
Abstract
The marked impairment in cerebrovascular endothelial nitric oxide synthase (eNOS) function that develops after ovariectomy may relate to the observation that the abundance of cerebral vascular eNOS and its endogenous inhibitor, caveolin-1, vary in opposite directions with chronic changes in estrogen status. The authors endeavored, therefore, to establish a link between these correlative findings by independently manipulating, in ovariectomized female rats, eNOS and caveolin-1 expression, while monitoring agonist (acetylcholine)-stimulated eNOS functional activity. In the current study, the authors showed that individually neither the up-regulation of eNOS (through simvastatin treatment), nor the down-regulation of caveolin-1 (through antisense oligonucleotide administration) is capable of restoring eNOS function in pial arterioles in vivo in these estrogen-depleted rats. Only when eNOS up-regulation and caveolin-1 down-regulation are combined is activity normalized. These results establish a mechanistic link between the estrogen-associated divergent changes in the abundance of caveolin-1 and eNOS protein and eNOS functional activity in cerebral arterioles.
The endothelial isoform of nitric oxide synthase (eNOS) plays an important protective role in several brain pathologies, including stroke. It is thought that a repressed cerebrovascular eNOS function contributes to the increased damage caused by stroke in estrogen-depleted females (Hurn and Macrae, 2000; Pelligrino et al., 2000). Accordingly, both the enhanced risk and eNOS functional impairment can be reversed with chronic 17β-estradiol supplementation. Nitric oxide generated in vascular endothelial cells by eNOS is ideally positioned to defend the brain through a variety of actions. One of the better characterized and potentially neuroprotective actions of eNOS-derived NO is its ability to promote relaxation of adjacent smooth muscle cells, thereby improving brain perfusion. Recent studies have shown that estrogen regulates the content of eNOS in cerebral microvessels, to the extent that chronic estrogen depletion (through ovariectomy) reduces eNOS content and activity, whereas estrogen replacement normalizes both (McNeill et al., 1999; Pelligrino et al., 2000). Nevertheless, the reductions in vascular eNOS expression appear to be relatively modest compared with the repression of eNOS function (Pelligrino et al., 2000). That is, in ovariectomized (OVX) rats, a complete loss of eNOS-dependent (acetylcholine [ACh]-induced) vasodilating function in pial arterioles was found (Pelligrino et al., 2000). Thus, it would seem that estrogen effects on eNOS functional activity go beyond simply modulating eNOS protein abundance. In the same study, it was observed that the pial arteriolar endothelial expression of the endogenous eNOS inhibitor, caveolin-1 (CAV-1), was increased in the OVX animals and was reduced toward normal with chronic estrogen treatment. Such evidence might have compelled one to propose that the “double-barreled” effect of chronic estrogen depletion—that is, to reduce eNOS while increasing CAV-1 expression—accounts for the loss of agonist-activated eNOS function in cerebrovascular endothelium. However, these findings can only be viewed as correlative.
In the current study, a strategy was devised to establish that a mechanistic link indeed does exist between eNOS/CAV-1 protein expression and eNOS functional activity. That strategy derived from the basic hypothesis that, in OVX females, normalization of eNOS expression, in the absence of any attempt to reduce CAV-1 expression, would be insufficient for restoring eNOS functional activity. The proper testing of that hypothesis, in estrogen-depleted rats, required the implementation of two key experimental manipulations: (1) up-regulation of eNOS without altering CAV-1 expression, and (2) an eNOS-independent specific down-regulation of CAV-1. For the former, chronic 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitor (simvastatin) treatments were used (Endres et al., 1998). The results of a preliminary study (Xu et al., 2000) revealed a normalization of eNOS expression in samples of cerebral cortical tissue taken from simvastatin-treated OVX rats, whereas CAV-1 abundance remained elevated. In the second case, a novel in vivo CAV-1 antisense delivery protocol was used. Thus, the ACh reactivities of pial arterioles, as well as the expression of CAV-1 and eNOS in pial tissue, were compared in the following groups of OVX rats: (1) CAV-1 antisense-treated, (2) simvastatin-treated, (3) combined CAV-1 antisense and simvastatin-treated, (4) CAV-1 missense-treated, (5) CAV-1 missense plus simvastatin-treated, and (6) untreated/vehicle-treated controls. Monitoring the reactivity of pial arterioles to topically applied ACh is a well-accepted strategy for examining in vivo cerebral vascular eNOS function in adult mammals (Wang et al., 1994; Meng et al., 1996; Pelligrino et al., 2000).
