
Abstract
A variety of recent studies suggest a role for both inflammatory cytokines such as interleukin-1 beta (IL-1β), and apoptosis in ischemic brain injury. Because IL-1β converting enzyme (ICE) is required for the conversion of proIL-1β to its biologically active form, and has homology with proteins that regulate apoptosis in invertebrates, we studied the effect of cerebral ischemia on brain injury in mutant mice deficient in the ICE gene (ICE knockout [KO] mice). Focal cerebral ischemia, produced by occlusion of the middle cerebral artery, resulted in brain edema (increased water and sodium content) at 4 hours and a histologically defined brain lesion at 24 hours. Both of these markers of brain injury were significantly reduced in the ICE KO mice as compared to wild-type C57BL/6 mice. Regional cerebral blood flow, determined using the flow tracer, N-isopropyl [methyl 1,3-14C] p-iodoamphetamine (14C-IMP), was similar in the two strains of mice, indicating that the reduced brain injury in the KO mice was not a result of a lesser degree of ischemia. These data show that ICE contributes to the development of ischemic brain damage, and that it plays a role at an early time in the pathologic process. Although the mechanism of this effect is uncertain, our results suggest that pharmacologic inhibition of ICE may be a useful treatment for stroke.
Recent evidence suggests that inflammatory processes in the brain play a crucial role in the pathogenesis of cerebral ischemia. Accordingly, animal studies of both global and focal cerebral ischemia have shown that proinflammatory cytokine expression is increased (Beamer et al., 1995; Liu et al., 1993; Minami et al., 1992; Szaflarski et al., 1995) and resident microglia are activated after an ischemic insult (Gehrmann et al., 1995; Gehrmann et al., 1992). Furthermore, infiltration of peripheral immune cells appears to be involved because the ischemic brain is protected by depletion of blood leukocytes and blocking vascular adhesion molecules (Chopp et al., 1994; Hallenbeck, 1996). A role for the proinflammatory cytokine, interleukin-1 beta (IL-1β) has been demonstrated by the findings that the endogenous IL-1 receptor antagonist (IL-1ra) (Betz et al., 1995; Relton and Rothwell, 1992) and a neutralizing antibody to IL-1β (Yamasaki et al., 1995) both reduce brain damage and edema when administered before a stroke in rats.
Biologically active IL-1β is formed by the enzymatic cleavage of inactive proIL-1β by the protease, IL-1β converting enzyme (ICE). ICE is a member of a family of cysteine proteases, the caspases, several of which are associated with the apoptotic cell death pathway. This pathway has been implicated in ischemic brain damage (Linnik et al., 1993). Thus, inhibition of ICE could be a viable strategy for reducing brain damage after a stroke due to its role in IL-1β formation and neuroinflammation, or apoptosis, or both. Testing this hypothesis has been difficult because selective ICE inhibitors have been unavailable. Recently, mutant mice, deficient in a functional gene for ICE have been produced (Kuida et al., 1995; Li et al., 1995a). In the present study we tested the hypothesis that these mutant mice, being unable to produce mature IL-1β, would have less brain damage than their wild-type controls in response to a focal ischemic insult.
MATERIALS AND METHODS
Ischemia in ICE knockout and C57BL/6 mice
Three independent studies, measuring histologic lesion size, regional CBF (rCBF) and brain edema formation after permanent middle cerebral artery occlusion (MCAO), were performed using male, 23 to 30 g, ICE knockout (KO) and C57BL/6 mice. The ICE-deficient mutant mice were created and characterized by the BASF Corporation, Worcester, MA (Li et al., 1995a). These mice contain a null mutation in the ICE gene that was generated by homologous recombination in embryonic stem cells. C57BL/6 mice were obtained from Jackson Labs, Bar Harbor, ME. Focal cerebral ischemia was induced by MCAO using the suture model as described previously (Yang et al., 1994).
