
Abstract
Cerebral ischemia-reperfusion leads to vascular dysfunction characterized by endothelial cell injury or death. In the present study, we used an in vitro model to elucidate mechanisms of human brain microvascular endothelial cell (HBMEC) injury after episodic ischemia-reperfusion. Near-confluent HBMEC cultures were exposed to intermittent hypoxia-reoxygenation (HX/RO) and, at different recovery time points, cell viability was assessed by the MTT assay, apoptotic death by fluorescence microscopy of terminal deoxynucleotidyl transferase-mediated 2'-deoxyuridine 5'-triphosphate-biotin nick end labeling (TUNEL)-positive cells, and nuclear translocation of apoptosis-inducing factor (AIF) and cleavage of poly(ADP-ribose) polymerase-1 (PARP-1) by immunoblotting of subcellular fractions. Reductions in HBMEC viability were proportional to the number of HX/RO cycles, and not the total duration of hypoxia. Using four cycles of 1-h HX with 1 h of intervening normoxic RO, cell viability was reduced 30% to 40% between 12 and 48 h. Treatment with the PARP-1 inhibitors 3-aminobenzamide or 4-amino-1,8-naphthalimide during the insult improved HBMEC viability at 24 h after insult, and resulted in dose-dependent reductions in TUNEL-positivity at 16 h after insult, but not if these treatments were delayed by 4 h. HX/RO-induced increases in nuclear AIF translocation, as well as PARP-1 cleavage, were also reduced dose-dependently at 4 h after insult by the inhibitors. The caspase inhibitor z-VAD-fmk blocked PARP-1 cleavage, but did not affect AIF translocation and was only modestly cytoprotective. These findings indicate that PARP-1 activation and a PARP-1-dependent, caspase-independent, nuclear translocation of AIF contribute to apoptotic cerebral endothelial cell death after ischemia-reperfusion, underscoring the potential for ischemic microvascular protection by inhibiting PARP activation or preventing AIF translocation.
Introduction
Cerebral hypoxia and ischemia can lead to injury of both parenchymal and vascular cells. The resultant cerebrovascular endothelial injury is manifested as impairments in autoregulation and vascular reactivity (Christopherson et al, 1993; Staunton et al, 1999), reduced expression of tight junction proteins (Fischer et al, 2002; Petty and Lo, 2002; Witt et al, 2003), increases in blood-brain barrier permeability (Petty and Lo, 2002; Witt et al, 2003), hypoperfusion, microvascular hemorrhage (Wang and Lo, 2003), and frank endothelial cell death. Moreover, endothelial cell ‘activation’ after hypoxia or ischemia is reflected by the production of proinflammatory mediators, adhesion molecule expression, and leukocyte and platelet adherence to endothelium with attendant microvascular plugging (del Zoppo and Mabuchi, 2003), all of which can impact significantly on neuronal injury and overall outcome after cerebral hypoxic and/or ischemic insults. Less is known about the cerebrovascular responses and consequences of intermittent hypoxia, although the neurocognitive and neurobehavioral dysfunction (Li et al, 2004; Row et al, 2002), neuronal injury (Gozal et al, 2001), and predisposition to stroke (Dyken et al, 1996; Hermann and Bassetti, 2003; Yaggi and Mohsenin, 2003) associated with chronic obstructive pulmonary disease, sleep apnea and sleep disordered breathing, asthma, pulmonary fibrosis, apnea of prematurity, and recurrent intrauterine asphyxia, are well described.
Poly(ADP-ribose) polymerase-1 (PARP-1; EC 2.4.2.30) is an abundant nuclear enzyme whose activity is rapidly stimulated by single- and double-stranded breaks in DNA. Poly(ADP-ribose) polymerase-1 normally functions in homeostatic protective and regulatory roles by repairing DNA and poly-(ADP-ribosyl)ating histones and other nuclear proteins, which in turn requires the ATP-dependent hydrolysis of NAD+ to nicotinamide (Nguewa et al, 2005). In the event of significant cellular stress accompanied by DNA damage, overactivation of the enzyme may irreversibly deplete the cell of NAD+ and ATP, leading to necrosis, a mechanism known as the PARP ‘suicide’ hypothesis (Ha and Snyder, 1999). Indeed, deletion of the PARP-1 gene (Eliasson et al, 1997; Endres et al, 1997) or pharmacological inhibition of PARP-1 (Endres et al, 1997; Takahashi et al, 1999) protects against cerebral ischemic injury. In vitro studies revealed that diverse apoptotic stimuli including oxidative stress with concomitant energy depletion (Du et al, 2003), serum deprivation (Simbulan-Rosenthal et al, 1998), and alkylating agents (Oliver et al, 1998) cause a caspase-mediated cleavage of the 113-kDa PARP-1 into 85- and 24-kDa fragments, thereby allowing the cell to preserve ATP levels required for energy-dependent apoptosis and to release the suppression of apoptosis by poly(ADP-ribosyl)ated histone H1 (Yoon et al, 1996); as such, the presence of cleaved PARP-1 is considered a hallmark of apoptosis (Kaufmann et al, 1993; Lazebnik et al, 1994). Studies in endothelial cell cultures from noncerebral tissues support an association between PARP activation and DNA fragmentation triggered by oxidant stress (Thies and Autor, 1991; Szabó et al, 1997; Walisser and Thies, 1999). With respect to brain endothelial cells, apoptosis of cultured bovine brain endothelial cells in response to bilirubin (Akin et al, 2002), oxyhemoglobin (Meguro et al, 2001), or the 16-kDa fragment of human prolactin (Martini et al, 2000) is associated with caspase activation (Martini et al, 2000) and PARP cleavage (Akin et al, 2002; Meguro et al, 2001). When cocultured with macrophages, rat brain endothelial cells succumb to a pneumococci-induced reduction in cell viability that is accompanied by PARP activity and blocked by a PARP inhibitor (Koedel et al, 2002). However, PARP activation and PARP cleavage after hypoxia/reoxygenation-induced injury of cerebral endothelial cells has not yet been investigated.