MATERIALS AND METHODS
Animal
This study's protocol was approved by the Institutional Animal Care and Use Committee. Female Sprague–Dawley rats (250 to 350 g), at ∼4 weeks after ovariectomy, were used (Pelligrino et al., 1998; Wang et al., 1999). The main groups studied were vehicle-treated OVX and simvastatin-treated OVX. In addition, parallel groups of intact and 17β-estradiol (E2)-treated (0.1 mg/kg/d, 1 week) (Pelligrino et al., 1998; Wang et al., 1999) OVX females were included. As shown in a previous report from the authors' laboratory (Wang et al., 1999), the 0.1 mg/kg/d dosage results in an average daily plasma E2 concentration that lies between the peak and nadir levels observed during the normal rat estrous cycle.
Simvastatin treatment
Simvastatin (Merck, West Point, PA, U.S.A.) was administered subcutaneously for 2 weeks at 20 mg/kg/d. Before injection, simvastatin was chemically activated by opening the lactones through alkaline hydrolysis (Laufs et al., 1997).
CAV-1 antisense and missense administration
Rats were anesthetized with halothane, intubated, paralyzed (with a short-acting muscle relaxant-vecuronium), and mechanically ventilated. For surgery, anesthesia was maintained with 0.8% halothane/70% N2 O/30% O2. Using established procedures (Wang et al., 1994), an acrylic cranial window equipped with three ports (inflow, outflow, and intracranial pressure monitoring) was fixed to the skull with cyanoacrylate gel. Either CAV-1 antisense or missense oligonucleotide (Sigma Genosys, St. Louis, MO, U.S.A.), at 10 or 25 μg in ∼300 μL of artificial cerebrospinal fluid (aCSF), was carefully introduced into the space underlying the cranial window and overlying the pia/arachnoid membrane, in vehicle or simvastatin-treated OVX females. The antisense (5′-TTTACCCCCAGACAT-3′), obtained in the unmodified phosphodiester form, was complementary to the initiation sequence of rat CAV-1 (GenBank accession no. Z46614), with only a single base difference compared with the mouse (#AB029931) or human (#XM004967) sequence (Ikezu et al., 1998a). The missense (5′-CAATCGGCTAACCTA-3′) also was a 15-mer unmodified oligonucleotide, with a similar distribution (but not sequence) of bases compared with the antisense. The 24-hour oligonucleotide exposure period was based on published information indicating an ∼80% reduction in CAV-1 protein abundance in endothelial cells at 24 hours after introduction of CAV-1 antisense (Griffoni et al., 2000). In additional females (intact, untreated OVX, or E2 -treated OVX), only 300 μL aCSF was introduced under the windows. Then the cranial window access ports were plugged, and the skin overlying the skull was sutured together. The animal was permitted to recover from anesthesia, extubated, and returned to its cage.
Pial arteriolar acetylcholine reactivity evaluations
On the day of study, 24 hours after window preparation, the animal was anesthetized with halothane/N2 O, paralyzed (curare), tracheotomized, and mechanically ventilated. Catheters were placed in the femoral artery to measure systemic arterial pressure and to obtain arterial blood samples, and also in the femoral vein for infusion of drugs. The cranial window was exposed and cannulae were placed into the three ports of the window. Halothane was discontinued and a loading dose of intravenous fentanyl (10 μg/kg) was given. Thereafter, the rat was maintained on 70% N2 O/30% O2 -fentanyl (25 μg/kg/h, intravenous). Rectal temperature was controlled at 37°C. The space under the window was filled with 37°C aCSF equilibrated with 10% O2 /5%CO2 and was suffused at 0.5 mL/min. Pial arterioles were observed using a microscope and video camera. Measurements of arteriole diameters were made using a calibrated video microscaler. In all experiments, initial diameter measurements of 25 to 50 μm pial arterioles were made after a 40-minute period of cortical suffusion with drug free aCSF. Hypercapnia (PaCO2 ≈ 70 mm Hg) then was imposed for 3 minutes, arteriolar diameters were again measured, and the CO 2 reactivity was calculated as the percentage diameter increase per mm Hg CO2 change. At 15 minutes after return to normal CO2, ACh was suffused (in aCSF) at 10 then 100 μmol/L, 5 minutes at each level. Diameter changes were expressed as a percentage increase from the baseline value measured just before introduction of ACh (or increased CO2).
