
Abstract
Poly(ADP-ribose) polymerase (PARP), or poly-(ADP-ribose) synthetase, is a nuclear enzyme that consumes NAD when activated by DNA damage. The role of PARP in the pathogenesis of traumatic brain injury (TBI) is unknown. Using a controlled cortical impact (CCI) model of TBI and mice deficient in PARP, the authors studied the effect of PARP on functional and histologic outcome after CCI using two protocols. In protocol 1, naïve mice (n = 7 +/+, n = 6 –/–) were evaluated for motor and memory acquisition before CCI. Mice were then subjected to severe CCI and killed at 24 hours for immunohistochemical detection of nitrated tyrosine, an indicator of peroxynitrite formation. Motor and memory performance did not differ between naïve PARP +/+ and –/– mice. Both groups showed nitrotyrosine staining in the contusion, suggesting that peroxynitrite is produced in contused brain. In protocol 2, mice (PARP +/+, n = 8; PARP –/–, n = 10) subjected to CCI were tested for motor and memory function, and contusion volume was determined by image analysis. PARP –/– mice demonstrated improved motor and memory function after CCI versus PARP +/+ mice (P < 0.05). However, contusion volume was not different between groups. The results suggest a detrimental effect of PARP on functional outcome after TBI.
Poly(ADP-ribose) polymerase (PARP) or poly ADP-ribose synthetase is a highly conserved, abundant nuclear enzyme that has been hypothesized to play a role in genomic surveillance and stability, cellular growth and differentiation, and repair of DNA damage (Althaus and Richter, 1987; de Murcia et al., 1994). After its activation by DNA strand breaks, PARP binds to damaged DNA and catalyzes the addition of ADP-ribose units to a number of nuclear proteins including PARP itself, using nicotinamide adenine-dinucleotide (NAD+) as substrate. ADP-ribosylation of chromosomal proteins may facilitate DNA repair by a number of mechanisms that include promotion of structural changes in chromatin and inhibition of inappropriate transcription and recombination of damaged DNA (Berger, 1985; Althaus and Richter, 1987; Satoh and Lindahl, 1992). However, overactivation of PARP by extensive DNA damage can lead to rapid consumption of NAD, depletion of ATP, and cell death by energy failure (Berger, 1985; Gaal et al., 1987). Hydroxyl radical and peroxynitrite (a reactive oxidant species produced from the reaction of superoxide and nitric oxide) are key endogenous triggers of PARP activation in numerous pathologic conditions (Szabo and Dawson, 1998).
DNA damage may be produced after traumatic brain injury (TBI) by direct mechanical injury, reactive oxygen species such as peroxynitrite, and by activation of endogenous endonucleases (Rink et al., 1995; Clark et al., 1997a; Nishio et al., 1997; Shohami et al., 1997). DNA damage has been reported in neurons in CA1, CA3, dentate gyrus, cortex, and other brain regions between 4 hours and 4 weeks after experimental TBI (Rink et al., 1995; Colicos and Dash, 1996; Clark et al., 1997a, b ; Conti et al., 1998; Yakovlev et al., 1997; Whalen et al., 1999b). DNA damage early after TBI would be a potent stimulus for PARP activation; DNA damage in cells injured after TBI could induce energy failure through extensive activation of PARP.
Exposure of neurons and other cell types to glutamate or reactive oxygen species in vitro induces DNA damage and cell death mediated by PARP activation and energy failure (Thies and Autor, 1991; Wallis et al., 1993; Cosi et al., 1994; Radons et al., 1994; Zhang et al., 1994; Cookson et al., 1998). Recent studies in experimental stroke show that induction of neuronal nitric oxide synthase and formation of peroxynitrite precede PARP activation, and that ischemic cell death is mediated in part by PARP activation in vivo (Endres et al., 1997; Takahashi et al., 1997; Endres et al., 1998a; Tokime et al., 1998). These studies have led to the “PARP suicide hypothesis” in the setting of ischemia when energy failure is a key initiator of necrotic cell death (Eliasson et al., 1997; Endres et al., 1997).