Recently, a caspase-independent cell death pathway was elucidated involving a proapoptotic flavoprotein termed apoptosis-inducing factor or AIF (Susin et al, 1999). Current evidence indicates that this protein may normally function as an oxidoreductase and mitochondrial antioxidant, but in response to apoptotic stimuli, it translocates from the mitochondria to the nucleus, where it triggers chromatin condensation and apoptosis (Hansen and Nagley, 2003; Klein et al, 2002). AIF is implicated in the apoptotic cell death of cultured neurons in response to glutamate toxicity, oxidative stress, and oxygen-glucose deprivation (Yu et al, 2002; Zhang et al, 2002; Plesnila et al, 2004), and in rodent models of transient global or focal ischemia, nuclear translocation of AIF occurs concomitant with or slightly before large-scale DNA fragmentation (Cao et al, 2003; Plesnila et al, 2004). In many of the aforementioned cell death models, caspase inhibitors are ineffective in blocking apoptosis (Cao et al, 2003; Yu et al, 2002), indicating that AIF-mediated apoptotic death is caspase-independent (Joza et al, 2001). Evidence can be found for AIF-mediated apoptosis of coronary endothelium in response to oxidized low-density lipoprotein (Zhang et al, 2004), but still unexplored is the role of AIF in hypoxic-ischemic injury of cerebral endothelium.
Under some conditions, AIF may be a key downstream effector of PARP-1-mediated cell death. Studies in cultured neurons and fibroblasts indicate that PARP activation secondary to DNA strand breaks leads to the poly(ADP-ribosyl)ation of nuclear proteins, which then translocate to the mitochondria to signal AIF release from this organelle; AIF, in turn, translocates to the nucleus to cause chromatin condensation and DNA fragmentation, thereby setting up a positive feedback loop (see Hong et al (2004) for a review). Indeed, AIF-induced apoptosis in vitro is blocked in PARP-deficient cells (Boulares et al, 1999; Yu et al, 2002) and by PARP inhibitors (Yoon et al, 1996; Yu et al, 2002), supporting the notion that AIF is a mediator of cell death secondary to PARP-1 activation. How these regulatory pathway operate in hypoxic or ischemic cerebral endothelium is not known.
Thus, the present study was undertaken to examine apoptotic injury mechanisms activated in cerebral endothelial cells subjected to HX/RO. Confluent cultures of human brain microvascular endothelial cells (HBMEC) were used to test the hypotheses that the demise of these cells after repetitive HX/RO insults is apoptotic, as evidenced by PARP-1 cleavage and terminal deoxynucleotidyl transferase-mediated 2'-deoxyuridine 5'-triphosphate-biotin nick end labeling (TUNEL)-positivity, and is accompanied by nuclear AIF translocation. The effect of pharmacological inhibition of PARP-1 and caspases on these end points was also investigated to confirm a link between caspase activation, PARP-1 activation, and AIF-induced cerebral endothelial cell cytotoxicity.
Materials and methods
In Vitro Human Brain Microvascular Endothelial Cell Injury Model
An SV-40 large T-antigen stably transfected HBMEC line (IT-1) that exhibits vigorous growth was used (Hess et al, 2000). We routinely documented that these cells stained positively for the endothelial cell-specific phenotypic marker von Willibrand factor VIII, and negatively for glial fibrillary acidic protein (data not shown). HBMECs were cultured in 24-well plates in complete medium M199 with 15% fetal bovine serum in a normoxic incubator (37°C, 5% CO2). For simulated ischemia-reperfusion, cultures at 90% confluence were washed twice with Hank's balanced salt solution (HBSS; in g/L: CaCl2 0.14, KCl 0.4, KH2PO4 0.06, MgCl2 0.1, MgSO4 0.1, NaCl 8.0, NaHCO3 0.35, Na2HPO4 0.048, and D-glucose 1.0) with 1% serum and placed in a hypoxic incubator (1% O2, 5% CO2, 94% N2) at 37°C. Reoxygenation was achieved by return of the cultures to the normoxic incubator in M199 (with 1% serum). Two episodic HX/RO protocols and one continuous hypoxia protocol were used for the HBMEC injury studies. In particular, HBMECs were subjected to four successive cycles of 1-h hypoxia followed by 1-h reoxygenation, or three successive cycles of 2-h hypoxia followed by 1-h reoxygenation, or 8 h of continuous hypoxia. After these challenges, cells were recovered under normoxic, normothermic conditions with 1% serum and 25 mmol/L glucose in M199 for 4 to 48 h depending on the end point studied.
Two structurally distinct PARP-1 inhibitors, 3-aminobenzamide (3-AB) (Banasik et al, 1992) and 4-amino-1,8-naphthalimide (4-AN) (Banasik et al, 1992; Schlicker et al, 1999; Thiemermann et al, 1997), were used. Three treatment regimens were used. In one, the drug was present throughout the episodic HX/RO treatments at a final concentration of 0.1 or 1 mmol/L for 3-AB, and 5 or 50 μmol/L for 4-AN, with fresh drug solution added at the start of each hypoxia cycle. HBMECs were not exposed to these drugs during the final normoxic recovery period (0 to 48 h). In the second treatment protocol, 3-AB (1 mmol/L) or 4-AN (50 μmol/L) were added once, in a delayed manner, at 4 h into the normoxic recovery period. A third protocol was used to assess the effects of pretreatment with the caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp fluoromethylketone (z-VAD-fmk; 40 μmol/L), which was administered 1 h before the episodic HX/RO insult.