Lesion size
The volume of ischemic brain damage after MCAO was assessed in age-matched KO and C57BL/6 mice. The mice were anesthetized with chloralhydrate (400 mg/kg, intraperitoneally) and their skulls exposed for measurement of pre-MCAO CBF by laser Doppler flowmetry (Model BMP2, Vasomedics Inc, St. Paul, MN). Immediately after MCAO, CBF was again measured to verify that the occlusion was successful. The mice were then allowed to recover from anesthesia and returned to their cages. Twenty-four hours later the mice were killed and their brains processed for histologic analysis of lesion size from hematoxylin/eosin-stained cryosections. Lesion volume was calculated as the difference in volumes of the nonischemic hemisphere and the normal tissue in the ischemic hemisphere, measured with a Nikon stereomicroscope and computer-assisted stereologic software (C.A.S.T. GRID Systems, Olympus DK). All measurements were made by a blinded observer.
Regional CBF
rCBF was determined in seven KO and seven C57BLK/6 mice 30 minutes after MCAO using a modification of the indicator fractionation technique described previously (Betz and Iannotti, 1983; Van Uitert and Levy, 1978). Mice were anesthetized with chloralhydrate and a cannula was placed in the femoral artery for blood sampling. Core body temperature was maintained at 37°C to 38°C by a feedback controlled heating pad. The middle cerebral artery (MCA) was occluded as described above. Thirty minutes later, 1 μCi of the flow tracer, N-isopropyl [methyl 1,3-14C] p-iodoamphetamine (14C-IMP), (Obrenovitch et al., 1987) in 100 μL of 0.9% saline was injected into the jugular vein and, simultaneously, a peristaltic pump was started to withdraw blood at a rate of 100 μL/min. After 1 minute of isotope circulation, the pump was stopped, the mouse killed immediately, and brain sampled by region for isotope counting. Cortex, basal ganglia, and blood samples were digested in methylbenzethonium hydroxide (Sigma, St. Louis, MO, U.S.A.), and prepared for liquid scintillation counting (Beckman 3801 scintillation counter, Beckman, Schaumburg, IL, U.S.A.). rCBF was calculated from tissue and blood radioactivity and expressed as mL/100 g/min.
Brain edema
Brain water content was measured in six KO and six C57BLK/6 mice, 4 hours after MCAO. Mice were anesthetized with isoflurane (1.5% to 2%) supplemented with oxygen. A cannula was placed in the femoral artery for measurement of blood pressure and sampling of blood for determination of pCO2, pO2 and pH. The MCA was occluded as described above and the mice maintained under anesthesia for 4 hours, after which the brains were sampled for determination of brain water content by the wet/dry weight method. Cortical and basal ganglion samples were weighed and then dried to constant weight for 24 hours at 100 °C. After reweighing, these samples were dissolved in nitric acid for determination of sodium and potassium content by flame photometry. Two wild-type mice were eliminated from the study because their blood pH was less than 7.10.
Statistical methods
Results are expressed as mean ± SD. Mean values were compared using the Student's t-test.
RESULTS
Brain lesion volume was determined in KO and C57BLK/6 mice 24 hours after induction of permanent focal ischemia. The lesion volume in the KO mice was 52% smaller than that of the wild-type mice (93 ± 25 mm3 versus 45 ± 27 mm3, respectively; P < .05; Fig. 1). Laser Doppler flowmetry measurement of CBF indicated that the MCAO was successful in both groups because the CBF was reduced similarly in the KO and control mice (92% versus 93% reduction, respectively).
