
Abstract
Nitric oxide from neuronal cells plays detrimental roles in glutamate neurotoxicity and in focal brain ischemia. Nitric oxide directly damages DNA, and breaks in the DNA strands activate poly(ADP-ribose) polymerase (PARP), which brings poly(ADP-ribosyl)ation of the nuclear proteins. The excessive activation of PARP is thought to cause depletion of ATP and the energy failure resulting in cell death. To clarify the involvement of poly(ADP-ribosyl)ation in ischemic insult, we examined poly(ADP ribosyl)ation by immunohistochemical methods and the protective effect of 3-aminobenzamide, which is a PARP inhibitor, on focal brain ischemia using an intraluminal permanent middle cerebral artery occlusion model in rats. Poly(ADP ribosyl)ation was widely and markedly detected 2 hours after the ischemic insult in the cerebral cortex and striatum in which infarction developed 24 hours later. The enhanced immunoreactivity of poly(ADP-ribose) gradually decreased, and 16 hours later, no immunoreactivity was detected. Intraventricular administration of 3-aminobenzamide (1 to 30 mg/kg) 30 minutes before the ischemic insult decreased infarction volume in a dose-dependent manner along with the immunohistochemical reduction of poly(ADP-ribosyl)ation. Pretreatment with 7-nitroindazole (25 mg/kg, intraperitoneally), a selective neuronal nitric oxide synthetase inhibitor, partially reduced poly(ADP-ribosyl)ation. These data suggest the involvement of poly(ADP-ribosyl)ation in the development of cerebral infarction.
Keywords
In cultured neurons, nitric oxide(NO) mediates glutamate neurotoxicity, but in ischemic circumstances, NO plays both beneficial and detrimental roles, and the effect of nitric oxide synthetase (NOS) inhibitors in focal brain ischemia has been controversial. This is mainly because NO is produced from various NOS isoforms (Iadecola et al., 1994). It has been shown that a specific neuronal NOS (nNOS) inhibitor, 7-nitroindazole(7-NI), is neuroprotective in the rat focal brain ischemia model (Yoshida et al., 1994), and that nNOS knockout mice are resistant to focal brain ischemia (Huang et al., 1994). These data suggest that NO produced from neurons is cytotoxic in focal ischemia. The free radical NO reacts with superoxide anion and produces peroxynitrite, which is one of the most powerful radicals. Peroxynitrite and NO directly damage DNA and enzymes involved in mitochondrial respiration (Wink et al., 1991). Studies indicate that one major pathway in which NO may exert toxicity is the activation of poly(ADP-ribose) polymerase (PARP) through damaging DNA. In neuronal cell cultures, NO and glutamate are able to activate PARP through damaging DNA (Cosi et al., 1994; Zhang et al., 1994, 1995). Once activated, PARP catalyzes poly(ADP-ribosyl)ation, the transfer of ADP ribose units from NAD to nuclear proteins. The excessive activation of PARP causes depletion of NAD and ATP, and leads to an energy failure. Furthermore, the inhibition of PARP reduces the neurotoxicity of NO and glutamate (Cosi et al., 1994; Zhang et al., 1994, 1995). These data indicate that the energy failure from excessive activation of PARP takes part in NO and glutamate neurotoxicity.
The current study clarifies-by using an intraluminal permanent middle cerebral artery (MCA) occlusion model in rats-the immunohistochemical involvement of poly(ADP ribosyl)ation and the neuroprotective effect of 3-aminobenzamide (3-ABA), which is a PARP inhibitor, as they affect the development of cerebral infarction.