Cell Viability Determinations
HBMEC viability after HX/RO was quantified in quadruplicate by reduction of 3-[4,5-dimethylthiazol]-2,5-diphenyltetrazolium bromide (MTT), wherein MTT is converted to a colored formazan precipitate in healthy, respiring cells (Hansen et al, 1989). After 12, 24, or 48 h of recovery, 40 μL MTT (5 mg/mL; Sigma, St Louis, MO, USA) was added to the medium in each well, mixed, and returned to the incubator for 5 h. Two hundred microliters of cell lysis buffer (10% sodium dodecyl sulfate (SDS), 0.01% HCl) was added to each well, and cells were again returned to the incubator for 12 h. Supernatant formazan content was quantified spectrophotometrically at 550 nm using a plate reader. Because HBMEC continued to grow and divide during the 12 to 48 h recovery period after the HX/RO intervention, cell viability was normalized to the formazan content in time-matched control cultures.
Fluorescence Microscopy for TUNEL-Positivity
Using HBMEC grown on gelatin-coated coverslips in 24-well plates, TUNEL staining was performed using a kit according to the manufacturer's instructions (Roche Applied Science, Indianapolis, IN, USA). Sixteen hours after HX/RO, cells were washed and fixed with paraformaldehyde, rinsed with phosphate-buffered saline (PBS), permeabilized briefly, and incubated in the dark with 50 μL TUNEL reaction mixture for 1 h at 37°C. The DNA-binding agent 4,6-diamidino-2-phenylindole (DAPI) was added (10 μg/mL) to counterstain all cell nuclei. After a final rinse, coverslips were reverse-mounted onto slides, and cells were observed by fluorescence microscopy. The number of TUNEL-positive cells, as a percentage of DAPI-positive cells, was determined in four fields at x 20 magnification.
Subcellular Fractionation
HBMECs were cultured to 90% confluency in 150-mm gelatin-coated dishes and subjected to the 4-cycle HX/RO protocol as described above. After 4 h of normoxic recovery, cells were collected and nuclear protein extracts were obtained as follows. All procedures were performed on ice or at 4°C. Cells were washed with ice-cold PBS, scraped from the dish, and centrifuged (600g for 5 mins) to obtain a cell pellet, which was then resuspended in lysis buffer (20 mmol/L HEPES-KOH (pH 7.4), 10 mmol/L NaCl, 1.5 mmol/L MgCl2, 1 mmol/L EDTA, 1 mmol/L EGTA, 250 mmol/L sucrose, 1 x protease inhibitor cocktail). After centrifugation (600g for 10 mins), the supernatant was discarded and the pellet was washed and resuspended in nuclear extraction buffer (20 mmol/L Tris-HCl) (pH 7.5), 1.5 mmol/L MgCl2, 420 mmol/L NaCl, 0.2 mmol/L EDTA, 25% glycerol, 0.5% 1 x Triton X-100, 0.1% NP-40, 1 x protease inhibitor cocktail), vortexed, and shaken for 30 mins. The suspension was then centrifuged (14,000g for 10 mins), and the supernatant containing the nuclear fraction was transferred to a prechilled microcentrifuge tube. Total protein was measured using the BCA Protein Assay Kit (Pierce, Rockford, IL, USA), and the nuclear extracts were aliquotted and kept at −80°C until immunoblotting.
Immunoblotting for Cleaved PARP-1 and Apoptosis-Inducing Factor Translocation
A 15 μg sample was loaded onto 4% to 15% SDS gradient gels (Bio-Rad, Hercules, CA, USA). Separated proteins were transferred to nitrocellulose membranes (Hybond ECL, Amersham Pharmacia Biotech, Piscataway, NJ, USA), immersed in 3% BSA in 1 x TBST for 1 h to block nonspecific antigen, and then exposed to primary antibody at 4°C overnight. Membranes were washed three times with 1 x TBST, and exposed to secondary antibody (1:2,000) for 1 h at room temperature. Rabbit anti-human antibodies were used for both PARP-1 and AIF (both at 1:1,000; BD Biosciences Pharmingen, San Diego, CA, USA). After multiple washes with 1 x TBST, membranes were immersed in ECL solution (Pierce) for 1 min at room temperature and exposed to film in a dark room. Protein abundance was quantified by measuring the relative optical density of the respective protein bands (Image Pro Plus, San Diego, CA, USA) and normalizing to β-actin to control for loading, and then normalizing again to normoxic control levels.
Statistics
All results are presented as means ± standard deviations. For the cell viability studies, five to eight independent experiments were conducted and for the TUNEL study, four independent experiments were conducted. Immunoblots were performed on samples from three independent experiments. Comparisons between experimental conditions were made by Mann-Whitney nonparametric ranksum tests, with significance defined as P<0.05.
Results
Episodic Hypoxia-Reoxygenation Injury of Human Brain Microvascular Endothelial Cells
Cell viability determinations revealed that episodic HX/RO injured cultured HBMEC to a greater extent than continuous hypoxia. Figure 1 shows that, relative to control cultures, both of the two episodic HX/RO protocols resulted in a rapid and sustained reduction in cell viability over the ensuing 12 to 48 h. These HX/RO interventions also slowed the growth of HBMEC over the 48-h observation period, with the most significant effect observed with the four-cycle protocol. Thus, overall, both cell viability and cell growth rate were affected by the HX/RO interventions. With regard to HBMEC injury, the protocol with four 1-h HX/RO insults was actually more injurious to HBMEC than the protocol defined by three 2-h HX/RO insults, even though the duration of hypoxia was decreased in the former (4 h) relative to the latter (6 h). Moreover, a continuous exposure to severe hypoxia for 8 h (twice the duration of hypoxia) was not significantly injurious to the cells until 48 h after insult. These findings indicate that the number of HX/RO episodes, and in particular the number of posthypoxic reoxygenation events (four versus three, respectively), is more directly related to HBMEC injury than the total duration of hypoxia per se. For the remaining set of experiments designed to assess the effects of PARP inhibition on cell viability, apoptosis, PARP-1 cleavage, and AIF translocation after HX/RO, the four-cycle HX/RO protocol was used given that it resulted in a moderate level of injury (30% to 40% loss of viability) 12 to 48 h after the intervention.