Excitotoxicity and production of DNA damaging agents such as oxygen free radicals and peroxynitrite are relevant to the pathogenesis of TBI (McIntosh, 1993; Palmer et al., 1993; Globus et al., 1995; Gong et al., 1995; Rink et al., 1995; Verweij et al., 1997; Yakovlev et al., 1997). Inhibitors of ADP-ribosylation protect hippocampal slices against percussion TBI-induced loss of CA1 evoked response in vitro, suggesting that PARP activation might contribute to loss of neuronal function after TBI (Wallis et al., 1996). Recent studies implicate PARP activation in the pathogenesis of CNS autoimmune and neurodegenerative diseases, and traumatic spinal cord injury (Hivert et al., 1998; Scott et al., 1999). Poly(ADP-ribose) polymerase activation has been observed in brain after fluid percussion in rats in a preliminary report (LaPlaca et al., 1998); however, the role of PARP in the pathogenesis of TBI is unknown.
The use of mice genetically deficient in PARP is a powerful strategy to study the role of PARP in the pathogenesis of TBI. The use of knockout mice completely eliminates PARP function and avoids the potential disadvantages of nonspecific PARP inhibitors. Based on evidence for a detrimental role of PARP activation in several models of CNS injury, we hypothesized that, compared to wild type mice (+/+), mice deficient in PARP (−/-) would have reduced histopathologic injury and improved functional outcome in a clinically relevant (controlled cortical impact [CCI]) model of TBI.
METHODS
Mice
All experiments were approved by the University of Pittsburgh Institutional Animal Care and Use Committee and complied with the NIH Guide for the Care and Use of Laboratory Animals. Mice were given free access to food and water and were housed in laminar flow racks in a temperature-controlled room with 12 hour day/night cycles.
Homozygous male (PARP –/–) and PARP +/+ litter mate controls consisting of a mixed genetic background of SV129 and C57BL/6 were generated as previously described (Wang et al., 1995), bred in an animal facility, and used at 12 to 13 weeks of age (23 to 38 g). PARP –/– mice were produced by deletion of part of the second exon and intron by standard techniques of homologous recombination in embryonic stem cells and the mice were genotyped as described in Wang et al. (1995). Poly-(ADP-ribose) polymerase –/– mice develop normally, are fertile, and are phenotypically normal (Wang et al., 1995).
Experimental protocols
Two experimental protocols were used. To exclude inherent differences in motor function and learning between PARP +/+ and –/– mice, the first protocol compared the performance of naïve mice (PARP +/+, n = 7 versus PARP –/–, n =6) in tests of motor (day 1) and memory (days 6 to 13) function described below. Naïve mice were then subjected to CCI and evaluated at 24 hours for immunohistochemical detection of nitrotyrosine, a marker for peroxynitrite, in contused brain regions. The second protocol compared the effect of CCI on motor and memory outcome in PARP +/+ (n = 8) versus PARP –/– (n = 10) mice. No preinjury training was used for Morris water maze (MWM) testing. Contusion volume in the injured (left) hemisphere, and left and right lateral ventricles and hemispheric volumes were also evaluated at 21 days in both groups.
Controlled cortical impact
The mouse CCI model was used as previously described (Smith et al., 1995) with minor modifications (Whalen et al., 1999a). Mice were anesthetized with 2% isoflurane (Anaquest, Memphis, TN, U.S.A.), N2O and O2 (2:1) and placed in a stereotactic frame. Brain temperature was monitored by a probe (0.009-inch diameter; Physitemp, Clifton, NJ, U.S.A.) inserted through a bur hole into the left frontal cortex, and body temperature was monitored by a rectal probe. A 5-mm craniotomy was performed over the left parietotemporal cortex, the bone flap was removed, and brain temperature was maintained at 37.5 ± 0.5°C for 5 minutes. Controlled cortical impact was then produced using a pneumatic cylinder (Bimba, Monee, IL, U.S.A.) with a 3-mm flat-tip impounder, velocity 6.0 ± 0.2 m/s, and depth of 1.2 mm. The bone flap was replaced and sealed immediately after CCI (Koldmount cement, Vernon Benshoff, Albany, NY, U.S.A.) and the scalp was sutured closed. Mice were allowed to recover from anesthesia in an oxygen hood for 30 minutes then returned to their cages.