Immunoblots
In ovariectomized rats, in the presence or absence of antisense and missense oligonucleotide exposure or simvastatin treatment, and intact females, pial tissue from under the cranial window was removed and CAV-1 protein was assessed through Western immunoblotting. In addition, pial tissue obtained from vehicle-treated and simvastatin-treated OVX rats not fitted with cranial windows was used for analysis of eNOS and CAV-1 expression. Also, pial tissue eNOS and CAV-1 expression in intact and E2 -treated OVX females was evaluated. In this instance, the rats were decapitated under anesthesia (halothane), the brains were removed, and the pia was stripped off. In all cases, pial tissue was placed into 0.4 mL of extraction buffer (45 mmol/L tris-HCl; 0.4% sodium dodecyl sulfate; 0.2 mmol/L phenylmethylsulfonyl fluoride; 1 mmol/L sodium vanadate; 0.5% Sigma protease inhibitor cocktail), sonicated, and centrifuged for 5 minutes at 12,000 g. A portion of the supernatant was used for total protein analysis and the remainder was combined with a Western loading buffer. Ten or 15 μg protein (diluted in buffer) was loaded onto each lane of the gel (sodium dodecyl sulfate-polyacrylamide gel electrophoresis [7.5% for eNOS or 11.5% for CAV-1] with 5% stacking gel and Tris-glycine running buffer). Each gel was presented with samples from each of the experimental groups, along with a standard human umbilical vein endothelial cell extract (Transduction Laboratories, Lexington, KY, U.S.A.). To confirm that each lane contained equivalent amounts of protein, one duplicate gel was run in parallel and stained for total protein using Gelcode Blue. The original gel was wet-transferred to a polyvinyl difluoride membrane using CAPS/methanol buffer. Membranes were blocked with 5% nonfat dry milk in phosphate-buffered saline/Tween 20, followed by overnight incubation at 4°C with mouse anti-eNOS or rabbit anti-CAV-1 antibody (Transduction Laboratories). After phosphate-buffered saline/Tween rinsing, specimens underwent a 2-hour incubation with the secondary antibody (goat anti-mouse or anti-rabbit IgG:horseradish peroxidase) and were rinsed again in phosphate-buffered saline/Tween. For quantitation of CAV-1 and eNOS expression, bands were visualized by incubation in enhanced chemiluminescence reagents and exposure to x-ray film. Quantitative assessment of band intensities (integrated optical densities) in the autoradiographs were performed using with an Alpha Innotech (Temucula, CA, U.S.A.) Imaging 2000 system. Background intensities (determined from an equal-sized area of the film adjacent to the band of interest) were subtracted.
Immunofluorescence analysis
Seven-micrometer coronal brain sections, obtained essentially from the midpoint of the region exposed by the craniotomy, were prepared from paraformaldehyde-fixed and paraffin-embedded brains (Pelligrino et al., 2000) harvested from simvastatin-treated and untreated OVX rats exposed to either antisense or missense oligonucleotide.
Statistics
Statistical comparisons of pial arteriole diameter changes between groups were made using a one-way analysis of variance, combined with a post hoc Tukey analysis. For comparisons of diameter values within a given experiment, a repeated measures one-way analysis of variance design and post hoc Tukey analysis was used. P < 0.05 was considered significant in all statistical tests. Values are presented as mean ± SD. All drugs and chemicals were obtained from Sigma, unless otherwise stated.