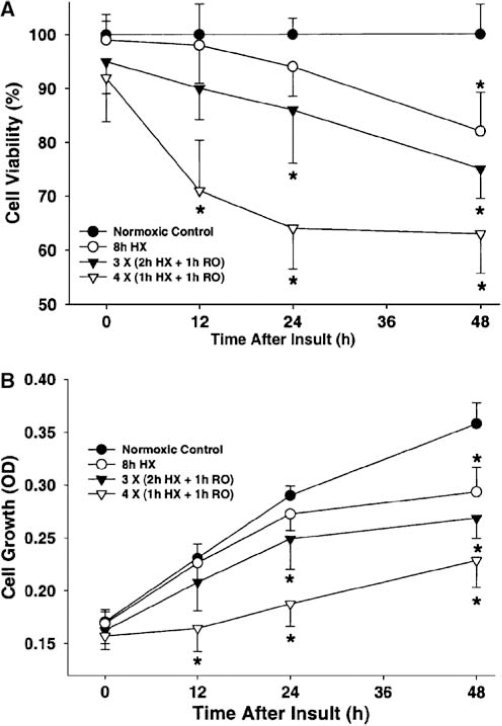
Effect of different periods of episodic HX and RO on cell viability and cell growth in cultured human microvascular cerebral endothelial cells. (
Effect of PARP-1 Inhibition on Human Brain Microvascular Endothelial Cell Viability after Episodic Hypoxia-Reoxygenation
Figure 2 shows that treatment of HBMEC with the PARP-1 inhibitors 3-AB or 4-AN improved cell viability after episodic HX/RO. When given concomitantly with the HX/RO stress, both doses provided relatively equal reductions in cell injury as assessed by MTT reduction at 24 h of recovery (Figure 2A). Nonsignificant trends towards protection were noted for both drugs 12 h after HX/RO (data not shown), but significant protection was still evident for both treatments 48 h after HX/RO (data not shown). However, when treatment was delayed until 4 h into the normoxic recovery period, improvements in cell viability in response to 3-AB (1 mmol/L) and 4-AN (50 μmol/L) were no longer evident (Figure 2A). In the absence of HX/RO, we determined that neither 3-AB nor 4-AN had any significant toxicity effects at the concentrations used (data not shown).
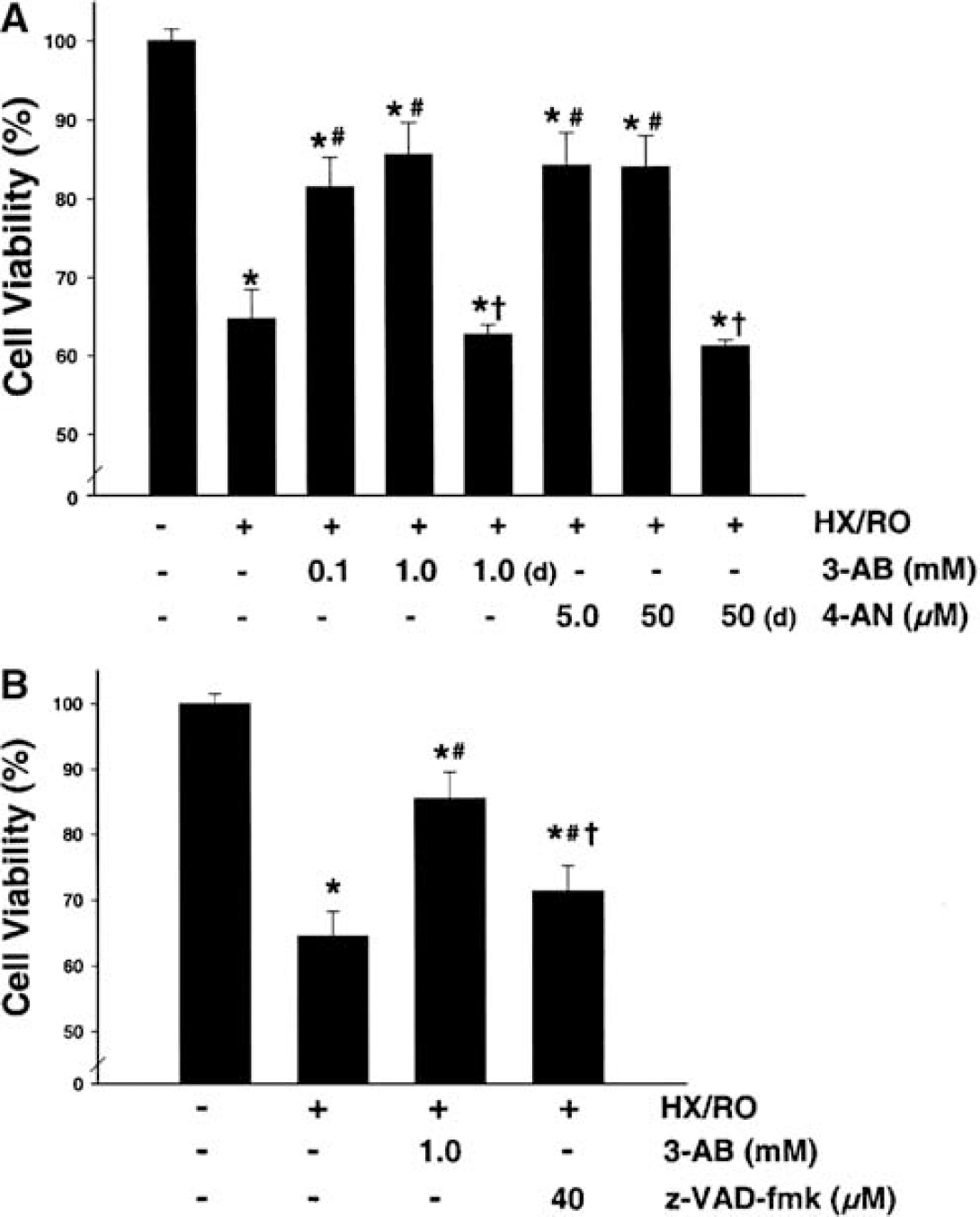
(
Pretreatment of HBMEC with the caspase inhibitor z-VAD-fmk was partially protective against episodic HX/RO-induced injury (Figure 2B).