MATERIALS AND METHODS
Induction of focal brain ischemia
Male Sprague-Dawley rats (Shimizu Laboratory Supplies Co., Ltd., Kyoto, Japan) weighing 280 to 320 g were used. After overnight fasting, rats were anesthetized with 4.0% halothane and maintained with 1.0% halothane in a gas mixture of 30% oxygen/70% nitrous gas using a face mask. The rectal temperature was maintained at 37.5 ± 0.5°C during surgery with a heating lamp and a heating pad connected to a rectal thermistor. The right femoral artery was cannulated to record blood pressure and to obtain arterial blood samples. Focal brain ischemia was induced under spontaneous respiration according to a method of intraluminal MCA occlusion with slight modification (Longa et al., 1989). Briefly, the right common carotid artery, external carotid artery, and internal carotid artery were isolated through a midline cervical skin incision under a microscope. Approximately 18 mm of 4-0 nylon monofilament, which was blunted by coating with silicone, was advanced from the lumen of the external carotid artery into the internal carotid artery to block the origin of the right MCA. Rats were allowed to survive for the indicated period before killing. Control animals were operated without MCA occlusion.
Pretreatment with 3-aminobenzamide and 7-nitroindazole
To examine the effect of 3-ABA (nacalai tesque, Kyoto, Japan), a PARP inhibitor, on brain ischemia, 3-ABA (0, 1, 3, 10, 30 mg/kg) was administered into the right lateral ventricle stereotactically 30 minutes before the ischemic insult under general anesthesia (described earlier). In a preliminary experiment, a dose of 1 to 100 mg/kg of 3-ABA was administered intraperitoneally 30 minutes before the ischemic insult with no reduction of infarction volume. Therefore, we used intraventricular injection of 3-ABA. The heads of rats were secured in a stereotactic frame, and a 26-gauge needle was inserted into the right lateral ventricle. Coordinates were 1.5 mm lateral, 0.8 mm posterior, and 3.3 mm ventral from the dural surface using bregma as a landmark. Thirty microliters of each drug solution were injected over 5 minutes. The 3-ABA was dissolved in distilled water, and pH was adjusted to 7.0. To examine the involvement of NO from nNOS in the induction of poly(ADP-ribosyl)ation, 7-NI(25 mg/kg) (Lancaster Co., Edinburg, U.K.) was administered intraperitoneally 30 minutes before the ischemic insult. The 7-NI was suspended in peanut oil (nakalai tesque, Kyoto, Japan).
Tissue preparation and immunohistochemistry
To clarify the temporal and spatial distribution of poly(ADP-ribosyl)ation after focal brain ischemia, rats were transcardially perfused with 4% paraformaldehyde in phosphate-buffered saline (pH 7.4) at 1, 2, 4, 8, 16, and 24 hours after the induction of ischemia (n= 3 at each time point). Control animals (n = 3) were killed 8 hours after operation. To examine the effect of 3-ABA (0, 1, 3, 10, 30 mg/kg) and 7-NI (25 mg/kg) on poly(ADP-ribosyl)ation after ischemia, rats (n = 3 for each dose) were perfused as described earlier at 2 hours after ischemia. The brains were rapidly removed and cryoprotected in 25% sucrose in phosphate-buffered saline overnight at 4°C. Frozen coronal sections(50-μm thickness) were prepared. After quenching endogenous peroxidase in 2% H2O2 in 60% methanol and blocking with 5% goat serum, sections were incubated overnight at 4°C with polyclonal antibody against poly(ADP-ribose) (Ikai et al., 1980). This antibody was a polyclonal antibody raised in rabbits against purified poly(ADP-ribose) (average chain length, 24); it was most reactive with polymers having the chain length of about 25 ADP-ribose units and weakly reactive with short oligomers. The specificity was confirmed by the disappearance of the immunostaining by preabsorption of the antiserum with purified poly(ADP-ribose) or by pretreatment of tissue sections with poly(ADP-ribose)-degrading enzymes. The sections were washed with phosphate-buffered saline, and then incubated with biotinylated anti-rabbit IgG antibody at 1:200 dilution for 2 hours and with an avidinbiotin complex (ABC kit from Vector) for 60 minutes. Peroxidase was demonstrated with a DAB substrate kit (Funakoshi, Tokyo, Japan). Negative control sections received identical treatment except for the primary antibody. We also used the commercially available polyclonal anti-poly(ADP-ribose) antibody prepared from guinea pig serum (Trevigen, Gaithersburg, MD, U.S.A.) to check interantibody differences in immunoreactivity. For evaluation of morphologic change, contiguous sections were stained with cresyl violet.