RESULTS
Consistent with previous results from the authors' laboratory (Pelligrino et al., 2000), pial tissue eNOS expression was less (Fig. 1A) and CAV-1 expression (Fig. 1B) was greater in samples from OVX versus intact females. Treatment of OVX rats with 17β-estradiol (E2) was accompanied by a restoration of eNOS and CAV-1 expression to levels observed in intact females (Figs. 1A and 1B). However, simvastatin treatment of OVX females was associated with a greater eNOS expression (Fig. 1C) than that seen in the untreated OVX group, but CAV-1 expression remained elevated (Fig. 1D). The three groups shown in each figure were run on the same gels, to permit a more accurate depiction of relative expression patterns.
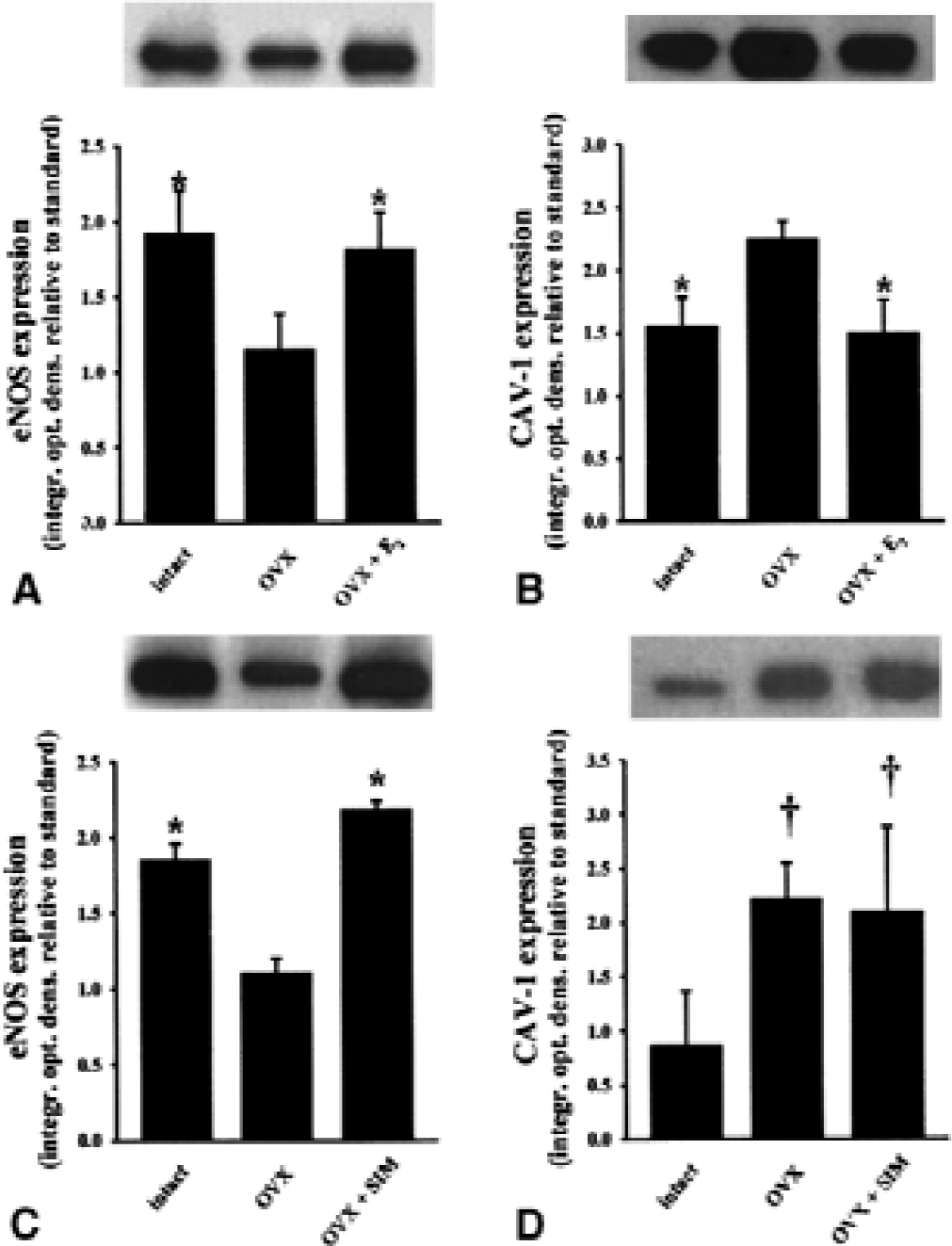
Relative expression of endothelial nitric oxide synthase (eNOS) and caveolin-1 (CAV-1) protein in pial tissue as a function of chronic estrogen manipulations or simvastatin treatment. Western blots were run by adding 10 μg protein onto each lane of the gel (7.5% for eNOS or 11.5% for CAV-1). Four different sets of immunoblots were run. The expression of eNOS
As previously reported (Pelligrino et al., 2000), OVX females exhibited a complete absence of ACh-induced vasodilating function (Fig. 2A) and a reduction in eNOS protein (Fig. 1A and 1C). However, chronic treatment of OVX rats with simvastatin did not restore agonist-activated eNOS function (Fig. 2B) despite the increase in eNOS protein (Fig. 1C). Recovery of the pial arteriolar response to ACh was observed only when simvastatin treatment was combined with 24-hour exposure to CAV-1 antisense oligonucleotide (Fig. 2C). The effect of the latter was dose-dependent and was not reproduced by missense oligonucleotide. Treatment with the antisense oligonucleotide alone had no effect (Fig. 2B). In a previous study, the authors showed that pial arteriolar ACh reactivity was lost in OVX females, but was restored with chronic E2 supplementation (Pelligrino et al., 2000). However, in that investigation, an acute cranial window preparation was used. Because, in the current study, pial arteriolar reactivities were measured 24 hours after window placement (and the introduction of 300 μL aCSF into the space under the windows), the authors thought it was important to repeat those previous evaluations. There were two objectives for these experiments. First, the authors sought to demonstrate that placement of the window and injection of aCSF 24 hours before study does not affect ACh responses. Second, the authors sought to confirm the findings summarized in Fig. 2B and 2C by using a previously-validated strategy, in OVX females, for up-regulating eNOS expression in combination with down-regulation of CAV-1 expression—that is, E2 treatment (Pelligrino et al., 2000). The current results (Fig. 2A) were virtually identical to those obtained in the authors' earlier study. Moreover, the pial arteriolar responses to 10 and 100 μmol/L ACh in the intact and E2 -treated females were similar to those seen in simvastatin-treated OVX rats given the higher dose of CAV-1 antisense. No significant differences in pial arteriolar CO2 reactivities were noted among the groups (data not shown). This essentially obviates the possibility that the differences in the ACh responses of pial arterioles were because of generalized alterations in vasodilating function.