Preparation of brain tissue for immunohistochemistry
At 24 hours after CCI, mice were anesthetized with isoflurane and transcardially perfused with 100 mL of 4% paraformaldehyde. The brain was rapidly removed intact and frozen at −70°C. Coronal sections (10 µm) were placed on poly-
Immunohistochemistry (nitrotyrosine detection)
Nitrotyrosine was detected immunohistochemically in brain sections as an indicator of the presence of peroxynitrite and other nitrosating agents, as previously described (Scott et al., 1999). Endogenous peroxidase was quenched for 15 minutes with 0.3% hydrogen peroxide in methanol. Nonspecific binding was minimized by incubating the sections in 2% normal goat serum in phosphate buffered saline for 1 hour. Sections were then incubated overnight with 1:500 dilution of primary anti-nitrotyrosine antibody (Upstate Biotechnology, Lake Placid, NY, U.S.A.), and specific labeling was detected with a biotin-conjugated goat antirabbit IgG and avidin-biotin peroxidase complex (Vectastain Elite ABC kit, Vector Laboratories, Burlingame, CA, U.S.A.).
Assessment of motor function
Gross vestibulomotor function was assessed at 1 day after injury using a beam balance (Feeney et al., 1981; Dixon et al., 1987) test. The beam balance task consisted of placing the mouse on a suspended 1.0-cm wide wooden beam and recording the time the mouse remained on the beam for up to 60 seconds. Mice were trained prior to CCI in three trials during which baseline measurements of time on the beam were made.
Spatial memory acquisition assessment
Experimenters blinded to mouse genotype evaluated spatial memory performance of mice using the MWM task as previously described (Hamm et al., 1992; Long et al., 1996). A white pool (83 cm diameter, 60 cm deep) was filled with water to 29 cm depth. Several highly visible extramaze cues that remained constant throughout the trials were located around the pool. Water temperature was maintained approximately 24°C. The goal platform (a round, white wooden platform 10 cm in diameter) was positioned 1 cm below the surface of the water approximately 15 cm from the southwest wall. A video tracking system mounted above the pool (Chromotrack 3.0, San Diego Instruments, San Diego, CA, U.S.A.) recorded the swimming movements of the mice. To ensure recovery from motor deficits, testing was performed on days 14 through 20 after CCI. Each mouse was subjected to a series of four trials per day. For each trial, mice were randomized to one of four starting locations (north, south, east, west) and placed in the pool facing the wall. Mice were given a maximum 120 seconds to find the submerged platform. If the mouse failed to reach the platform by 120 seconds, it was placed on the platform by the experimenter and allowed to remain there for 30 seconds. Mice were placed in a 37°C incubator for 4 minutes between trials. To exclude differences in visual acuity between PARP +/+ and PARP –/– mice, testing was performed on a platform 1 cm above the surface of the water on days 19 and 20 after injury. Mouse performance in the MWM was quantitated by latency to find the platform. Probe trials were used to measure place memory. The mice were placed in the maze without the platform and the video tracking system was used to measure the percent time that the animal swam in the quadrant that previously contained the platform.
The swim speeds of PARP +/+ and PARP –/– mice were determined for each mouse on the first day of MWM testing by dividing the sum of the total distance that the mice swam during the four trials by the total swim time.
Lesion volume
Morphometric image analysis (MCID, Imaging Research, St. Catherines, Ontario, Canada) was used to determine contusion, ventricular, and hemispheric volumes at 21 days after CCI. Mice were anesthetized with isoflurane and transcardially perfused with 4% paraformaldehyde. Coronal sections (20 µm) were cut at 0.5 mm distances from the anterior to the posterior brain and mounted on poly-
Statistical analyses
Data are mean ± SD. Body weight, brain temperature, swim speed, and volumetric data were analyzed by t-test. Morris water maze acquisition times in naïve and injured PARP +/+ and –/– mice were analyzed by analysis of variance for repeated measures (group x time). Between group differences were analyzed by post hoc comparisons at each timepoint. Probe trial results were analyzed by one-way analysis of variance. Performance on the beam balance test was analyzed by analysis of variance followed by the appropriate post hoc test. For each test, P < 0.05 was considered significant.