Episodic Hypoxia-Reoxygenation Injury of Human Brain Microvascular Endothelial Cells is Apoptotic
Episodic HX/RO resulted in a robust increase in the number of TUNEL-positive cells at 16 h of recovery, as observed by fluorescence microscopy, whereas very few cells were TUNEL-positive under normoxic conditions (Figures 3 and 4). At this time, nearly 80% of HBMEC were intensely TUNEL-positive, and were notable morphologically for their shrunken nuclei. PARP-1 inhibition by 3-AB or 4-AN significantly and dose-dependently decreased the number of TUNEL-positive cells at this time point (Figures 3 and 4).
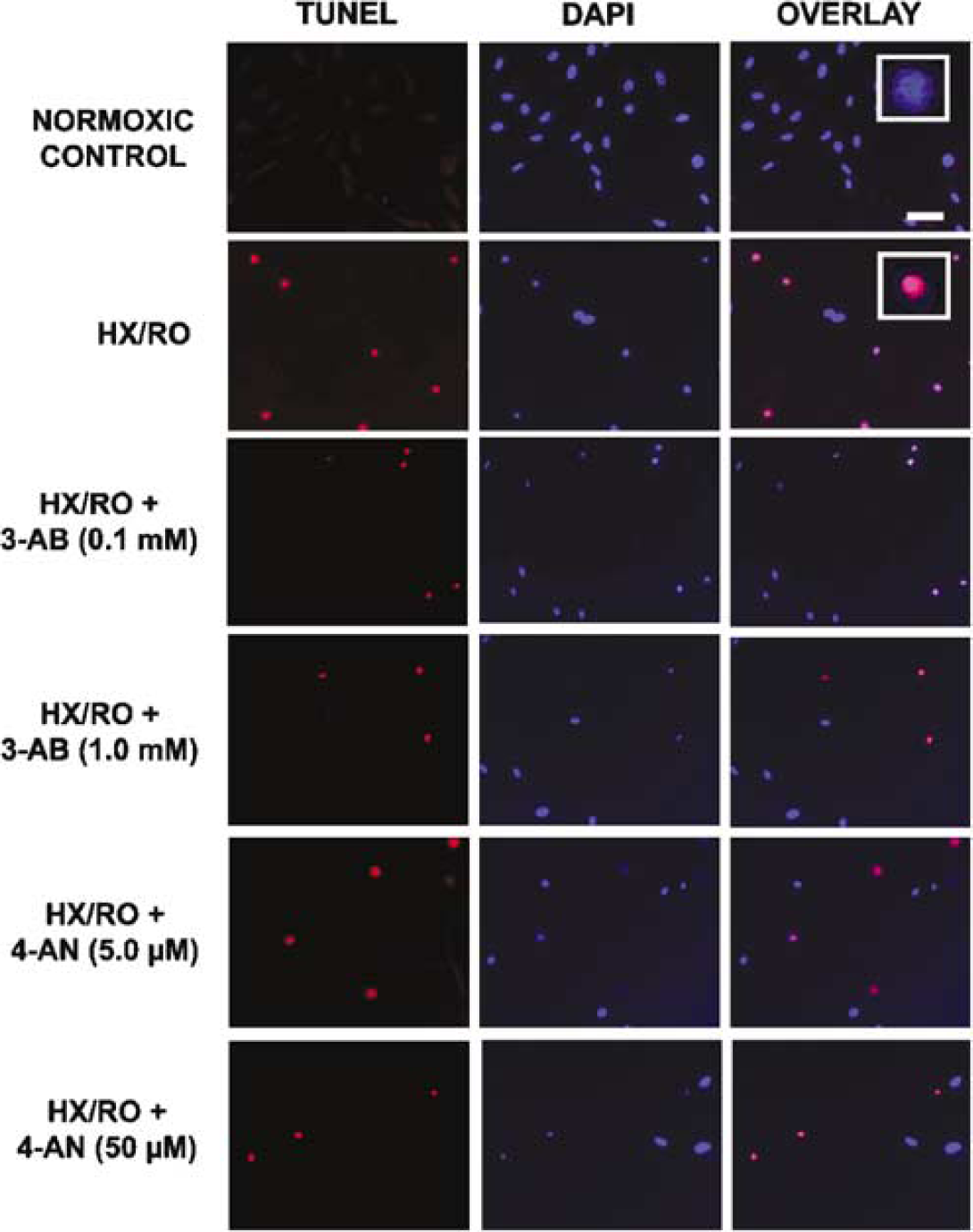
Effect of PARP-1 inhibition with 3-AB or 4AN on TUNEL staining of cultured human microvascular cerebral endothelial cells measured 16 h after episodic HX/RO. TUNEL staining for apoptotic nuclei (red, left panel), DAPI staining for all cell nuclei (blue, middle panel), and overlay (right panel) reveals increased TUNEL-positivity after HX/RO was reduced by both inhibitors. Inset: TUNEL-positive cells exhibited smaller, condensed nuclei after HX/RO insult relative to normoxic controls. Scale bar in top right panel = 40μm.
