
Abstract
In this study the authors addressed the hypothesis that estrogen (i.e., 17β-estradiol) acts to repress leukocyte adhesion, The experiments involved comparing leukocyte adhesion in cerebral venules in vivo, in intact ovariectomized and 17β-estradiol-treated (100 μ/kg/day for 1 week) ovariectomized female rats using topical applications of the adhesion-promoting drug, phorbol 12-myristate 13-acetate (PMA), Adherent Rhodamine-6G-labeled leukocytes were viewed through a closed cranial window using intravital microscopy/videometry, Leukocyte dynamics were recorded at baseline and after each dose of PMA, The PMA was suffused (1.0 mL/min) at increasing concentrations (0,01, 0,1, and 1.0 μmol/L, 15 minutes at each level), A videotape record of each experiment was made for subsequent analysis of leukocyte adhesion, The data showed that the percentage venular area occupied by adherent leukocytes at baseline was significantly greater in the ovariectomized compared to the intact and 17β-estradiol—treated groups (12,2%, 3.4%, and 4,2% respectively), That relationship was maintained during PMA treatments to the extent that the percentage venular area occupied by adherent leukocytes increased to 26.4% in the untreated ovariectomized group compared to 14.4% and 11.3% in the intact and 17β-estradiol—treated groups, respectively, In conclusion, the authors found chronic estrogen depletion enhances leukocyte adhesion in the rat cerebral circulation. Estrogen repletion in such animals is accompanied by a significant reduction in leukocyte adhesion, These findings could, at least in part, account for the ischemic brain damage seen in ovariectomized versus intact females, and the restored neuroprotection observed upon 17β-estradiol treatment reported in earlier studies.
Keywords
There is extensive evidence that the leukocyte adhesion and infiltration that occurs after cerebral ischemia and reperfusion can exacerbate neuropathology (Garcia et al., 1994; Heinel et al., 1994; Juurlink and Sweeney, 1997; Kochanek and Hallenbeck, 1992; Lefer and Lefer, 1996). Studies have also shown that estrogen, particularly 17β-estradiol (E2), has protective effects against ischemic neuropathology (Alkayed et al., 1998; Dubal et al., 1998; Pelligrino et al., 1998; Simpkins et al., 1997; Wang et al., 1999). The mechanisms responsible for estrogen-related neuroprotection are not well understood, but they appear to be diverse, First, chronic estrogen replacement therapy promotes cerebral vasodilation, an action that is seemingly related to upregulation of endothelial nitric oxide synthase (eNOS). This increases the capacity of the brain to resist the reductions in perfusion accompanying ischemia (Pelligrino et al., 1998), Second, estrogen also provides ischemic neuroprotection via perfusion-independent mechanisms (Wang et al., 1999). The possibilities for perfusion-independent estrogen-related brain protection include antioxidant actions (Gridley et al., 1997), and an antiapoptotic effect (Garciasegura et al., 1998). Relevant to this article, estrogen has been reported to reduce leukocyte accumulation in cardiac tissue after ischemia (Squadrito et al., 1997), and in cultured human umbilical vein (Caulinglaser et al., 1996), Whether this effect occurs via direct or indirect actions has not been established. However, an indirect pathway related to eNOS upregulation remains a viable possibility. Nitric oxide (NO) has also been linked to repression of leukocyte adhesion during ischemia and reperfusion in different tissues including the brain (Gidday et al., 1998) and the heart (Fukuda et al., 1995).
In the current study we addressed the hypothesis that estrogen represses leukocyte adhesion in the brain. Therefore, we compared the magnitude of leukocyte adhesion, as it relates to chronic depletion and repletion of estrogen, in pial venules in vivo under resting and “stimulated” conditions. For the latter we suffused the established adhesion-promoting drug, phorbol 12-myristate 13-acetate (PMA) (Okayama et al., 1999).