RESULTS
Protocol I. Behavior assessment in naïve mice and posttraumatic immunohistochemistry
Naïve PARP +/+ and –/– mice performed equally well in the beam balance (Fig. 1) test. The data in Figs. 1 and 2 contain results for both naïve and injured (see below, protocol II) mice to facilitate comparison. Naïve PARP +/+ and –/– mice also performed equally well in the spatial memory acquisition paradigm (Fig. 2). Also, performance in the probe trials, measured by the percentage of time spent by the mice in the target quadrant, did not differ between naïve PARP +/+ (32.8 ± 4.0 %) and PARP –/– (32.4 ± 1.9 %) mice. Similarly, swim speed did not differ between naïve PARP +/+ (20.7 ± 1.9 cm/s) and PARP –/– (21.3 ± 1.3 cm/s) mice.
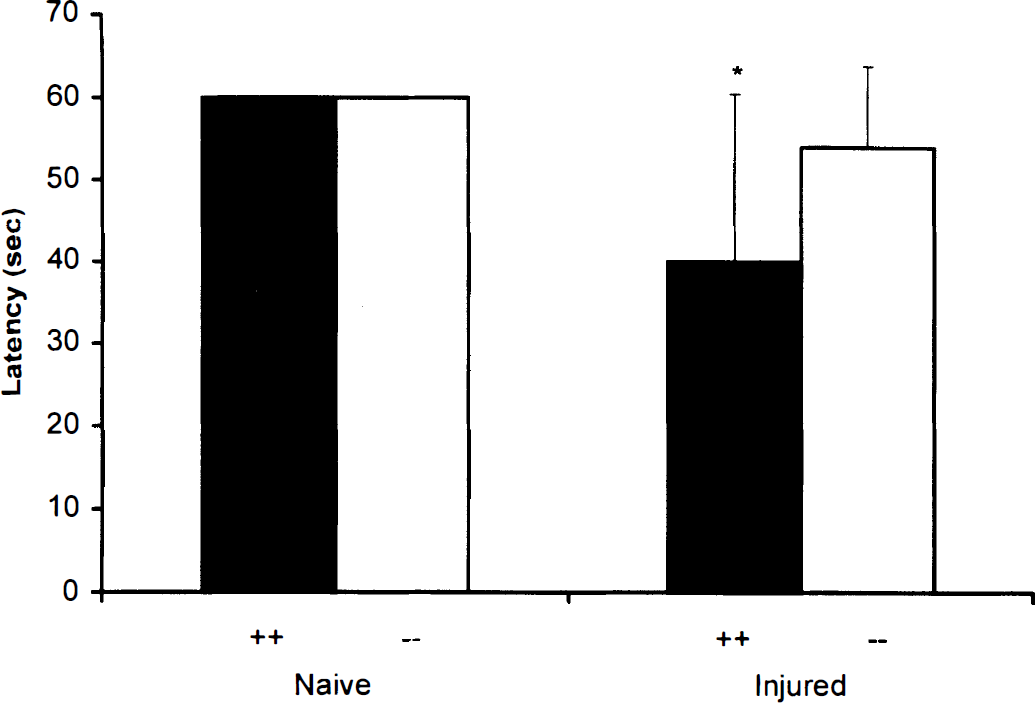
Motor function assessed by latencies (mean ± SD) to balance on a beam at 1 day after controlled cortical impact (CCI) in poly(ADP-ribose) polymerase (PARP) +/+ and –/– mice. Mice were tested for their ability to balance up to 60 seconds on a 1-cm wide wooden beam. Latency on the beam balance was measured in both groups of mice after CCI. No differences were observed between naïve PARP +/+ and PARP –/– mice. Injured PARP +/+ mice had significantly shorter (impaired) latencies versus PARP –/– (*P < 0.05).