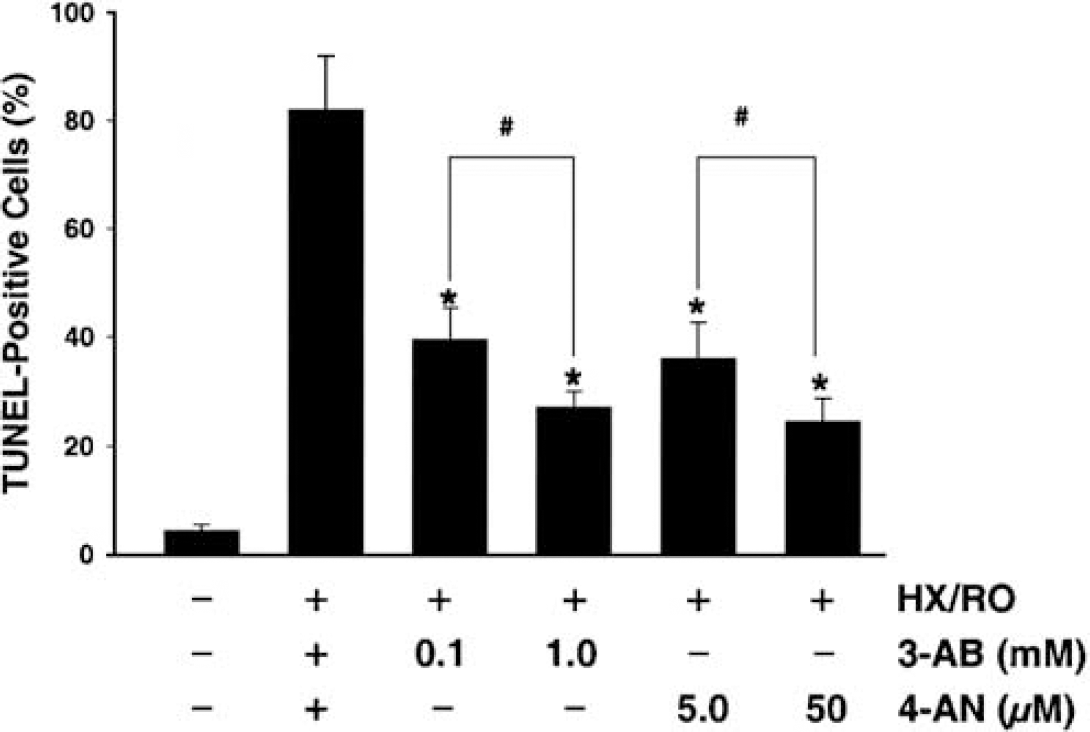
Quantification of the effect of PARP-1 inhibition by 3-AB or 4-AN on TUNEL-positive human microvascular cerebral endothelial cells measured 16 h after episodic HX/RO. *P<0.05 versus untreated HX/RO; #P<0.05 versus decreased dose of respective inhibitor.
PARP-1 Cleavage and Apoptosis-Inducing Factor (AIF) Translocation
Four hours after episodic HX/RO, total nuclear PARP-1 protein (116 kDa) expression remained unchanged (quantification of immunoblot results not shown) but a 3-fold increase in its 85-kDa cleavage product was measured (Figure 5). The PARP-1 inhibitors 3-AB and 4-AN attenuated this HX/RO-induced increase in PARP-1 cleavage, the magnitude of which was dose-dependent (Figure 5).
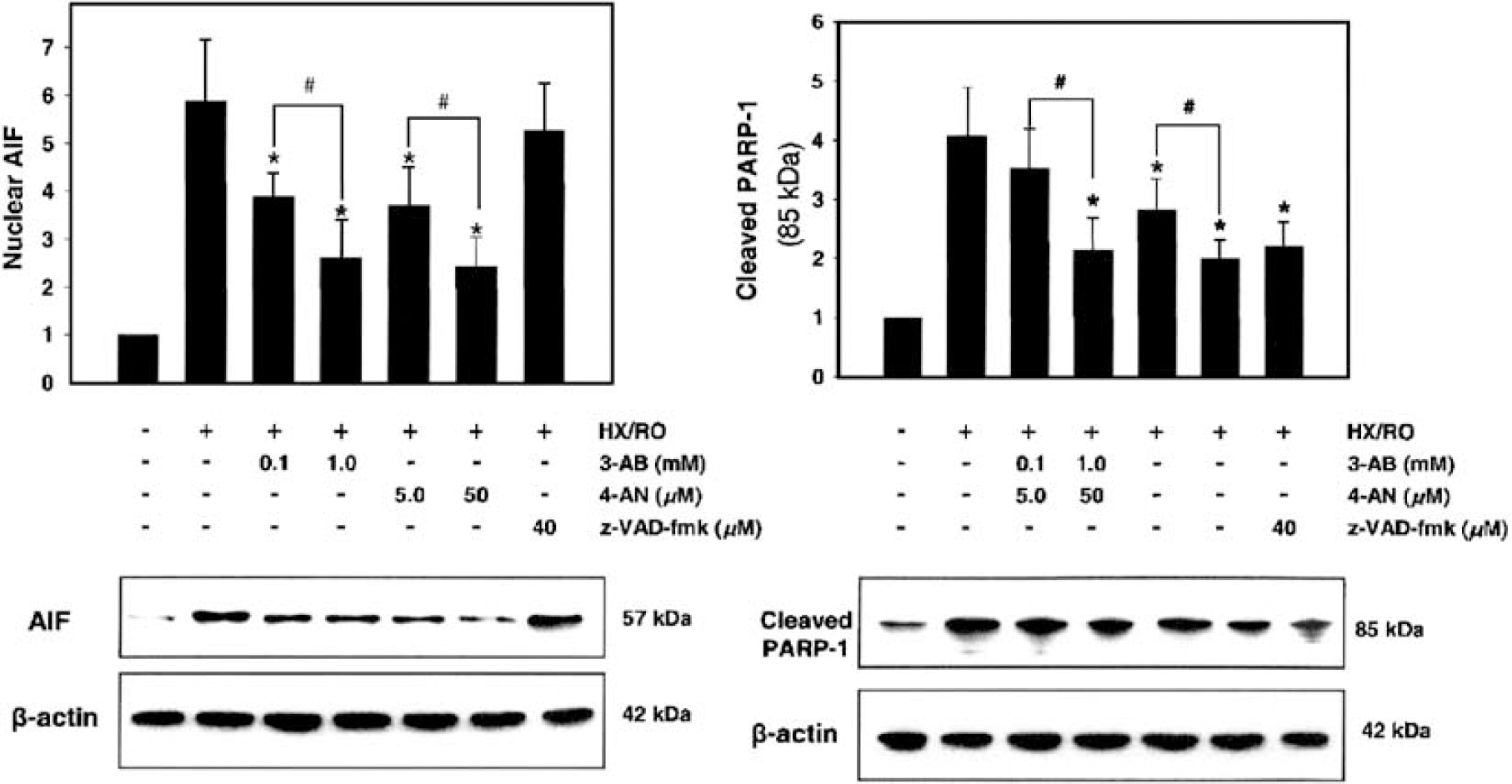
Immunoblotting for nuclear AIF and the nuclear, 85-kDa PARP-1 cleavage product (quantified in histograms) 4 h after exposure of human microvascular cerebral endothelial cells to episodic HX/RO. The dose-dependent effects of PARP-1 inhibition with 3-AB or 4-AN on expression of nuclear AIF translocation and PARP-1 cleavage is shown, along with the selective effect of caspase inhibition on PARP-1 cleavage but not AIF translocation. Representative blots are shown below the histograms. Expression changes measured in nuclear subfractions are normalized for each protein to normoxic controls; β-actin was used to document equal loading. *P<0.05 versus untreated HX/RO; #P<0.05 versus decreased dose of respective inhibitor.