Measurement of infarction volume
To examine the effect of intraventricular administration of 3-ABA 30 minutes before the ischemia on the development of cerebral infarction, rats were killed 24 hours later after ischemic insult for 2,3,5-triphenyltetrazolium chloride (nacalai tesque) staining (Bederson et al., 1986) to measure infarction volume. Twenty-seven rats were used (n = 7, 4, 6, 6, 4 for vehicle and doses of 1, 3, 10, and 30 mg/kg, respectively). Rats were decapitated, the brains were removed immediately, and seven coronal slices 2, 4, 6, 8, 10, 12, and 14 mm distal from the frontal pole were dissected using a brain slicer. Infarction areas were calculated for each coronal slice by NIH Image (ver. 1.56), and infarction volumes were determined by numeric integration of infarction areas. Data were presented as mean ± SD and statistically analyzed using analysis of variance followed by post hoc Bonferroni test using Stat View II (Abacus Concepta, Berkeley, CA, U.S.A.), and when P is less than 0.05, differences were considered significant.
RESULTS
Immunohistochemistry of poly(ADP-ribose)
Two hours after the intraluminal right MCA occlusion, neuronal damage was detected only in the medial part of right striatum in cresyl violet staining. Neuronal damages gradually expanded thereafter, and 24 hours later a damaged area expanded to most of the right striatum and right frontoparietal cortex (Fig. 1).
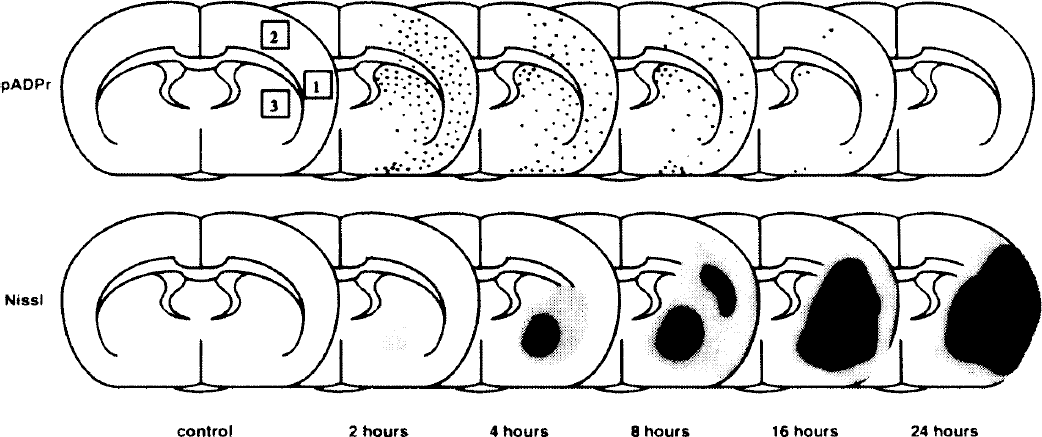
Schematic illustration of positive immunoreactivity for poly(ADP-ribosyl)ation and neuronal degeneration. Top: Temporal profile and distribution of immunoreactive cells for poly(ADP-ribosyl)ation. One dot indicates approximately five strongly immunopositive cells. The photographs were taken as shown in Fig. 2 at the site of squares 1 through 3. Square 1 is the center of ischemic core in the cortex. Square 2 is periinfarct area(ischemic penumbra). Square 3 is the lateral part of striatum (ischemic core). Areas 1, 2, and 3 are on the same section, 7.2 mm anterior to interaural line. Bottom: Temporal profile and distribution of neuronal damage. Neuronal degeneration was evaluated by Nissl stain. Bright shadow indicates about 30% of neurodegeneration. Dark shadow indicates more than 70% of neurodegeneration.
