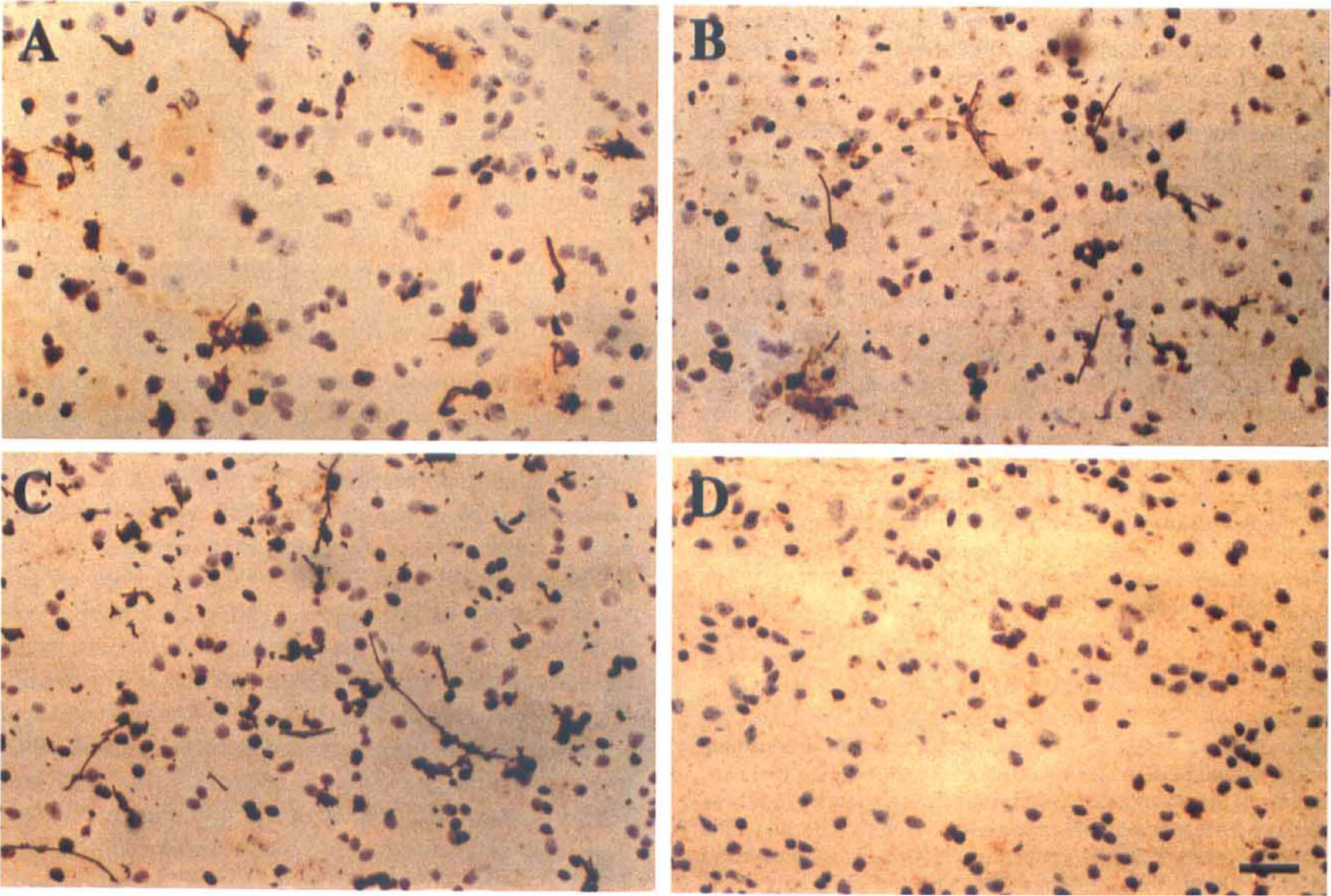
Photographs showing tumor necrosis †actor alpha (TNF-α) immunostaining in the ischemic core area in the mouse brain after 3
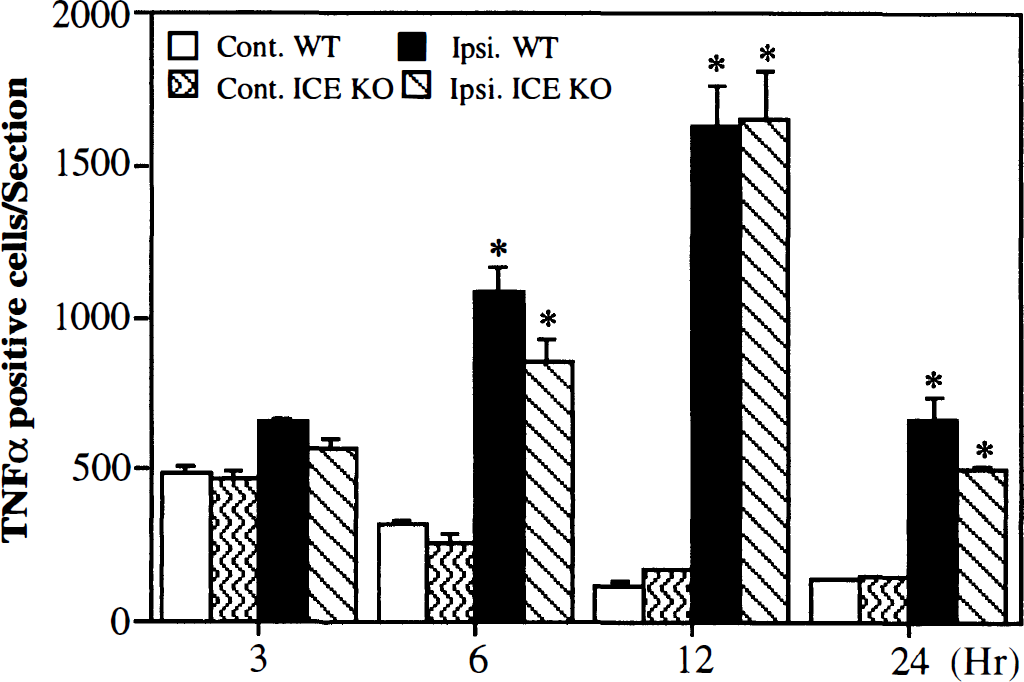
Bar graph showing tumor necrosis factor alpha (TNF-α) immunopositive cells in the ischemic brain both in the interleukin-1β converting enzyme knockout (ICE KO) mice and wild-type (WT) mice, which received 3, 6, 12, and 24 hours of middle cerebral artery occlusion (MCAO). Immunopositive stained cells in the three coronal sections (+0.86, +0.38, and −0.10 mm related to Bregma) per animal were counted and summed. The immuno positive stained cells in all the contralateral and all the ipsilateral cross-section were evaluated separately. There were few TNF-α immunopositive cells in either the sham-operated mice or the contralateral hemisphere of the experimental animals, and there were no significant differences between the ICE KO and W mice at any time point (P > .05). The TNF-α-positive cells in the ipsilateral hemisphere increased at 6 hours, peaked at 12 hours, and decreased at 24 hours after MCAO; there were also no significant differences between the ICE KO and WT mice (P > .05). Values are mean ± SD; N = 4 in each group; *P < .05, the ipsilateral hemisphere compared to the contralateral hemisphere.
ICAM-1 immunohistochemistry
There were few ICAM-1 immunopositive vessels in either the sham-operated mice or in the contralateral hemisphere of the experimental animals after MCAO. Increased expression of ICAM-1-positive vessels was observed in the ischemic core at 3 and 6 hours of MCAO; however, these ICAM-1-positive vessels were much more apparent in the perifocal area after 12 hours of MCAO. The negative control section showed no ICAM-1 immunopositive staining. ICAM-1 expression was significantly decreased in the ipsilateral hemisphere in the ICE KO mice compared to the WT mice (Fig. 3). Statistical analysis showed no significant differences in ICAM-1 expression in the contralateral hemisphere between the two groups at different time points (Fig. 4, P > .05). As with TNF-α expression, lCAM-1 expression increased in the ischemic hemisphere after 3 hours of MCAO, peaked at 12 hours (peaked at 12 hours in the WT mice), and decreased at 24 hours of MCAO. There were fewer ICAM-1–positive vessels in the ICE KO mice compared to the WT mice after 3, 6, 12, and 24 hours of MCAO (Fig. 4, F = 3.5, df = 26, P < .05).