Expression of the 57-kDa processed form of the full-length AIF, normally found in the mitochondrial inner and outer membrane interspace, was low in nuclear fractions from normoxic cultures. However, 4 h after episodic HX/RO, nuclear AIF expression was elevated 4-fold (Figure 5). In parallel with their effect on PARP cleavage, both PARP-1 inhibitors dose-dependently decreased nuclear translocation of AIF (Figure 5).
However, while caspase inhibition with z-VAD-fmk prevented PARP-1 cleavage, it was without effect on AIF translocation (Figure 5).
Discussion
Endothelial dysfunction and injury resulting from episodes of cerebral hypoxia or ischemia affects vascular homeostasis and indirectly influences glial and neuronal injury. The present study used cell cultures of HBMEC subjected to intermittent periods of simulated HX/RO and documented that PARP-1 activation, nuclear AIF translocation, and PARP-1 cleavage occurs before the loss of cell viability; ultimately, the apoptotic demise of the cells was reflected by extensive TUNEL-positive staining. Early inhibition of PARP-1 activation dose-dependently abrogated the extent of AIF translocation, PARP-1 cleavage, TUNEL staining, and cell death, whereas cytoprotection was lost if inhibition of PARP-1 activation was delayed. Inhibition of caspase activation blocked PARP-1 cleavage, but was without effect on AIF translocation and ultimately was only marginally cytoprotective. These results indicate that, after intermittent HX/RO, HBMECs are vulnerable to a caspase-independent, AIF-mediated apoptosis that is triggered by PARP-1 activation.
Apoptosis involving the nuclear translocation of the death execution protein AIF occurs in neurons subjected to ischemia (Cao et al, 2003; Plesnila et al, 2004), but to our knowledge, this is the first report to examine the involvement of this cell death pathway in hypoxic-ischemic cerebral endothelium. Accumulating evidence supports the hypothesis that AIF-mediated cell death results from a stimulus-induced loss of mitochondrial integrity, after which AIF is released from the mitochondria and translocates to the nucleus to activate endonucleases that induce chromatin condensation and large-scale DNA fragmentation (Cao et al, 2003; Susin et al, 1999). This translocation can occur independent of alterations in overall AIF gene expression at either the message or protein level (Cao et al, 2003). We observed AIF nuclear translocation at 4 h of recovery after episodic HX/RO, before the presence of significant endothelial cell death and TUNEL-positive staining. This temporal sequence of events is consistent with the notion that AIF translocation to the nucleus is an early step in the progression to apoptotic HBMEC death after HX/RO.
The mechanisms responsible for AIF release from mitochondria in response to different death-promoting stimuli have yet to be clarified, but appear to vary depending on the cell type, the apoptosis trigger, and the metabolic state of the cell (Szabó et al, 1997; Szabó and Dawson, 1998; Nagayama et al, 2000). Studies in PARP-1 knockout mice and PARP-1-null neuronal cultures indicate that PARP-1 activation may signal this translocation step secondary to the poly(ADP-ribosyl)ation of unidentified nuclear proteins that translocate from the nucleus to the mitochondria (Hong et al, 2004; Yu et al, 2003). Studies in cultured astrocytes and other cells indicate that it is also possible that depletion of ATP and/or NAD+ in mitochondria, secondary to PARP-1 activation, signals mitochondrial AIF release (Alano et al, 2004). In any event, it is now well established that pharmacologic or genetic interruption of PARP-1 activation is cytoprotective in a wide variety of in vivo and in vitro models in response to an equally wide variety of injurious stimuli. Prevailing evidence suggests that the initial extent of DNA injury dictates the extent of PARP-1 activation, which in turn determines whether the cell can repair itself, or will die by apoptosis or necrosis. Moderate activation of PARP-1 can promote apoptotic cell death by signaling AIF translocation to the nucleus (Yu et al, 2002) as well as facilitating the accessibility of nuclear chromatin to endonucleases (Duriez and Shah, 1997). But overactivation of PARP-1 in response to severe DNA damage will deplete the cell of NAD+ and ATP, leading to energy failure and necrotic cell death (Ha and Snyder, 1999).
In our HBMEC model of HX/RO that results in a 30% to 40% loss of cell viability, treatment with 3-AB and 4-AN, two mechanistically distinct PARP-1 inhibitors (Banasik et al, 1992; Schlicker et al, 1999; Thiemermann et al, 1997), during HX/RO dose-dependently blocked AIF translocation and reduced apoptotic cell death, supporting the concept that early PARP-1 activation is an upstream trigger of mitochondrial AIF release in cerebral endothelium. While a number of investigations in cultured endothelium (Andreoli, 1989; Thies and Autor, 1991) and other cell types (Shiokawa et al, 1997; Simbulan-Rosenthal et al, 1999; Yoon et al, 1996) reported cytoprotective effects of PARP-1 inhibition or PARP-1 gene deletion against proapoptotic stimuli, the possibility that this protective effect resulted from the subsequent blockade of nuclear AIF translocation was not realized at the time these studies were published. Similarly, the earlier successes of PARP-1 inhibition or deletion strategies in reducing ischemic brain injury in vivo (Ding et al, 2001; Eliasson et al, 1997; Endres et al, 1997; Takahashi et al, 1999) may have their mechanistic basis in the secondary reduction in nuclear AIF translocation (Cao et al, 2003; Plesnila et al, 2004).