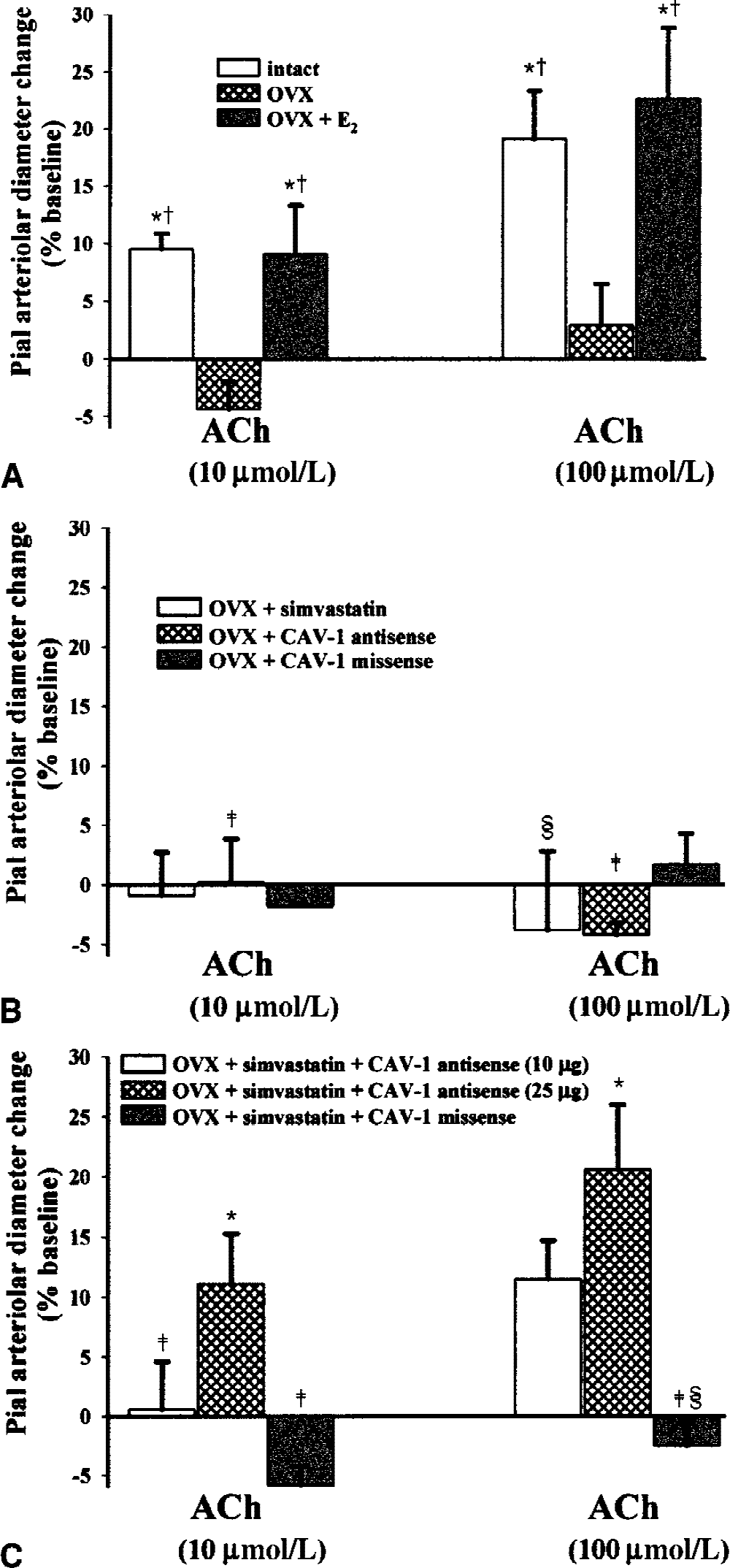
Recovery of endothelial nitric oxide synthase (eNOS)-dependent vasodilating function in ovariectomized (OVX) females requires both an increase in eNOS expression and a decrease in CAV-1 expression. Endothelial NOS function was determined by monitoring the dose-related effects of acetylcholine (ACh) on 25 to 50 μm pial arterioles. Results are expressed as the percentage change in diameter from the baseline value (means ± SD).
The effectiveness of the CAV-1 antisense treatment was confirmed by Western immunoblot analysis of CAV-1 expression in pial tissue harvested from the area under the cranial windows. Thus, CAV-1 abundance was much lower in tissue obtained from rats exposed to antisense compared to tissue from missense-treated animals (Fig. 3). In fact, CAV-1 expression was perhaps even more robust in the missense group as it was in “window” pial tissue taken from OVX or OVX + simvastatin-treated rats not exposed to any oligonucleotide. The specificity of the antisense treatment was additionally supported by the finding that pial tissue eNOS expression (via Western) was similar in simvastatin-treated rats exposed to antisense vs missense oligonucleotides (not shown). The CAV-1 immunoblot results were corroborated when viewing CAV-1 immunofluorescence in coronal brain sections that included the region under the cranial window. In a simvastatin-treated OVX rat exposed to 25 μg of the CAV-1 oligonucleotide, only faint and sparse immunoreactivity was observed in pial vessels and parenchymal vessels within 300 μm from the cortical surface (Fig. 4A and 4B). The intensity of the immunoreaction increased at greater distances from the surface, which probably is a reflection of the limited extent of oligonucleotide penetration. On the other hand, in a simvastatin-treated OVX rat exposed to 25 μg of the missense oligonucleotide, a robust and uniform pattern of vascular CAV-1 expression was observed throughout the field of view (Fig. 4C and 4D).