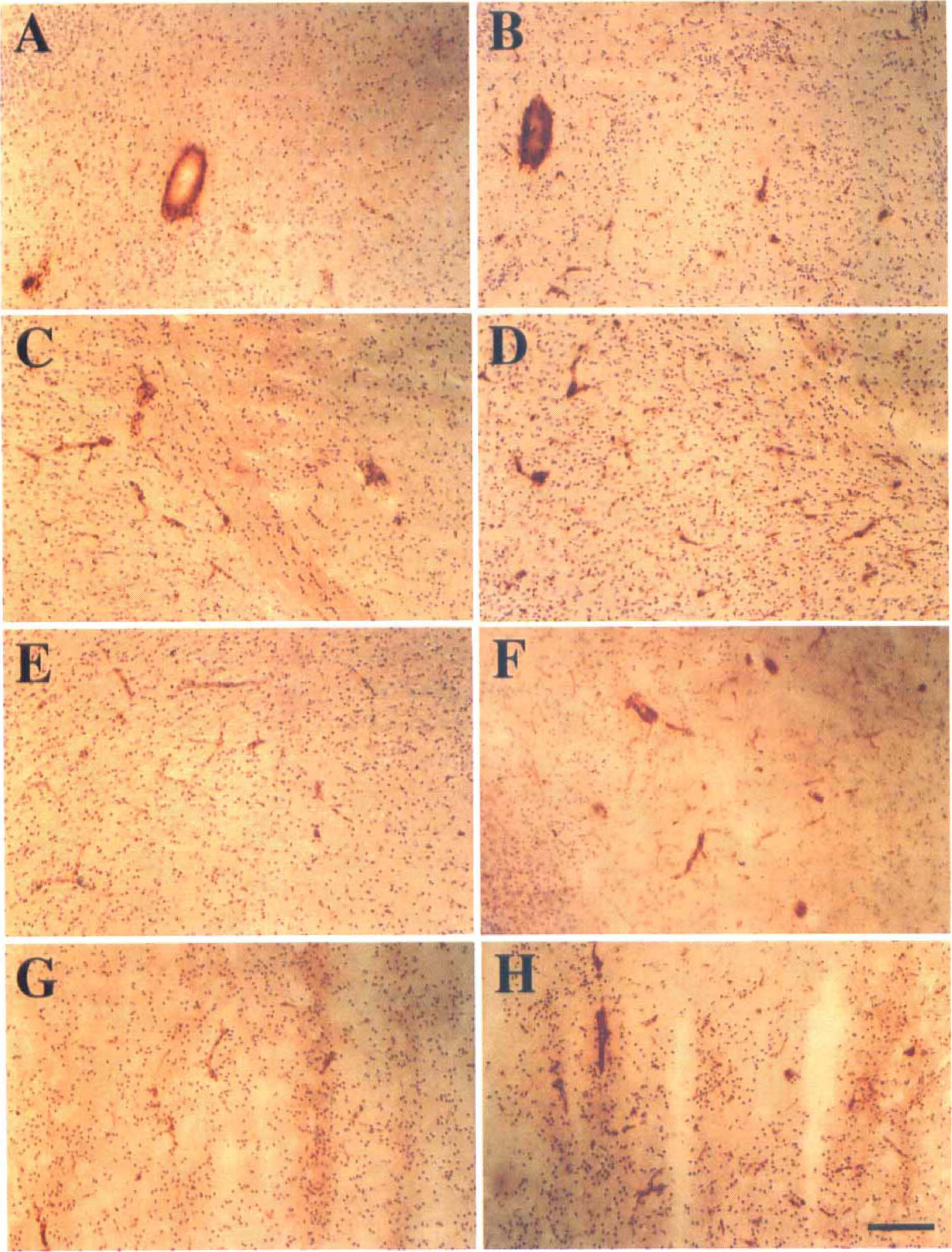
Photographs showing intercellular adhesion molecule-1 (ICAM-1)–positive vessel staining in ipsilateral hemisphere in the interleukin-1
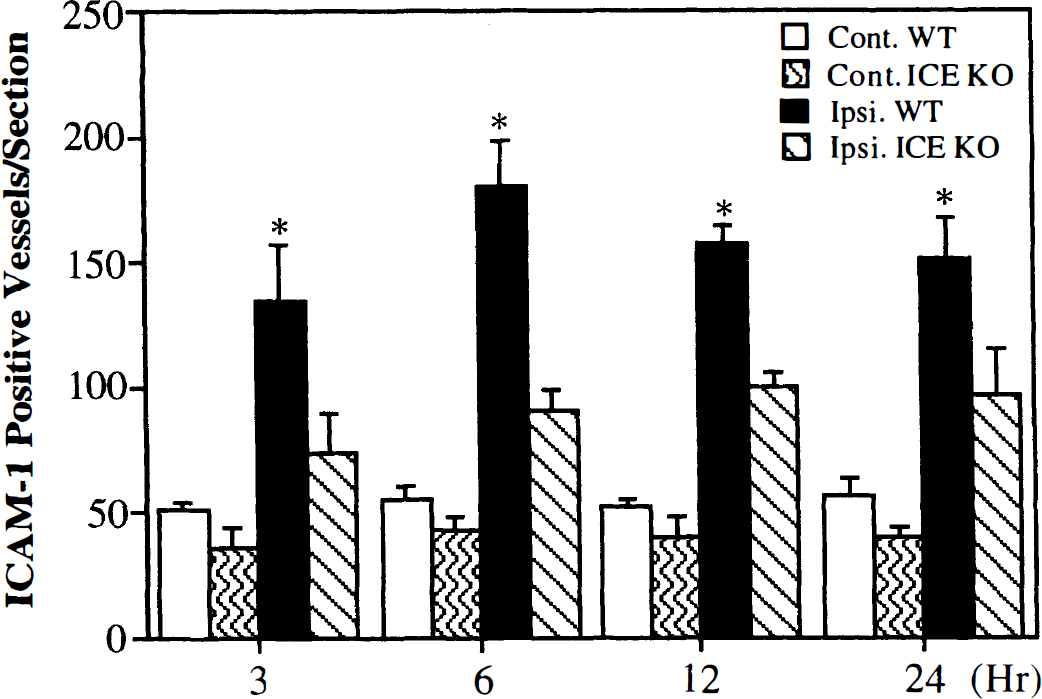
Bar graph showing the numbers of intercellular adhesion molecule-1 (ICAM-1)–positive vessels in the ischemic brain in both the interleukin-1β converting enzyme knockout (ICE KO) mice and wild-type (WT) mice after 3, 6, 12, and 24 hours of middle cerebral artery occlusion (MCAO). Immunopositive stained cells in the three coronal sections (+0.86, +0.38, and −0.10 mm related to Bregma) per animal were counted and summed. The immunopositive stained cells in all the contralateral and all the ipsilateral cross-section were evaluated separately. There were few ICAM-1–immunopositive vessels in either the sham-operated or the contralateral hemisphere of the experimental animals, and there were no significant differences between the ICE KO and WT mice in these control hemispheres (P > .05). ICAM-1–positive vessels in the ipsilateral hemisphere gradually increased from 6 hours, peaked at 12 hours (peaked at 6 hours in the WT mice), and decreased at 24 hours after MCAO. ICAM-1–immunopositive vessels in the ischemic hemisphere were significantly less in the ICE KO mice than in the WT mice after MCAO (F = 11.2, df = 26, P < .05). Values are mean ± SD; N = 4 in each group; *P < .05 ICE KO group versus WT group mice.