METHODS
The study protocol was approved by the Institutional Animal Care and Use Committee. Twenty-four adult female Sprague Dawley rats (Charles River, Wilmington, MA, U.S.A.l weighing 300 to 400 g were used. Rats were divided in three groups: intact females, ovariectomized (OVX) females, and 17β-estradiol (E2)–treated OVX females. Ovariectomies were performed by the supplier 4 to 6 weeks before the study. The E2 was prepared in dimethyl sulfoxide and 100 μg/kg/day were administered intraperitoneally for 1 week before the study. On the day of the study, rats were anesthetized with halothane, tracheostomized, paralyzed with curare and ventilated with 0.8% halothane in 70% N2O:30% O2. Femoral arterial and venous catheters were placed for monitoring of mean arterial blood pressure and arterial blood gases, and for drug infusion, respectively. Rats were secured in a head holder in prone position to facilitate placement of a closed cranial window. Details of this procedure are provided in previous publications (Koenig et al., 1993; Wang et al., 1994). Briefly, a 10 mm diameter craniotomy is performed over the skull midline, the underlying dura was carefully removed, and an 11 mm diameter acrylic window—outfitted with three ports for inflow, outflow, and intracranial pressure monitoring—was placed and fixed to the skull with cyanoacrylate gel. After window placement, halothane was discontinued and a bolus of intravenous fentanyl was administered (10 μg/kg) followed by a maintenance dose of intravenous fentanyl of 25 μg/kg/h and ventilation with 70% N2O:30% O2, These conditions were maintained throughout the study. Cannulae were inserted in the ports, and the space under the window was filled with artificial cerebrospinal fluid, which was suffused at a rate of I mL/min and maintained at a temperature of 37°C, P
Pial venules were viewed through a microscope (Nikon, Fryer Co. Inc., Huntley, IL, U.S.A.) equipped with a color video camera (Sony, Fryer Co. Inc., Huntley, IL, U.S.A.). An epi-illumination system with a mercury lamp was used. Magnifications of 800x were displayed on a video monitor. Pial venules were localized and precise diameter measurements were made using a calibrated video microscaler (Optech, Imagen Corporation, Trenton, NJ, U.S.A.). Leukocytes were labeled with Rhodamine 6G (200 μg/mL in 0.9% saline) given initially as an intravenous bolus (1 mL), and followed by continuous infusion at a rate of 1 mL/h (Lindauer et al., 1996). An appropriate Rhodamine filter set was inserted into the light path and baseline leukocyte dynamics were recorded before suffusion of the established adhesion-promoting drug, PMA (Sigma, St. Louis, MO, U.S.A.). The PMA was suffused at 1 mL/min at increasing concentrations (0.01, 0.1, and 1.0 μmol/L). Each dose was maintained for 15 minutes before recording adhesion. In all cases, illumination was limited to 60 seconds at a time to avoid photoquenching. A videotape record of each experiment was made for subsequent analysis of leukocyte adhesion. This analysis was performed by capturing multiple frames of taped images in a computer and measuring the percentage of the venular area occupied by adhering leukocytes using the Image Pro Plus analysis system (Media Cybernetics, Silver Spring, MD, U.S.A.). These images were representative of the dynamics observed during each step of the experiments. Each experimental group was divided into 2 subgroups: one subgroup receiving PMA and the other used as a time control. For the latter, artificial cerebrospinal fluid was suffused continuously, and leukocyte dynamics were recorded at baseline and after 15, 30, and 45 minutes of suffusion.
Statistical analyses were performed using one way analysis of variance with a post hoc Tukey, repeated measures analysis of variance, and paired t-test. All values are reported as mean ± SD. Statistical significance was considered at P < 0.05.
RESULTS
The arterial blood variables measured at baseline are summarized in Table 1. There were no significant differences in pH, P
Arterial blood variables at baseline
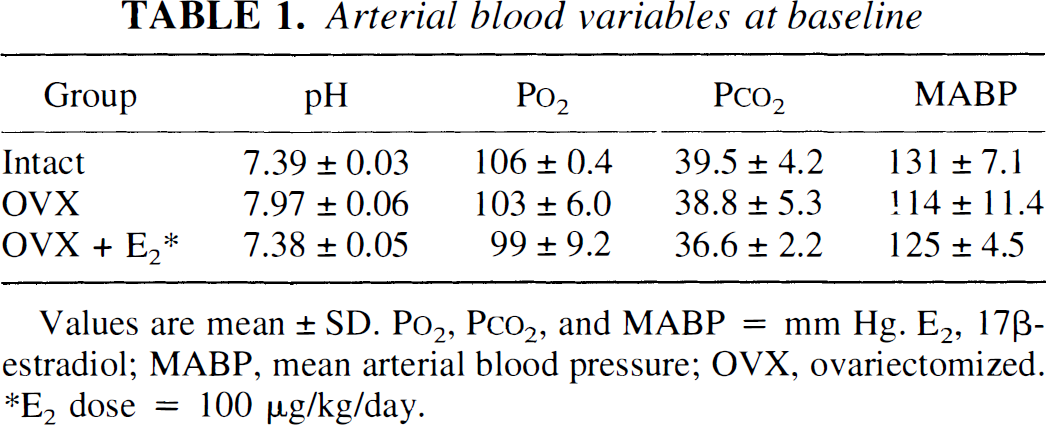
Values are mean ± SD. P
E2 dose = 100 μg/kg/day.