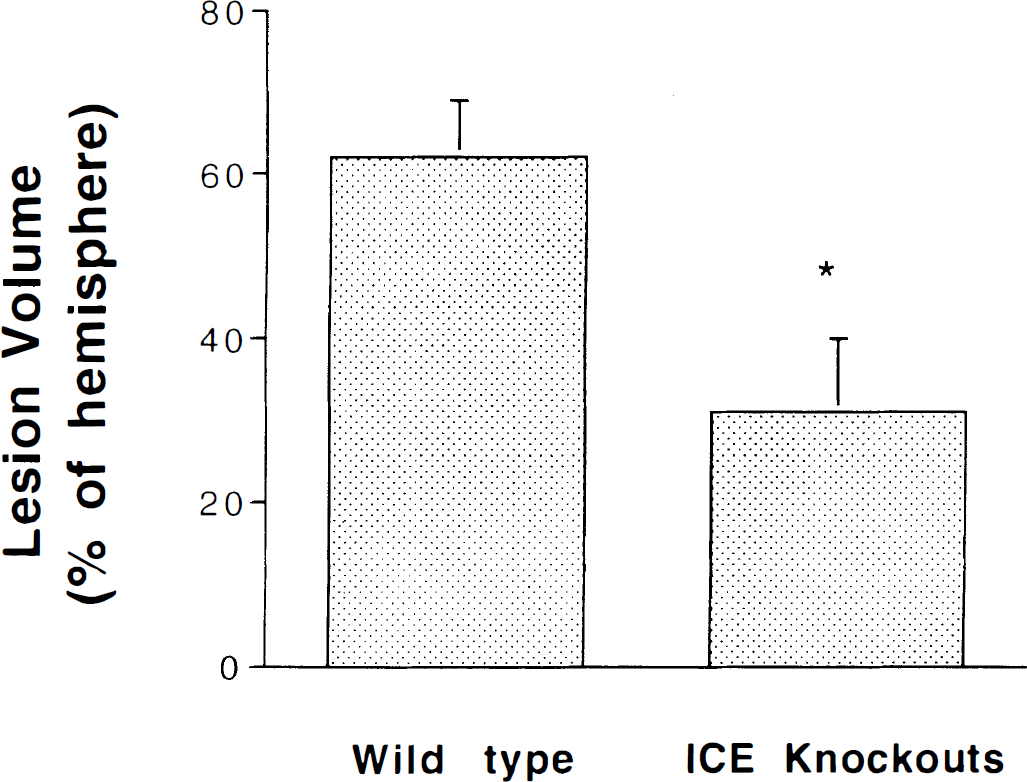
Brain lesion volume estimated from hematoxylin/eosin-stained sections 24 hours after permanent MCAO and expressed as percent of hemispheric volume. ICE KO mice (n = 5) had 52% smaller lesions compared to C57BLK/6 mice (n = 5). *P < .03, unpaired Student's t-test. Values are means ± SD.
To determine if the protection afforded the KO mice was the result of a smaller reduction in CBF after MCAO, we used a more quantitative measure of the regional CBF in the cortex and basal ganglia, 30 minutes after MCAO. Figure 2 shows that KO and C57BLK/6 mice had similar rCBF in the nonischemic hemisphere. In the ischemic hemisphere, rCBF was reduced to 20 ± 14 and 25 ± 6 mL/100 g/min in the cortex and 41 ± 28 and 53 ± 32 mL/100 g/min in the caudate of the controls and KO mice, respectively. These data show that rCBF was reduced to a similar extent in the mutant and control strains. Thus, the smaller brain lesions in the KO mice were not due to incomplete occlusion of the MCA or greater collateral flow capacity.
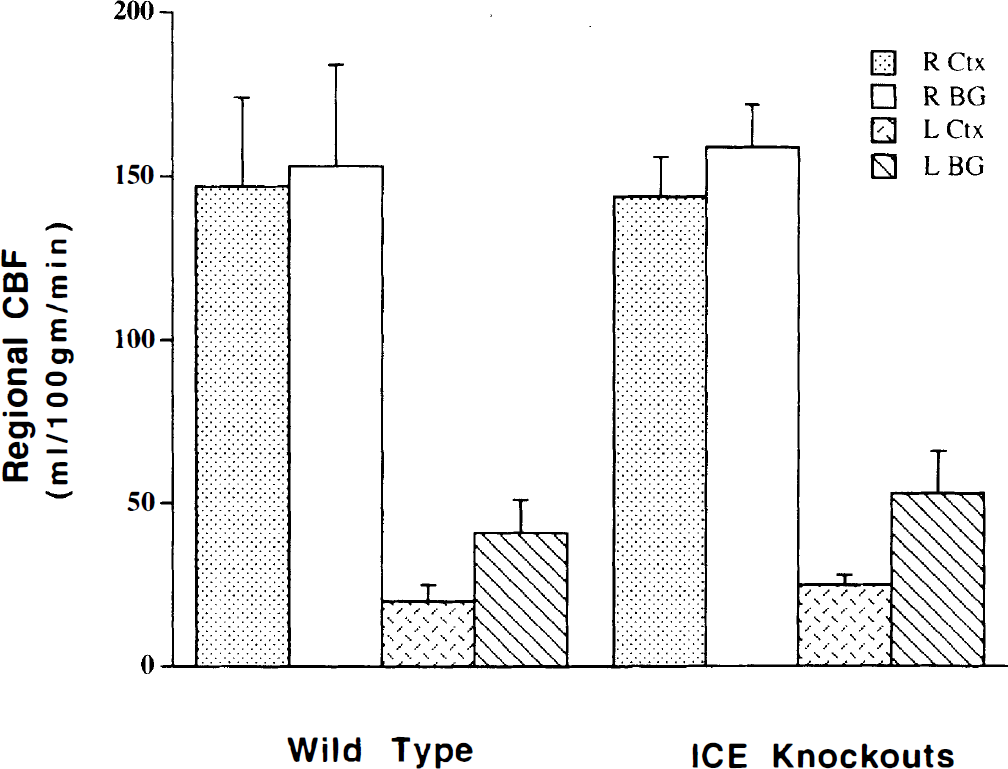
CBF was measured 30 minutes after MCAO in the right and left cortices and basal ganglia using the flow tracer, 14C-IMP. C57BLK/6 (n = 7) and ICE KO (n = 7) mice had similar CBF in both the nonischemic right hemisphere and the ischemic left hemisphere. Values are mean rCBF ± SD.