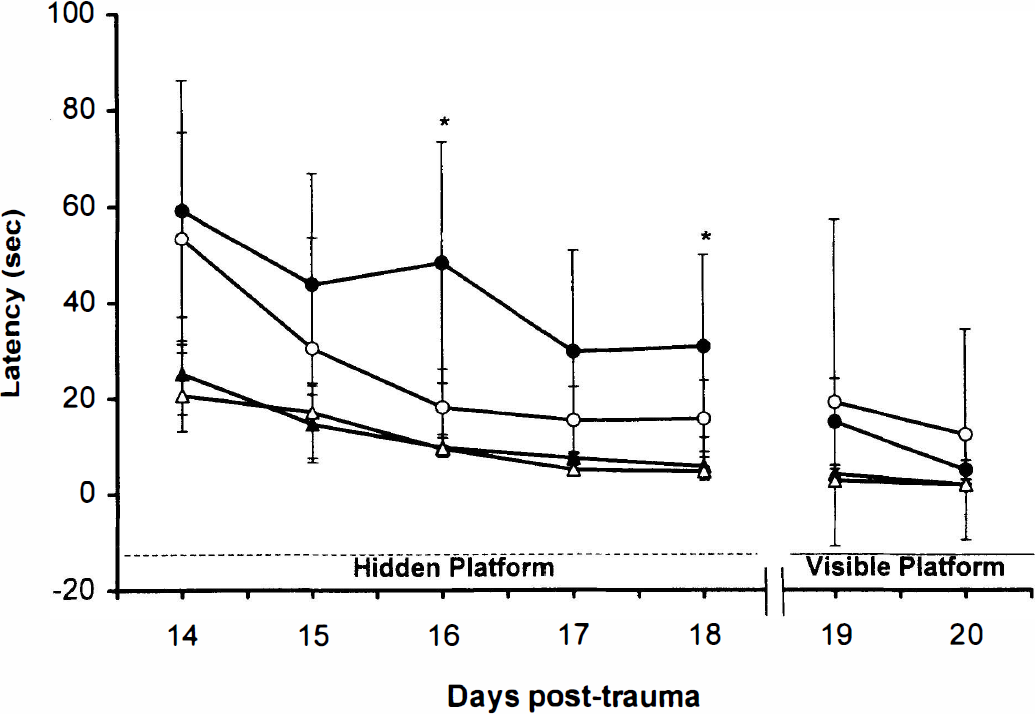
Results of spatial memory testing in mice using a Morris water maze acquisition paradigm (latency [mean ± SD) to find a hidden [submerged] or visible platform). Injured mice were tested on days 14 to 20 after controlled cortical impact (CCI). Naïve mice were tested on 5 consecutive days before CCI. Naïve poly-(ADP-ribose) polymerase (PARP) +/+ (▲) and –/– (Δ) mice performed equally well on all 5 days. Injured PARP +/+ (●) mice exhibited impaired performance compared to PARP –/– (ˆ) for this task (*P < 0.05). Performance in both injured and uninjured groups of mice improved on days 19 to 20, indicating that visual impairment did not explain the differences between groups.
All mice survived TBI with 24-hour outcome. Figure 3 shows the results of immunohistochemical detection of nitrotyrosine in representative brain sections from PARP +/+ and –/– mice at 24 hours after CCI. Nitrotyrosine, an indicator of peroxynitrite formation, was detected in the brain of both PARP +/+ and PARP –/– mice in the area adjacent to the contusion, whereas no staining was observed in uninjured brain regions. There were no qualitative differences in the degree of staining between PARP +/+ and PARP –/– mice. Omission of the primary nitrotyrosine antibody resulted in complete absence of immunoreactivity (data not shown).