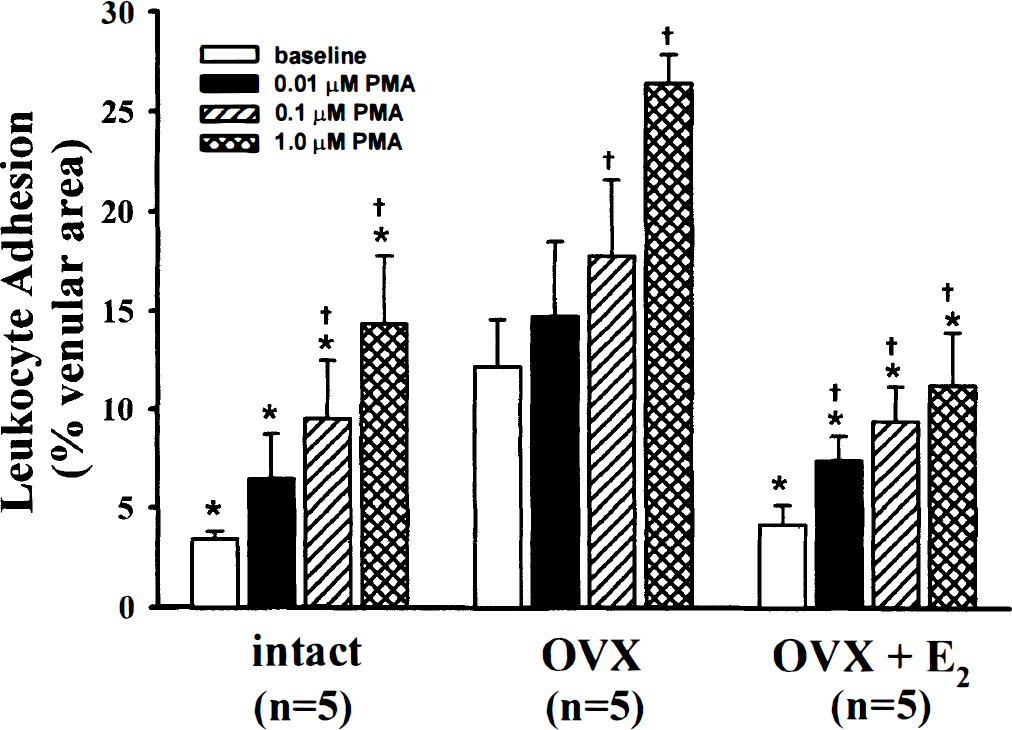
Baseline leukocyte adhesion was considerably greater in ovariectomized (OVX) versus intact females and OVX females treated for 1 week with 17β-estradiol (E2). Topical applications of phorbol 12-myristate 13-acetate (PMA), as expected, produced a dose-dependent increase in leukocyte adhesion in all groups. Values are mean ± SD. *P < 0.05 versus OVX, †P < 0.05 versus baseline.
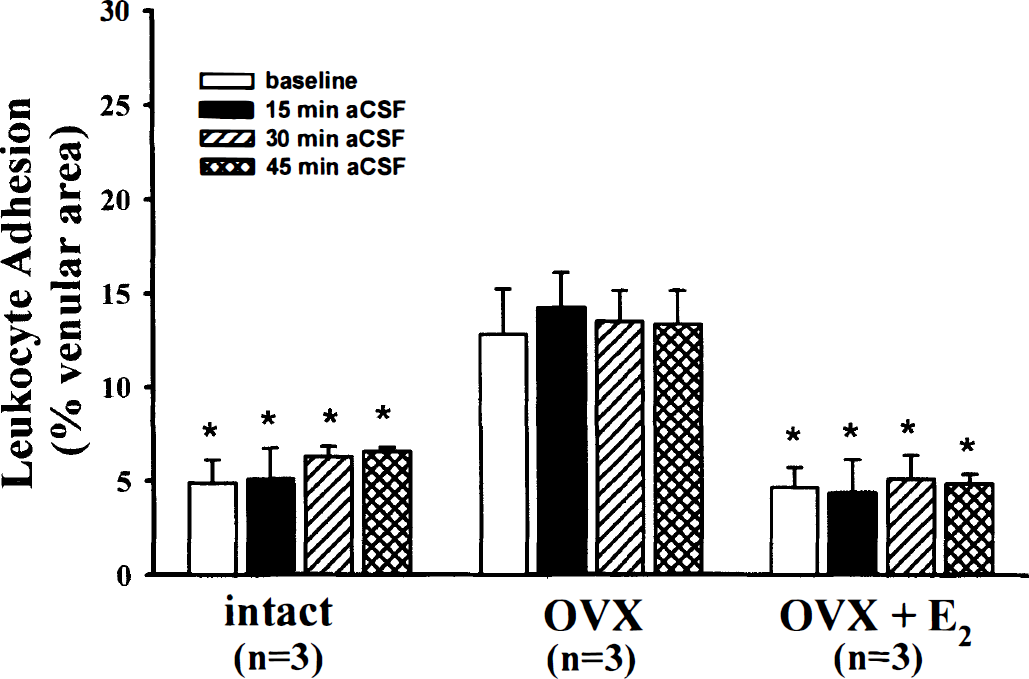
The percentage venular area occupied by adhering leukocytes was three times greater in the ovariectomized (OVX) compared to the other groups. That relationship was maintained throughout 45 minutes of suffusion with drug-free artificial cerebrospinal fluid (aCSF). Values are mean ± SD. *P < 0.05 versus OVX. E2, 17β-estradiol.