The extent of ischemic brain damage can be influenced by physiologic variables such as blood pressure and body temperature. An altered response of such critical processes to the ischemic event could be responsible for the smaller lesions we observed in the mutant strain at 24 hours. Therefore, we measured brain edema formation in control and ICE-deficient mice at 4 hours after MCAO. Blood pressure, although unstable, was similar in the two groups. Body temperature was maintained at 36.5°C to 38°C. Brain water, sodium, and potassium content in the nonischemic hemisphere of control and mutant mice were similar. Water and sodium content increased and potassium content decreased in the ischemic cortex and basal ganglia of the wild-type mice. These changes were significantly blunted in the KO mice (Table 1). The degree of brain edema observed at 4 hours likely reflects the volume of tissue that is compromised and eventually becomes infarcted. The reduced brain edema at 4 hours in the ICE-deficient mice suggests that ICE activity is contributing early in the sequence of events leading to ischemic brain injury.
Brain water, sodium, and potassium content 4 hours after MCAO
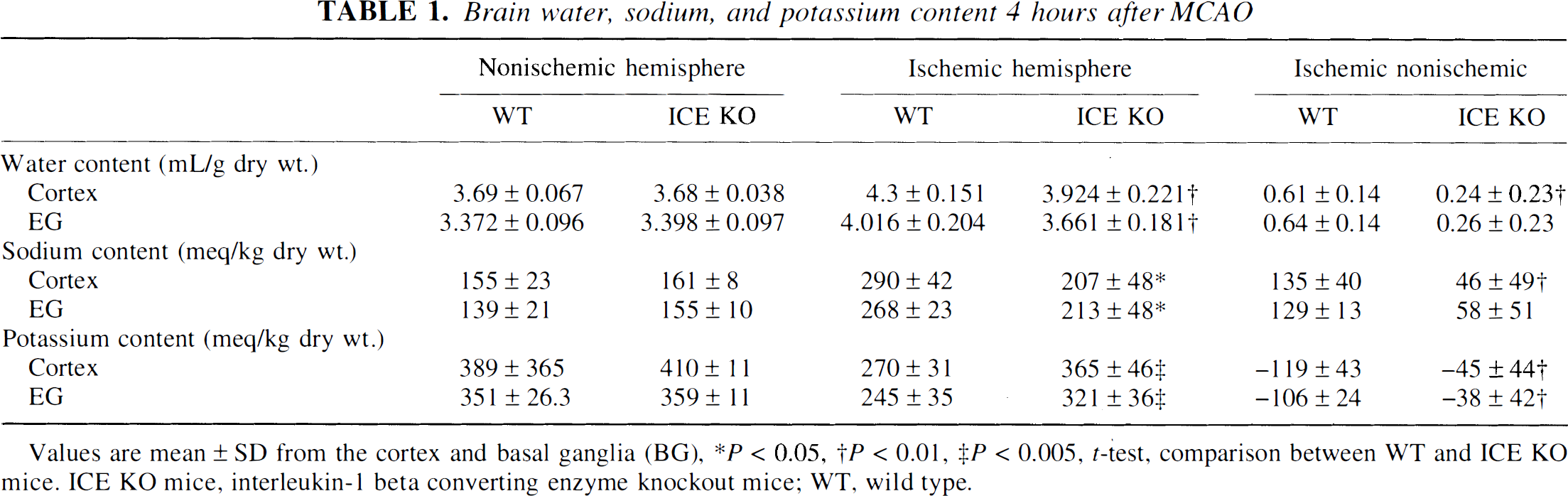
Values are mean ± SD from the cortex and basal ganglia (BG)
P < 0.05
P < 0.01
P < 0.005, t-test, comparison between WT and ICE KO mice. ICE KO mice, interleukin-1 beta converting enzyme knockout mice; WT, wild type.