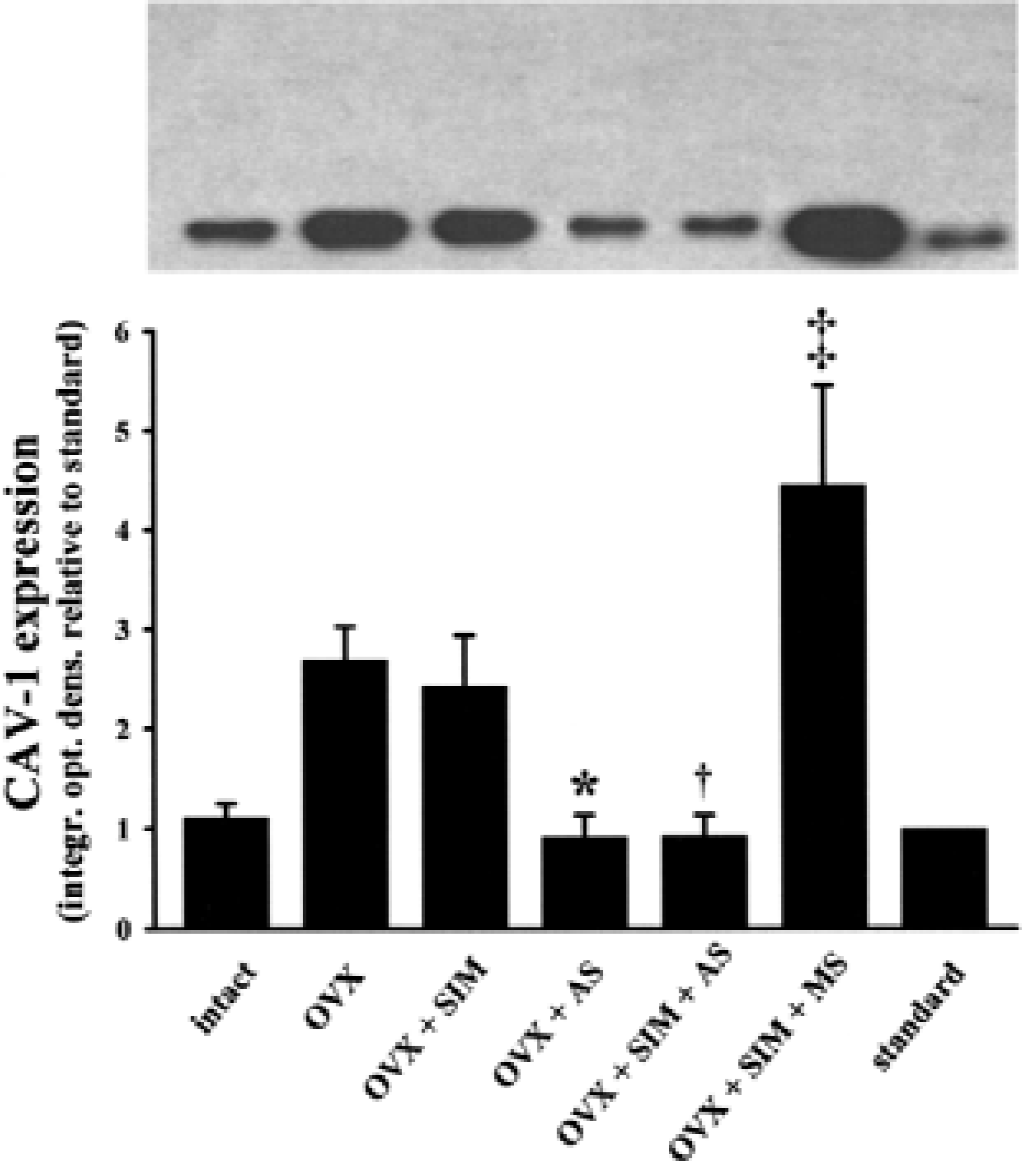
Expression of caveolin-1 (CAV-1) protein in pial tissue harvested from the space under the cranial windows in intact females; untreated ovariectomized (OVX) females; and OVX females given chronic simvastatin treatment (OVX + SIM), 25 μg CAV-1 antisense oligonucleotide (OVX + AS), chronic simvastatin plus 25 μg CAV-1 antisense (OVX + SIM +AS); or chronic simvastatin plus 25 μg CAV-1 missense (OVX + SIM + MS). For comparison, a HUVEC standard is also shown. Western blots were run by adding 15 μg protein onto each lane of the 11.5% gel. Similar results were obtained in three separate sets of experiments, as summarized in the bar graph. Data represent the means ± SD of three separate experiments. Band intensities (integrated optical densities) are expressed relative to a standard (HUVEC extract). * P < 0.05 versus OVX; †P < 0.05 versus OVX + SIM; ‡P < 0.05 versus OVX + SIM + AS.