ICAM-1 Western blotting analysis
Immunoprecipitation and Western blot analysis for ICAM-1 detected a major band at approximately 90 kd in all samples tested. ICAM-1 protein was present at a very low level in the sham-operated and contralateral hemisphere after 3, 6, and 24 hours of permanent MCAO. ICAM-1 protein expression was lower in the ipsilateral hemisphere in the ICE KO mice than in the WT mice after 3, 6, and 24 hours of permanent MCAO (Fig. 5). However, statistical analysis shows that the decrease of ICAM-1 protein expression is only significant at 24 hours after MCAO (P < .05).
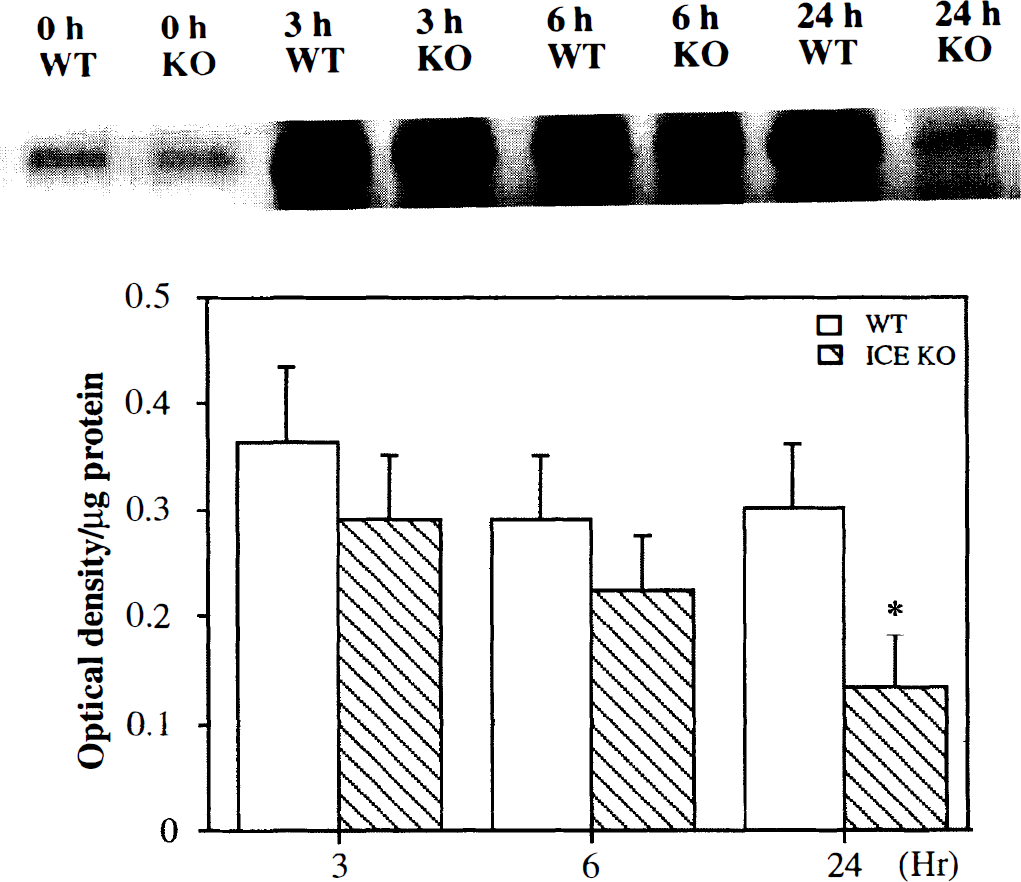
Immunoprecipitation and Western blot analysis of intercellular adhesion molecule-1 (ICAM-1) protein from the ipsilateral hemisphere of ischemic brain shows a band of ICAM-1 at 90 kd (upper). The immunoprecipitates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (7.5% acrylamide), transferred, blotted and detected by antibody and chemiluminescence. ICAM-1 protein expression was lower in the ipsilateral hemisphere in the interleukin-1β converting enzyme knockout (ICE KO) mice than in the wild-type (WT) mice after 3, 6, and 24 hours of permanent middle cerebral artery occlusion (MCAO) (upper). The bar graph shows optical density with equal amounts of isolated protein from the ipsilateral hemisphere in the ICE KO and the WT mice after 3, 6, and 24 hours of MCAO. (N = 3 in each group, lower). Values are mean ± SD; *P < .05, the ICE KO group versus the WT group mice.