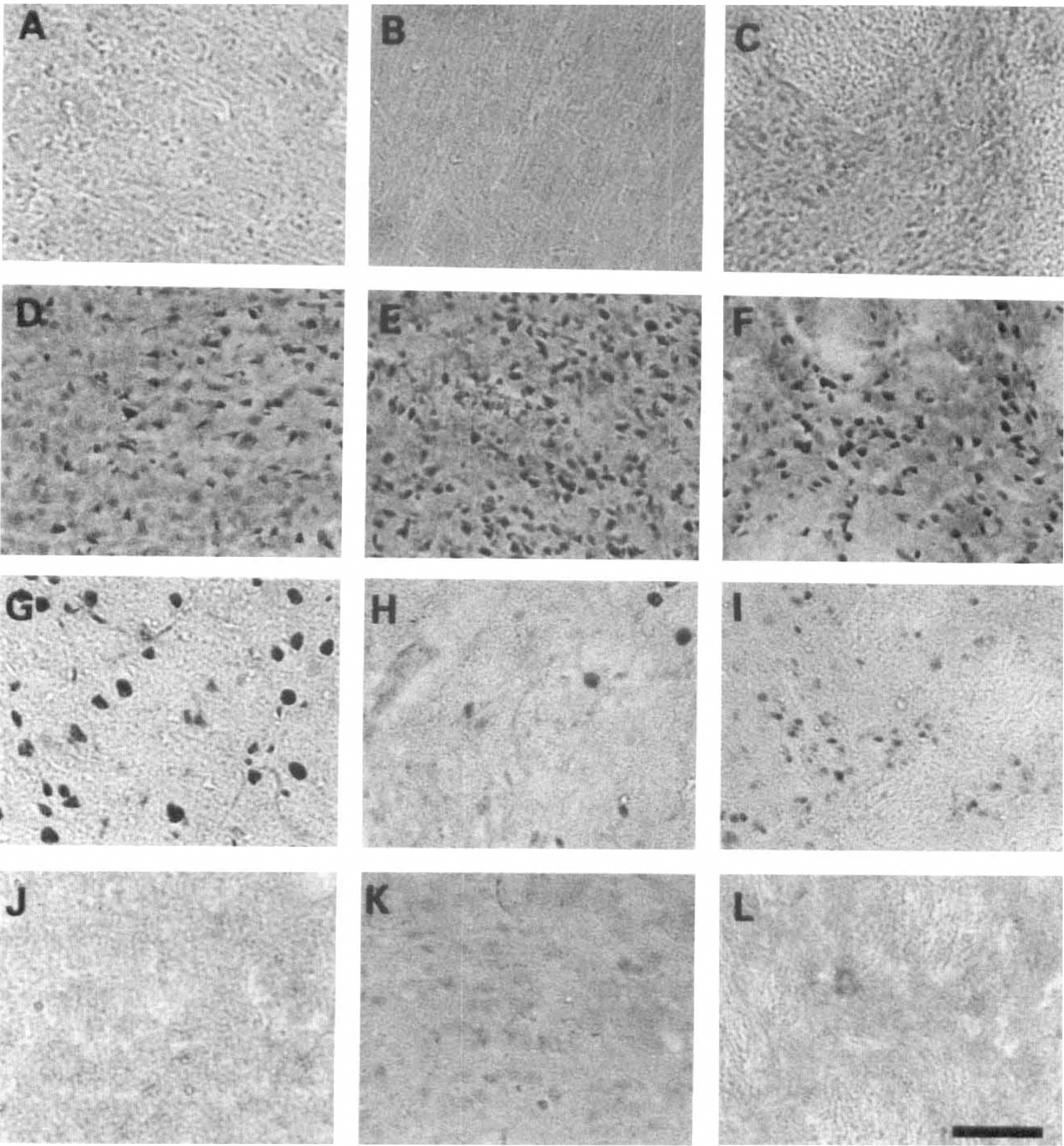
Representative microphotographs of poly(ADP-ribose) immunohistochemical study at 0
We used two polyclonal antibodies, and temporal and spatial distribution of poly(ADP-ribosyl)ation was essentially the same. The main difference between the immunoreactivities in two antibodies was the intracellular distribution. The antibody from by Ikai and others produced strong immunoreactivity, mainly in the nuclei with faint staining in the cytoplasm (Figs. 2 and 3), but the antibody from Trevigen produced diffuse immunoreactivity, both in the nuclei and cytoplasm (see Sugino et al, 1997). In the current study, we demonstrated the results obtained using the former, since the antibody from Trevigen produced higher background than the polyclonal antibody by Ikai and colleagues.