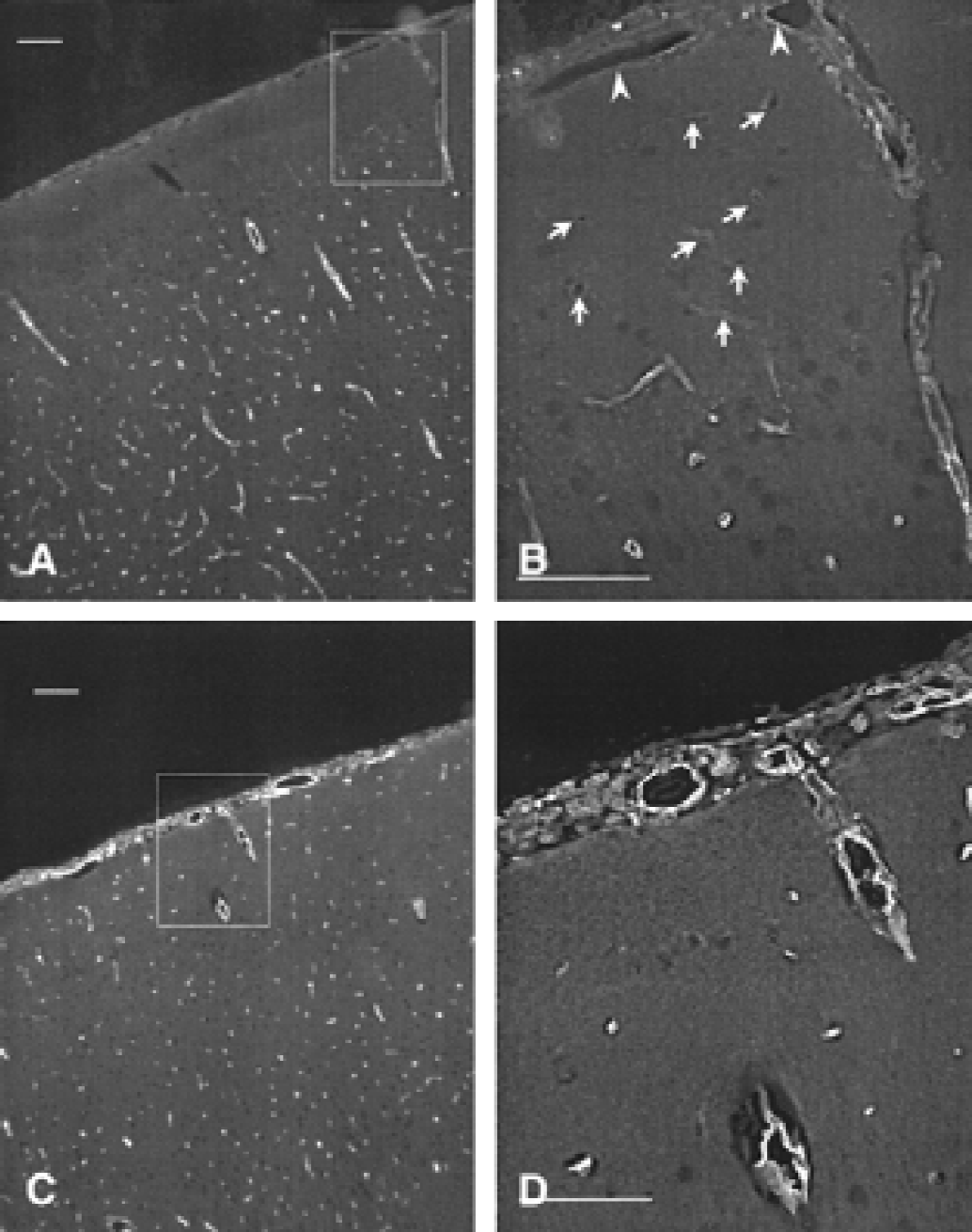
Immunofluorescence analysis of caveolin-1 (CAV-1) expression (using a rabbit polyclonal antibody and fluorescein isothiocyanate as the fluorphore) in 7-μm coronal sections of ovariectomized (OVX) rats chronically treated with simvastatin and exposed for 24 hours to topically applied
DISCUSSION
These findings demonstrated that to recover cerebral arteriolar agonist-activated eNOS function in OVX females, both an increase in eNOS expression and a reduction in CAV-1 expression were needed. Individually restoring either eNOS or CAV-1 expression to “normal” levels was insufficient. The current study, therefore, provides important mechanistic evidence to support the correlative findings obtained in the authors' previous article (Pelligrino et al., 2000). In that article, it was suggested that the marked eNOS functional sensitivity of cerebral arterioles to chronic E2 manipulations was related to the parallel observations of concomitant, but opposite, variations in the expression of eNOS and its endogenous inhibitor, CAV-1. Although many articles have been published regarding the ability of CAV-1 ability to regulate eNOS activity (Feron et al., 1998; Smart et al., 1999), the current study represents the first demonstration that CAV-1 can influence the functional activity of eNOS in vascular tissue in vivo. However, reducing the expression of CAV-1 does not, under all circumstances, ensure that eNOS functionality will improve—that is, the results of this study also suggested that a primary diminution in CAV-1 abundance can not increase agonist-activated eNOS function if eNOS protein levels are too low.