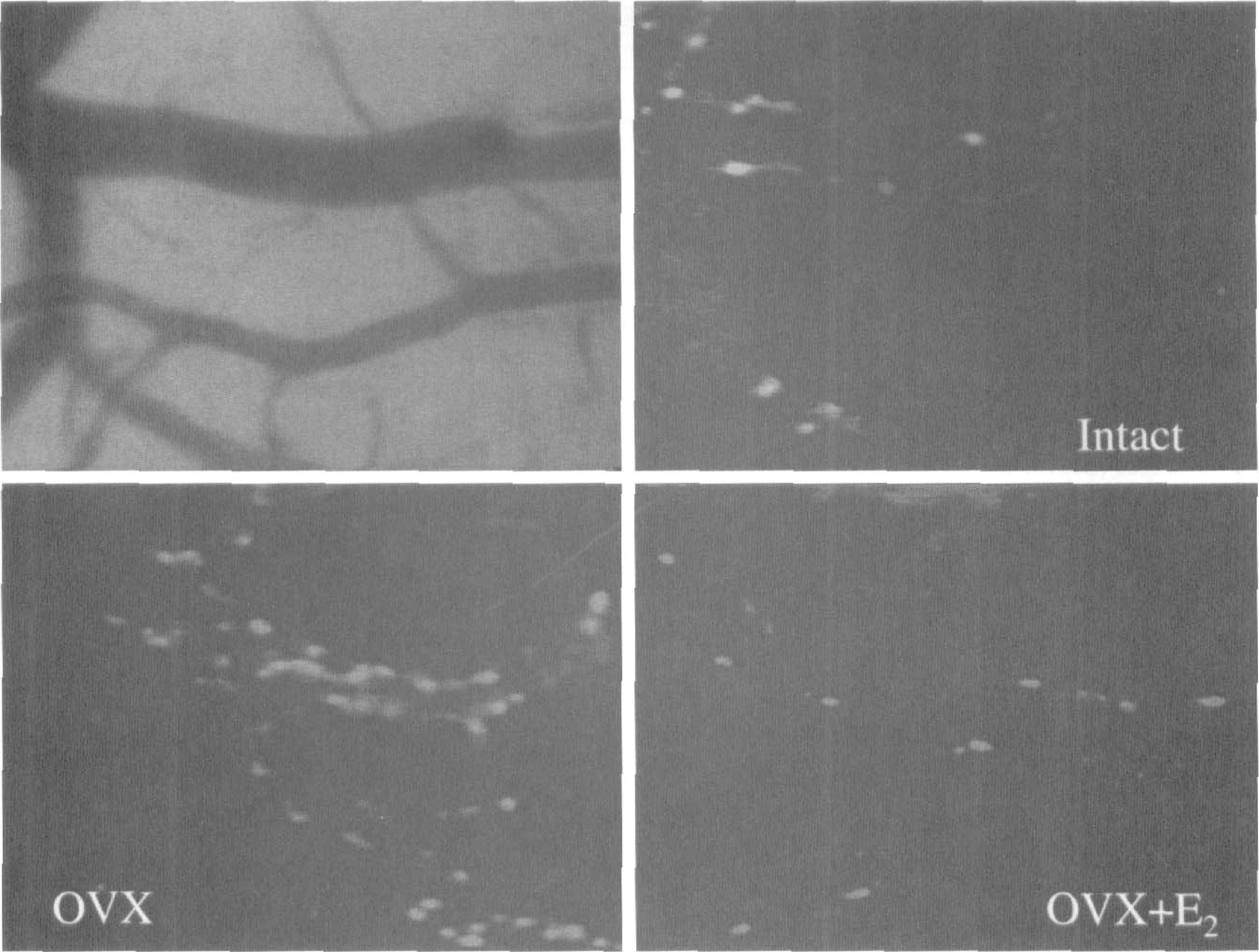
Representative baseline images of pial venules with adherent Rhodamine 6G-labeled leukocytes are shown. Notice the increased adhesion in the ovariectomized (OVX) group versus intact and 17β-estradiol (E2)-treated OVX. The left upper panel shows an image that corresponds to the intact panel before placement of a Rhodamine filter.