DISCUSSION
This study shows that the adhesion molecule ICAM-1 and the pro inflammatory cytokine, TNF-α, are expressed in ICE KO and WT mice after focal cerebral ischemia. However, in mice which are deficient in the ICE gene and not able to process proIL-1β, the expression of ICAM-1, but not TNF-α, was reduced. These findings suggest that after ischemia, ICAM-1 expression is dependent on IL-1β whereas the expression of TNF-α is independent of IL-1β. Considerable evidence points to ICAM-1 as an important mediator of ischemic brain damage. Thus, reduced expression of this adhesion molecule may be one of the mechanisms through which ICE KO mice are protected during cerebral ischemia.
Gene knockout mice are useful tools for defining pathways leading to ischemic cell death (Huang et al., 1994; Yang et al., 1994). In a previous study we showed that after permanent focal ischemia, ICE KO mice had less brain edema at 6 hours and smaller lesions at 24 hours compared to their WT controls (Schielke et al., 1998). This is in concert with studies showing that lesion size was reduced after MCAO in mutant mice expressing a dominant inhibitory ICE protein, or mice treated intraventricularly with peptide ICE inhibitors (Hara et al., 1997a, b ). A possible mechanism underlying these observations is reduced IL-1β production by the ischemic brain. ICE is a member of the caspase family of cysteine proteases, which are associated with both proinflammatory and apoptotic actions (Dinarello, 1992; Dinarello, 1996; Hara et al., 1997a; Schielke et al., 1998). Also known as caspase 1, ICE is critical for processing pro-IL-1 to the biologically active cytokine, IL-1β. Previous studies have shown that macrophages isolated from the ICE KO mice are not capable of producing IL-1β. Similarly, in a model of endotoxic shock, no IL-1β is detected in the plasma of the ICE KO mice, and survival is greatly enhanced. IL-1α production is also reduced in these mice, although not to the extent of IL-1β (Dinarello, 1997).
Recently studies have shown that pro-IL-18 (also known as interferon-τ inducing factor) is processed to its active form by ICE. IL-18, constitutively expressed in the rat brain, is structurally similar to IL-1β (Culhane et al., 1998). In addition to stimulating the production of interferon-τ, it may directly upregulate ICAM-1 expression (Kohka et al., 1998). Although the role of IL-18 in the ischemic brain has not been established, IL-18 or other as yet unidentified ICE-dependent cytokines may contribute to the reduction in ischemic brain injury as shown in the ICE KO mice.
The physiologic mechanism underlying the reduction of brain injury in ischemic ICE KO mice should be considered. Our data showed that both the ICE KO and WT mice developed normally. The changes of surface CBF and body temperature in ICE KO and WT mice were not different during and after MCAO (Table 1). Several studies showed the susceptibility to cerebral ischemia between different strains related to the structural and functional differences in the patency of the circle of Willis (Barone et al., 1993; Connolly et al., 1996b). Strains such as SV/129, commonly used to generate genetically modified mice, are resistant to cerebral ischemia as compared to C57/BL6. Both the ICE KO and WT mice used in our study were derived from the same progenitors, a mixed SV/129 and C57/BL6 background. Thus, our result suggests that the resistance to cerebral ischemia in the ICE KO mice is not likely to be due to the differences in strains or changes in cerebrovascular anatomy.
Inflammatory mechanisms have been implicated as important mediators of ischemic brain damage. IL-1β is a cytokine that exerts an array of proinflammatory actions, including induction of adhesion molecules, other cytokines, chemokines, arachadonic acid, amyloid precursor protein and nitric oxide (Betz et al., 1996). TNF-α has many of the same actions (Feuerstein et al., 1994). Expression of both IL-1β and TNF-α mRN As is upregulated after cerebral ischemia in rats (Wang et al., 1994b). The first evidence that IL-1β contributes the pathophysiology of stroke was the observation that intraventricular injection of the endogenous IL-1 receptor antagonist (IL-1ra) reduced the extent of ischemic damage after MCAO in rats (Relton and Rothwell, 1992). This was confirmed in studies where IL-1ra was administered intravenously or overexpressed in the brain using an adenoviral vector in a gene therapy approach (Betz et al., 1995). In addition, IL-1β neutralizing antibody, administered intraventricularly, reduced brain edema after MCAO (Rothwell and Hopkins, 1995).