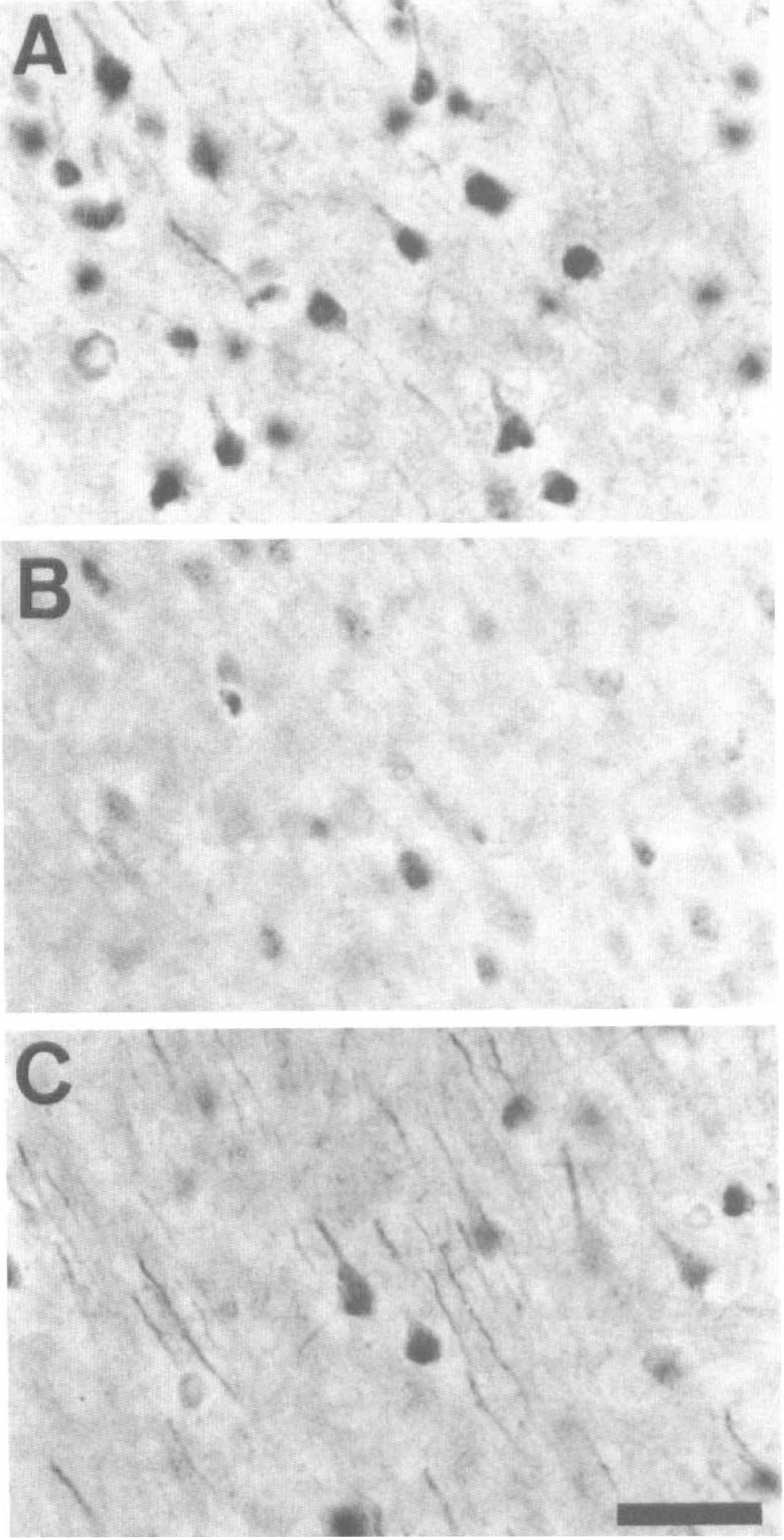
Representative photomicrographs of poly(ADP-ribose) immunohistochemical study at the area of square 1 (indicated in Fig. 1) 2 hours after ischemic insult in animals that received intraventricular injection of vehicle
In the control group and 0 hours after ischemic insult, no poly(ADP-ribosyl)ation was detected in the striatum or frontoparietal cortex (Figs. 1 and 2A through C). Faint poly(ADP-ribose) immunoreactivity was detected only in neurons of the piriform cortex and cingulate gyrus. One hour after ischemic insult, poly(ADP-ribosyl)ation was weakly detected in the medial and lateral striatum and in the frontoparietal cortex (data not shown). Two hours after ischemic insult, poly(ADP-ribosyl)ation peaked in the lateral striatum and throughout the frontoparietal cortex (Fig. 2D through F). We confirmed that the positive cells were mostly neurons by using morphologic study and a double staining method (data not shown). Among intracellular organelles, nuclei were stained prominently with occasional faint staining in the cytoplasm (Fig. 3A). In the medial part of striatum, in which neurodegeneration was severe, the staining was less dense. Ischemic penumbra in the cortex, which is indicated as square 2 in Fig. 1, showed moderate immunoreactivity. The piriform cortex, in which neuronal degeneration could not be found 24 hours later in this model, also showed moderate immunoreactivity. Four hours after ischemic insult, the number of positive cells gradually decreased. Eight hours after ischemic insult (Fig. 2G through I), the positive cells decreased rapidly in the striatum, and the positive cells were prominent only in the fifth layer of the cortex, although the piriform cortex was stained moderately. At this time, the staining showed a larger and rounder shape, probably because of degeneration of nuclei (Fig. 2G). Sixteen hours and 24 hours after intraluminal MCA