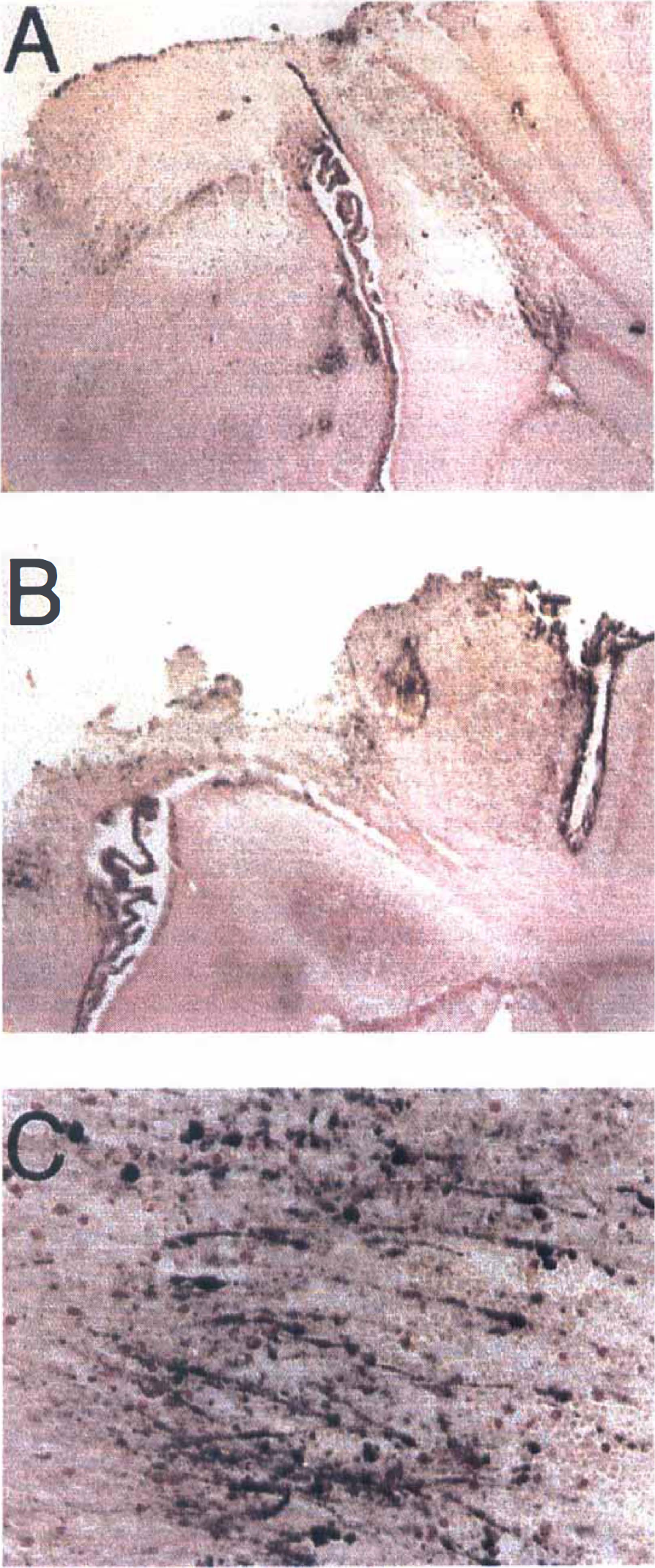
Nitrotyrosine staining in mouse brain at 24 hours after controlled cortical impact (CCI).
Protocol II. Behavior and histopathologic outcome after controlled cortical impact
Seventeen of the eighteen mice survived the 21-day trauma protocol. Body weight did not differ between PARP +/+ (29.6 ± 4.4 g) and PARP –/– (30.6 ± 4.1 g) mice before CCI.
Beam balance (Fig. 1) performance was significantly impaired in PARP +/+ and –/– mice compared to naïve mice. In addition, PARP +/+ mice performed significantly worse than PARP –/– on the beam balance test after CCI (P < 0.05; Fig. 1).
Latency to find the hidden platform in the MWM test was increased in both PARP +/+ and –/– mice compared with naïve mice (P < 0.05). In addition, MWM performance was significantly worse in PARP +/+ versus –/– mice (P < 0.05; Fig. 2). In contrast to the results with naïve mice, performance in the probe trials was impaired in injured PARP +/+ (22.0 ± 4.1%) compared with PARP –/– (32.3 ± 6.4%) mice (P = 0.33). Acquisition of spatial memory using the visible platform was similar in injured PARP +/+ and PARP –/– mice (days 19 to 20), indicating that impaired performance in the PARP +/+ mice was not caused by differences in visual acuity between groups (Fig. 2). Swim speeds (cm/s) did not differ between injured PARP +/+ (18.7 ± 1.1) and PARP –/– (20.1 ± 1.0) mice.