DISCUSSION
This study shows that brain edema and the extent of brain damage resulting from MCAO are significantly reduced in the ICE-deficient mice compared to C57BLK/6 mice, despite similar reductions in blood flow in these two strains. In a preliminary study we also compared ischemic lesions in ICE KO and 129/SV mice, the strain from which the embryonic stem cells were derived and, thus, contributed to the genetic make-up of the KO mice. We found a similarly reduced histologic lesion in the KO mice (Betz et al., 1996). Because others have reported similar lesion sizes after MCAO in these two background strains (Huang et al., 1994), it is unlikely that our findings are due to variability in the background strain's responses to MCAO, but rather are a result of the elimination of the ICE gene. Supporting this is the recent finding that a mutant ICE gene expressing an ICE inhibitory protein reduces lesion volume after transient focal ischemia in mice, although inhibition of other caspases cannot be ruled out in these dominant negative mutants (Friedlander et al., 1997; Hara et al., 1991a). Additionally, peptide fluoromethyl and chloromethyl ketones, which are nonspecific irreversible inhibitors of the ICE protease family, reduce ischemic and excitotoxic brain injury (Hara et al., 1991b). Our findings in mutant mice, in which the ICE gene has been selectively eliminated, indicate that ICE (rather than its homologues) plays an important role in ischemic pathology in the brain. However, the precise mechanisms by which ICE contributes to the pathogenic process are still uncertain.
Our observations are consistent with the hypothesis that the KO mice are protected from an ischemic insult because they have lower brain IL-1β levels. There is considerable evidence that ICE and IL-1β/IL-1ra mRNA levels are acutely increased in cerebral ischemia (Buttini et al., 1994; Liu et al., 1993; Wang et al., 1994), in the microglia and astrocytes and endothelial cells but not neurons (Bhat et al., 1996; Lynch et al., 1997). The first demonstration that IL-1β mediates ischemic brain damage was the finding that intracerebroventricular injection of the antagonist protein, IL-1ra, reduced stroke size in rats by 50% (Loddick and Rothwell, 1996; Relton and Rothwell, 1992). This was confirmed in a subsequent study in which over-expression of IL-1ra in the brain was induced by intracerebroventricular injection of a recombinant adenovirus vector carrying the human IL-1ra cDNA, and the lesion was reduced by 60% (Betz et al., 1995). More recently intravenous administration of a large quantity of human recombinant IL-1ra was also found to be neuroprotective (Garcia et al., 1995; Relton et al., 1996). Additionally, intracerebroventricular administration of a polyclonal IL-1β neutralizing antibody reduced brain edema after focal ischemia with reperfusion in the rat, thus indicating IL-1β as an important mediator of ischemic damage (Yamasaki et al., 1995). Although it is reasonable to assume that in the ICE KO mice little proIL-1β is processed, it is conceivable that the brain could produce biologically active IL-1β through an alternative pathway induced during ischemia. Brain tissue IL-1β levels were not measured in this study because the available antibodies are not sufficiently selective for biologically active mature IL-1β over proIL-1β and Western blot analysis is not sensitive enough to detect brain levels (unpublished data, K. Wang and R. Nath, November 1996). However, macrophages isolated from these ICE KO mice do not produce IL-1β when stimulated by the proinflammatory endotoxin, lipopolysaccharide (Li et al., 1995a). Additionally, KO mice are protected from the shock induced by this endotoxin and no IL-1β is detected in their plasma.