Notably, the authors chose to use unmodified phosphodiester oligonucleotides rather than the nuclease-resistant, phosphorothioate-modified forms. There were several reasons for this decision. First, the absence of nucleases in CSF (and, of course, aCSF;Whitesell et al., 1993), coupled with a rather slow turnover of the fluid under the cranial window, should restrict the loss of oligonucleotides in tissue that is in immediate contact with the periarachnoid CSF. Obviously, this includes the tissue targeted for CAV-1 “knockdown”—that is, the pial vessels. Second, the authors sought to avoid potential problems that might arise as a result of phosphorothioate-modified oligonucleotides binding nonspecifically to proteins and causing cytotoxicity (Wahlestedt, 1994). The fact that 60% to 80% reductions in CAV-1 expression were observed in the presence of the 25-μg antisense dose supports the effectiveness of the treatment. Moreover, no changes in eNOS expression were associated with the CAV-1 antisense exposure; this diminishes the likelihood that oligonucleotide degradation products (through nuclease actions) elicited nonspecific reductions in protein expression.
Caveolin-1 is a 22-to 24-kDa membrane-associated protein (which, in the brain, is concentrated in vascular endothelial cells [Ikezu et al., 1998b]) that is capable of binding eNOS through interactions between a selected amino acid sequence on the CAV-1 molecule (the “scaffolding domain”—a.a. 82–101) and a specific binding motif on eNOS (Smart et al., 1999). That particular interaction, which appears to be largely localized to plasma membrane structures called caveolae, results in loss of enzyme function. Upon Ca2+ -stimulated calmodulin binding, CAV-1 is competitively displaced and enhanced NO generation ensues. There is remarkably little information in the literature regarding the effects of a primary increase or decrease in CAV-1 expression, by itself, on eNOS function in endothelial cells. Nevertheless, Feron et al. recently reported that increasing CAV-1 expression (over 24 hours) diminished eNOS activity in cultured aortic endothelial cells (Feron et al., 1999).
Current evidence indicates that controlling CAV-1 expression represents an important mechanism for estrogen regulation of eNOS activity. However, evidence from studies in other tissues suggests that actions of estrogen, indirectly related to CAV-1, may also contribute to enhancement of eNOS function. One interesting possibility relates to estrogen-associated increases in calmodulin availability (Hayashi et al., 1994), another is serine phosphorylation of eNOS (through Akt;Simoncini et al., 2000). This has been reported to increase eNOS sensitivity to Ca2+ /calmodulin (McCabe et al., 2000), in essence, reducing CAV-1 inhibitory interactions with eNOS. Whether estrogen acts similarly in cerebral endothelium—that is, to increase calmodulin availability or sensitivity—remains to be established.
The mechanism involved in the apparent negative correlation between chronic estrogen status and CAV-1 expression is unknown. No estrogen response elements have been identified on the CAV-1 promoter. However, the possibility that estrogen affects CAV-1 expression through intermediary genomic regulators merits consideration. This includes the capacity of estrogen to reduce cholesterol levels (Liu and Bachmann, 1998), coupled with the reported link between high cholesterol and increased CAV-1 expression (Feron et al., 1999). A second possibility relates to estrogen's reported capacity to increase mitogen-activated protein kinase pathway activity, coupled with the documented effect of increased MAP kinase activity to reduce CAV-1 expression (Smart et al., 1999). A third may be linked to the purported ability for estrogen to activate adenylyl cyclase and protein kinase A (PKA) (Lagrange et al., 1997), along with indications that PKA activation interferes with CAV-1 transcription (Smart et al., 1999). Although these possibilities have yet to be tested in cerebral endothelium, they may provide a useful starting point when formulating experimental strategies designed to clarify the mechanisms involved in estrogen regulation of cerebral CAV-1 expression and eNOS function.
Footnotes
Acknowledgments:
The authors thank Susan Anderson and Dr. Shu Hua Ye for their expert technical assistance.