Contusion volume expressed in mm3 (PARP +/+, 21.01 ± 3.16; PARP –/–, 17.57 ± 1.57) or as percent injured hemisphere (PARP +/+, 9.84 ± 1.45; PARP –/–, 9.37 ± 0.86) did not differ between groups. Similarly, left ventricular volume did not differ between PARP +/+ (2.01 ± 0.25 mm3) and –/– (2.41 ± 0.21 mm3) mice. However, right (noninjured) hemispheric volume was less in PARP –/– (222.62 ± 6.56 mm3) compared with PARP +/+ (254.19 ± 11.79 mm3) mice (P < 0.05).
DISCUSSION
The main finding of this study is that mice deficient in PARP show protection from functional deficits after severe CCI. The effect of PARP deletion was evident early after TBI because PARP –/– mice performed significantly better than wild-type mice (PARP +/+) in the motor function tests on day 1 after injury. In addition, the beneficial effect of PARP depletion on memory acquisition was observed on days 16 and 18 after TBI, suggesting a sustained effect. The data are not explained by innate differences in learning, motor performance, visual acuity, or swim speed in naïve PARP –/– mice versus +/+ mice. The beneficial effect on functional outcome was seen despite no significant benefit of PARP deletion on histopathologic outcome.
This is the first examination of the role of PARP in an in vivo model of TBI using measures of functional outcome. A single study showing a modest beneficial effect on motor function at 24 hours after cerebral ischemia in PARP –/– versus +/+ mice and in mice treated with pharmacologic inhibitors of PARP has been reported (Endres et al., 1997). However, several studies have shown a dramatic decrease in infarct size in PARP –/– versus +/+ mice, suggesting a powerful detrimental effect of PARP after experimental stroke (Eliasson et al., 1997; Endres et al., 1997; Endres et al., 1998a). In vitro studies using hippocampal slices suggested a role for PARP activation in the loss of CA1 evoked potentials after fluid percussion injury because addition of PARP inhibitors restored electrophysiologic function (Wallis et al., 1996). Recent studies in rats have shown PARP activation at 24 hours after spinal cord injury (Scott et al., 1999) and within 2 hours after fluid percussion brain injury (LaPlaca et al., 1998); however functional outcome was not reported in these studies. In our study, there seemed to be a dissociation between histologic and functional outcome in PARP –/– mice after CCI. Contusion volume is a refractory therapeutic endpoint that is relatively difficult to manipulate in the CCI model (Clark et al., 1997b; Dixon et al., 1998; Whalen et al., 1998,1999a).
One mechanism by which PARP –/– mice may be protected from functional deficits is by inhibition of posttraumatic energy failure. After TBI, endogenous endonucleases and reactive oxygen species contribute to DNA damage. DNA single strand breaks are detectable in injured brain regions early after CCI in rats (Whalen et al., 1999b). DNA damage-initiated activation of PARP could lead to consumption of NAD and depletion of ATP, ultimately causing energy failure and cell death (Yamamoto et al., 1981; Schraufstatter et al., 1986). Numerous in vitro studies implicate PARP activation and energy failure in the necrotic death of neurons and astrocytes exposed to excitotoxic or oxidizing agents, as inhibition or deletion of PARP prevents depletion of NAD and cell death (Wallis et al., 1993; Cosi et al., 1994; Zhang et al., 1994,1995; Eliasson et al., 1997; Szabo, 1997; Endres et al., 1998b).
We demonstrated the formation of nitrotyrosine at 24 hours after CCI in contused brain. Robust nitrotyrosine staining in and around the contusion site in both PARP +/+ and PARP –/– mice supports a role for peroxynitrite, a known PARP activator (Szabo et al., 1998), in the pathogenesis of cell death and/or injury after CCI. Nitrotyrosine is a relatively specific marker of oxidative damage to proteins by peroxynitrite. Thus, it is possible that peroxynitrite generation is also producing oxidative DNA damage after TBI. In early stroke, poly ADP-ribose formation was abolished by the genetic inactivation of the neuronal isoform of NOS (Endres et al., 1998a). Peroxynitrite formation via neuronal NOS activity may also be involved in the pathogenesis of TBI. However, these results must be interpreted with caution because recent data suggest that nitration of tyrosine residues may also occur by reaction with oxygen radicals other than peroxynitrite (Sampson et al., 1998).