IL-1β is a proinflammatory cytokine with numerous actions in the brain that could account for its influence on ischemic brain damage. For example, brain temperature is a critical variable in ischemia studies and IL-1β is pyrogenic (Klir et al., 1994; Smith and Kluger, 1992). However, in the present study, and in the rat MCAO studies with IL-1ra, reduced body temperature appears not to be the cause of the observed neuroprotection. Other actions of IL-1β include microglial activation, induction of leucocyte adhesion molecules, stimulation of infiltration of inflammatory cells, and induction several other cytokines and mediators such as IL-6, arachidonic acid, nitric oxide, amyloid precursor protein, and corticotrophin-releasing factor (Dinarello, 1996). Many of the central actions of IL-1β, such as its effects on temperature regulation, are mediated through the prostaglandins and are blocked by cyclo-oxygenase inhibitors. Several of these biochemical pathways may generate potentially harmful free radicals that could contribute to ischemic brain damage. Thus, there are many mechanisms that could explain how decreased ICE activity and the subsequent reduction in brain IL-1β could influence the outcome from an ischemic insult (Rothwell and Hopkins, 1995). In addition, other substrates for ICE may exist (Gu et al., 1997) whose product may contribute to ischemic brain injury. Moreover, our studies in ICE KO mice cannot exclude as candidate protective mechanisms, compensatory changes that may have occurred during development.
Because ICE is a member of a family of cysteine proteases (caspases) that has been implicated in apoptotic cell death (Yuan et al., 1993) and apoptosis may occur in brain ischemia (Li et al., 1995b; Linnik et al., 1993; van Lookeren Campagne and Gill, 1996), the possibility that our findings in the ICE KO mice might be due to interference with an apoptotic process should be considered. The two known functions of ICE itself are cleaving of the inactive 31-kd proIL-1β to the active 17-kd IL-1β and processing pro—interferon gamma—inducing factor. Nonetheless, ICE has been implicated in apoptosis by several in vitro studies in non-neuronal and neuronal models of apoptotic cell death. For example, when ICE is over-expressed in rat fibroblasts, apoptosis is stimulated (Miura et al., 1993). This and several other in vitro apoptosis models can be affected by pharmacologic ICE inhibitors (Enari et al., 1995; Milligan et al., 1995). However, these inhibitors lack selectivity between ICE and other members of the caspase family, making it difficult to pinpoint ICE in the apoptotic process. Such specificity is achieved in the ICE KO mice that have normal CNS structure and cell number. This apparently normal phenotype suggests that ICE is not required for the apoptosis that occurs naturally during development. In vitro, macrophages from KO and wild-type mice both undergo apoptosis when stimulated with ATP (Li et al., 1995a). Similarly, KO thymocytes that are treated with dexamethasone or gamma irradiation undergo normal apoptosis. The exceptions to this pattern are that Fas-mediated killing of thymocytes is reduced in thymocytes isolated from KO mice (Kuida et al., 1995) as is apoptotic death in dorsal root ganglion cultures after growth factor withdrawal (Friedlander et al., 1997). In addition, some studies show that ICE is not expressed in neurons (Bhat et al., 1996; Lynch et al., 1997), even when the brain is challenged by global forebrain ischemia or lipopolysaccharide and expression is increased in astrocytes and microglia (Vasilakos et al., 1995). However, it is still unknown whether or not ICE is expressed in neurons after a focal ischemic insult.
Thus, it is still uncertain if ICE-deficient mice are protected from ischemic damage by a reduced apoptotic response, and therefore our findings should not be interpreted as evidence that apoptosis is important in ischemic brain injury. However, considering the evidence showing a role for IL-1β in cerebral ischemia, it is possible that IL-1β permits or stimulates apoptosis. A recent study supports a role for IL-1β as a potentiating factor in apoptotic death in culture. (Friedlander et al., 1996).
In conclusion, our results show that ICE plays an important role in ischemic brain damage. It is likely that this effect is due to a reduced production of IL-1β, although the precise mechanism is not known. Regardless of the mechanism, this study suggests that selective inhibitors of ICE would be valuable for the treatment of stroke.