The studies of TNF-α in stroke models have provided evidence that TNF-α also contributes to ischemic brain damage, although apparently contradictory findings exist (Feuerstein et al., 1994). Direct administration of TNF-α enhanced, and neutralizing antibodies to TNF-α reduced the lesion size after MCAO (Yang et al., 1998). However, mice deficient in the TNF-α receptor have larger lesion volumes after MCAO (Bruce et al., 1996).
The present study showed that the TNF-α protein is expressed in the ischemic brain. It appeared in the ischemic core region 3 hours after MCAO and gradually increased at 6 hours, peaked at 12 hours, and then subsided. This time course is similar to that of IL-1β mRNA expression described previously (Wang et al., 1994b). Studies have shown that TNF-α mRNA and protein were expressed in ischemic rat brain, and the early expression was found in cells with a neuronal morphology. Buttini et al.(1996) identified a rapid upregulation of TNF-α mRNA and protein in microglia and macrophages after ischemia in the rat. Furthermore, Siren et al. (1992) found that spontaneously hypertensive rats had higher levels of TNF-α production in the brain as compared to normotensive rats. TNF-α is produced by neurons, microglial cells, macrophages, and astrocytes (Liu et al., 1994; Medana et al., 1997). We have shown that TNF-α protein was expressed in ischemic CD-1 mice, and double labeled immunofluorescence localized the TNF-α to neurons, astrocytes, and ependymal cells (Gong et al., 1998). The present study shows that TNF-α can be produced in ICE KO mice, suggesting that TNF-α production is not dependent on IL-1b.
ICAM-1 is a member of the immunoglobulin family of adhesion molecules, which are expressed on the surface of endothelial cells. ICAM-1 is important for establishing adhesion of leukocytes before their movement across the endothelium into the tissue (Wang et al., 1994a). There is considerable evidence supporting the idea that leukocyte infiltration into the brain contributes to the development of the ischemic lesion. Blocking ICAM-1 with an antibody reduces both neutrophil accumulation and the size of the lesion (Zhang et al., 1994). IL-1β and TNF-α can induce ICAM-1 expression in cultured endothelial cells (Feuerstein et al., 1997). Endothelial cell adhesion molecules including ICAM-1, endothelial leukocyte adhesion molecule-1, and P-selectin are important in establishing endothelial adhesion to the leukocyte before infiltration (Feuerstein et al., 1997; Sharma and Kumar, 1998). Proinflammatory cytokines IL-1β and TNF-α are known to be induced earlier than the adhesion molecules (Wang and Feuerstein, 1995). IL-1β and TNF-α can induce ICAM-1, endothelial leukocyte adhesion molecule-1, and P-selectin in vitro (Feuerstein et al., 1997). The development of an inflammatory response in focal ischemia, therefore, may appear in the following sequence: expression of IL-1β and TNF-α, upregulation of adhesion molecules, then migration of leukocytes. TNF-α mimics every known IL-1β action on endothelial cells. In our study, however, the expression of ICAM-1 protein is significantly less in the ICE KO mice as compared with WT mice after MCAO. This result suggests that mature IL-1β is a critical mediator in the upregulation of ICAM-1 expression during cerebral ischemia.
In summary, we showed that TNF-α was expressed after permanent MCAO in both the ICE KO and the WT mice, suggesting that the overexpression of TNF-α during focal ischemia is not dependent on IL-1β. The lower ICAM-1 expression in the ICE KO mice suggests that IL-1β may play an important role in the upregulation of ICAM-1.
Footnotes
Acknowledgment
The authors thank Ms. Kathleen Donahoe for editorial assistance.