Severe TBI, both experimentally and clinically, initiates a cascade of events that result in acute energy failure, including the release of excitatory amino acids, direct cellular disruption, and local ischemia (Hayes et al., 1992; Hovda et al., 1995; Adelson et al., 1997). This results in sustained depression of bioenergetic status (Headrick et al., 1994) and formation of purine degradation products (Bell et al., 1996). It is likely that these events are additive in the setting of NAD depletion by PARP activation after TBI.
Poly(ADP-ribose) polymerase inhibition or deletion reduces infarction volume up to 80% in models of cerebral ischemia (Eliasson et al., 1997; Endres et al., 1997; Takahashi et al., 1997; Endres et al., 1998b). Energy failure is an important component of the pathophysiology of both cerebral ischemia and TBI. Poly(ADP-ribose) polymerase has also been hypothesized to play a role in neurodegenerative diseases mediated by the formation of oxygen free radicals and cellular energy depletion, such as excitotoxic neuronal degeneration (Meldrum and Garthwaite, 1990) and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced catecholamine depletion in brain (Cosi et al., 1997). Mechanisms other than energy failure, such as inhibition of enzymes by ribosylation, may also play a role in PARP-mediated neural injury.
Interpretation of our results depends on the demonstration that functional PARP activity and alternate pathways of ADP-ribosylation are absent in PARP –/– mice. Poly(ADP-ribose) polymerase –/– mice used in this study lack normal length PARP transcripts and PARP activity in brain and other tissues (Wang et al., 1995). In addition, poly (ADP-ribose) polymers were not detected after DNA damage in fibroblasts from PARP –/– mice (Wang et al., 1995). Nevertheless, interpretation of any results using the PARP –/– mice will be complicated by the fact that PARP –/– cells can produce significant, albeit small levels of poly ADP-ribose polymers and novel, additional PARP-like enzymes have been recently identified (Shieh et al., 1998; Smith et al., 1998).
One potential confounding result of this study is the previously unreported finding that hemispheric volume is approximately 10% less in PARP –/– versus +/+ mice. However, physical deformation of the brain by 1.2 mm via CCI in both groups would be expected to bias the insult against the PARP –/– group. The smaller hemispheric volume in PARP –/– mice is not explained by differences in body weight between PARP –/– and +/+ mice used in this study. We have previously shown that this injury does not lead to volume loss in the contralateral hemisphere (Whalen et al., 1999a). Despite the observed differences in brain volume, no differences in motor, visual, or cognitive testing were observed between naïve PARP –/– and +/+ mice.
Our study shows a detrimental role for PARP in the pathogenesis of TBI. This finding is impressive in light of the lack of an effect on functional outcome of other treatment strategies in the CCI model targeting a variety of other mechanisms in our laboratory. No effect on functional outcome or contusion volume was observed after CCI in mice deficient in intercellular adhesion molecule-1 (Whalen et al., 1999a) and interleukin-8 receptor homologue versus respective wild-type mice (Whalen et al., 1998). In addition, motor and memory function were actually worse after CCI in mice deficient in inducible nitric oxide synthase versus wild-type, indicating an endogenous neuroprotectant role for inducible nitric oxide synthase (Sinz et al., 1997). Similarly, TNF-α knockout mice exhibited worse cognitive outcome versus wild-type at 7 to 28 days after CCI (Scherbel et al., 1997).
In conclusion, we have shown for the first time a detrimental effect of PARP on functional outcome after TBI. Further studies are needed to confirm these results and define the mechanisms responsible for the observed effects. Pharmacological inhibition of PARP may represent a therapeutic strategy for reducing neurologic injury after TBI in patients.
Footnotes
Acknowledgments
The authors thank Bradley Stezoski for preparation of the figures and Marci Provins for preparation of the manuscript.