In addition to its effects on DNA fragmentation, AIF also triggers the release of mitochondrial cytochrome c and, ultimately, caspase activation, which may amplify the apoptosis machinery in a feed-forward cascade (Susin et al, 1999; Yu et al, 2003). In some models, AIF translocation appears to be caspase-dependent (Arnoult et al, 2002; Guo et al, 2004); however, our finding that caspase inhibition, while blocking PARP-1 cleavage, did not affect AIF translocation and had only a modest cytoprotective effect is consistent with related findings in other cell injury models that caspase inhibitors do not abrogate AIF-dependent apoptotic death (Cao et al, 2003; Cregan et al, 2002; Susin et al, 1999; Yu et al, 2002; Zhang et al, 2002). Rather, ours and the latter studies support a mechanism whereby AIF and caspases may act in a parallel, synergistic manner, with the cleavage of PARP-1 by activated caspases preventing overactivation of the enzyme, so that the cell can maintain levels of ATP and NAD+ sufficient for apoptosis. Our finding that the low dose of 3-AB reduced the extent of PARP-1 activation enough to reduce AIF translocation and endothelial cell death, but did not significantly affect PARP-1 cleavage suggests that the mechanisms for PARP-1-mediated AIF translocation and caspase activation may exhibit different thresholds, or may be differentially regulated. The PARP-1 cleavage we observed ‘concomitantly’ with AIF translocation at 4 h of recovery from HX/RO may be the net result of several overlapping cycles of sequential PARP-1 activation, AIF translocation, and caspase activation given the protracted episodic HX/RO stimulus we studied. More directed temporal profiling after a singular proapoptotic stimulus indicates that AIF translocation and DNA fragmentation precedes the release of cytochrome c and caspase activation that follows (Soldani et al, 2001; Susin et al, 1999; Yu et al, 2002).
Our documentation of an abrogation of AIF translocation and apoptotic cell death by early administration of PARP-1 inhibitors, and the lack of AIF translocation and loss of robust cytoprotection by delayed treatment with the PARP-1 inhibitors, implicates PARP-1 activation as a key upstream event responsible for initializing an apoptotic death cascade in cerebral endothelial cells in response to episodic HX/RO. Apoptotic cell death of cultured cerebral endothelium in response to hypoxia or simulated ischemia is not a novel finding (Beetsch et al, 1998; Bresgen et al, 2003; Fischer et al, 2002), but to our knowledge, this is the first report mechanistically linking the demise of these cells to PARP-1 activation. Although PARP-1 activation may also be triggered by phospholipase C (Homburg et al, 2000) and other signals (Koedel et al, 2002), if DNA strand breaks are the primary activator of the enzyme (de Murcia and de Murcia, 1994), then HBMECs must be vulnerable to significant chromatin damage by HX/RO stress. It is likely that the oxidative stress associated with reoxygenation (simulated reperfusion), particularly the repetitive reoxygenation used in our protocol, contributes importantly to DNA damage. We showed previously in cultured porcine cerebral endothelium that it is not the period of anoxia per se but the free radical production during reoxygenation, primarily superoxide from the xanthine oxidase pathway, that is causal to cerebral endothelial cell death (Beetsch et al, 1998). And the evidence for oxygen radical-induced activation of PARP-1, particularly by peroxynitrite, is strong (Szabó, 1996).
The validity of our cell culture findings is dependent on the specificity of 3-AB and 4-AN as PARP-1 activation inhibitors. In fact, there are a number of studies supporting this contention. While relatively low in potency, the long history of using the nicotinamide analog 3-AB as a PARP-1 inhibitor (Ding et al, 2001; Endres et al, 1997; Heller et al, 1995; Koedel et al, 2002; Nagayama et al, 2000; Shiokawa et al, 1997; Szabó et al, 1997; Takahashi et al, 1999; Thiemermann et al, 1997; Walisser and Thies, 1999; Yoon et al, 1996) derives support prospectively from studies in PARP-1-null cells and PARP-1 knockout mice, which documented no effects of 3-AB on stimulus-induced formation of reactive oxygen species, including peroxynitrite, even at levels up to 5 mmol/L (Heller et al, 1995; Szabó and Dawson, 1998). There is also immunohistochemical evidence that 3-AB attenuated ischemia-induced increases in cerebral poly(ADP-ribosyl)ation and attenuated ischemia-induced reductions in cerebral NAD+ levels (Endres et al, 1997). The use of 4-AN as a more specific and potent PARP-1 inhibitor is also well supported (Banasik et al, 1992; Schlicker et al, 1999; Thiemermann et al, 1997). Moreover, these structurally dissimilar compounds resulted in nearly identical effects on molecular apoptotic events downstream of PARP-1 activation, consistent with a common and specific mechanism of action on PARP-1.
In conclusion, ischemia-reperfusion, and particularly intermittent ischemia-reperfusion, renders cerebral endothelium vulnerable to oxidative DNA injury, which leads successively to PARP-1 activation, translocation of AIF from mitochondria to nucleus, a caspase-mediated PARP-1 cleavage, and the apoptotic demise of these cells. Our demonstration that pharmacological inhibition of PARP-1 activation abrogated each of these deleterious molecular events and protected human cerebral endothelial cells indicates that PARP-1 inhibitors may be useful clinically for neuroprotection in the setting of cerebral ischemia or intermittent hypoxia, secondary to the direct and indirect protective effects of preserving microvascular endothelial cell viability.
Footnotes
Acknowledgements
The authors thank Aarti Shah for helpful technical assistance. They are grateful to Charles Cheng and David C Hess for the donation of the human brain microvascular endothelial cell line.