DISCUSSION
The findings in this study clearly show that (1) chronic estrogen depletion enhances leukocyte adhesion in the cerebral circulation in rats; (2) this effect was reversed by estrogen treatment; and (3) PMA increased leukocyte adhesion in intact, OVX, and E2-treated OYX rats.
Leukocyte rolling and sticking results from interactions between receptor-ligand pairs on both the leukocyte and the endothelial cell (Hickey and Kubes, 1997; Sharar et al., 1995). There are different families of adhesion molecules acting at each step. Leukocyte rolling is mediated primarily by the selectin family of adhesion molecules (P-, E-, and L-selectin), as they bind to their respective counter-ligands (Lasky, 1992). Subsequent firm adhesion is mediated by the interaction between members of the integrin family, on leukocytes, with adhesion molecules on the endothelial surface (ICAM-1, ICAM-2, VCAM-l). Of particular importance in this step is the stable bond mediated by the (12 integrins LFA-l (CD11a/CD18) and Mac-1 (CD11b/CD18) to ICAM-1 (Smith, 1993). The last step in the adhesion cascade is the leukocyte transendothelial migration, or diapedesis, and involves the participation of an adhesion molecule present in both endothelium and leukocytes (i.e., PECAM-1).
The finding that baseline leukocyte adhesion was increased in OVX rats, and reversed by E2 treatment, suggests that estrogen acts to repress leukocyte adhesion. The mechanism involved in that action has not been established. It is known, however, that PMA increases leukocyte adhesion through activation of protein kinase C, which results in rapid mobilization of P-selectin from Weibel-Palade bodies in the endothelial cell (Okayama et al., 1999). Mobilization of P-selectin, as mentioned above, is an important step in initiating leukocyte rolling in the vessel wall. In this study, the PMA-mediated increase in leukocyte adhesion was proportional in all groups, but reached higher levels in estrogen-depleted rats. This suggests that estrogen may decrease leukocyte adhesion by modulating expression of adhesion molecules. Evidence from cultured human umbilical vein endothelial cells points to a mechanism whereby estrogen inhibits cytokine (interleukin-1β)-mediated endothelial cell adhesion molecule transcriptional activation. The adhesion molecules modulated by E2 were E-selectin, ICAM-1, and VCAM-1 (Caulinglaser et al., 1996). In another study, treatment with E2-reduced leukocyte accumulation in myocardium, after ischemia and reperfusion, by reducing ICAM-1 (Squadrito et al., 1997). A similar mechanism could explain, at least in part, the phenomenon observed in the current study. However, data on cerebral expression of adhesion molecules in estrogen-deprived rats must await further investigation.
There is also compelling evidence that NO, in particular eNOS-derived NO, reduces leukocyte adhesion (Lefer, 1997). The major action of NO, in this regard, apparently is to diminish the expression of adhesion molecules, particularly E-selectin, ICAM-1, and VCAM-1 (Decaterina et al., 1995; Kupatt et al., 1997). In some instances that effect of NO, like that of E2, may be caused by interference with cytokine-induction mechanisms. Another possibility is a NO-associated inhibition of NFkB, a transcription factor involved in the regulation of most proinflammatory genes, including adhesion molecules (Collins et al., 1995; Kupatt et al., 1997). Because both E2 and NO have similar effects on adhesion molecules, this suggests that, in this model, estrogen may decrease leukocyte adhesion by increasing NO release from the endothelium. This possibility is supported by recent findings that E2 upregulates brain eNOS (as well as neuronal NOS) (Pelligrino et al., 1998; Wang et al., 1999).
In summary, we found that estrogen reduces cerebral leukocyte adhesion in the female rat. Whether estrogen modulates leukocyte adhesion through NO-dependent, or NO-independent mechanisms, remains to be established. Recent findings showing that estrogen decreases ischemic damage, coupled with results indicating that ischemic damage can be exacerbated by leukocyte infiltration, suggests that decreased leukocyte adhesion may be an important mechanism in estrogen-mediated neuroprotection.
Footnotes
Acknowledgment
The authors thank Anthony Sharp for expert technical assistance.