occlusion, the immunoreactivity of poly(ADP-ribose) almost completely disappeared (Figs. 1 and 2J through L). Contralateral hemisphere showed no poly(ADP-ribose) immunoreactivity during the ischemic period, except faint staining in the piriform cortex and cingulate gyrus. Negative control without primary antibody did not show poly(ADP-ribose) immunoreactivity, either.
The effect of 3-aminobenzamide and 7-nitroindazole on poly(ADP-ribosyl)ation
Intraventricular administration of 3-ABA (0, 1, 3, 10, 30 mg/kg) 30 minutes before the ischemic insult reduced poly(ADP-ribose) immunoreactivity 2 hours after ischemia in a dose-dependent manner, and the maximum reduction of the poly(ADP-ribose) immunoreactivity was obtained with a dose of 10 mg/kg (Fig. 3A and B). The reduction of immunoreactivity was more prominent in the cortex than in the striatum (data not shown). Intraperitoneal administration of 25 mg/kg of 7-NI moderately reduced poly(ADP-ribose) immunoreactivity 2 hours after ischemia (Fig. 3C), but less effectively than 3-ABA.
The effect of 3-aminobenzamide on infarction volume
Twenty-four of 27 rats were survived for 24 hours after 3-ABA treatment. Mortality rates were 5/7, 3/4, 6/6, 6/6, and 4/4 for vehicle and doses of 1, 3, 10, and 30 mg/kg of 3-ABA, respectively. Intraventricular administration of 3-ABA (0, 1, 3, 10, and 30 mg/kg) 30 minutes before the ischemic insult significantly reduced infarction volume 24 hours after the ischemia in a dose-dependent manner, and the maximum effect was obtained at a dose of 10 mg/kg(Figs. 4 and 5). The reduction of infarction volume was more prominent in the cortex than in the striatum. Systemic blood pressure, arterial gas analysis (pH, Po2, Pco2), hematocrit, and blood glucose were not significantly different between groups 5 minutes before the ischemia (Table 1).
Physiologic parameters before intraluminal middle cerebral artery occlusion under halothane anesthesia
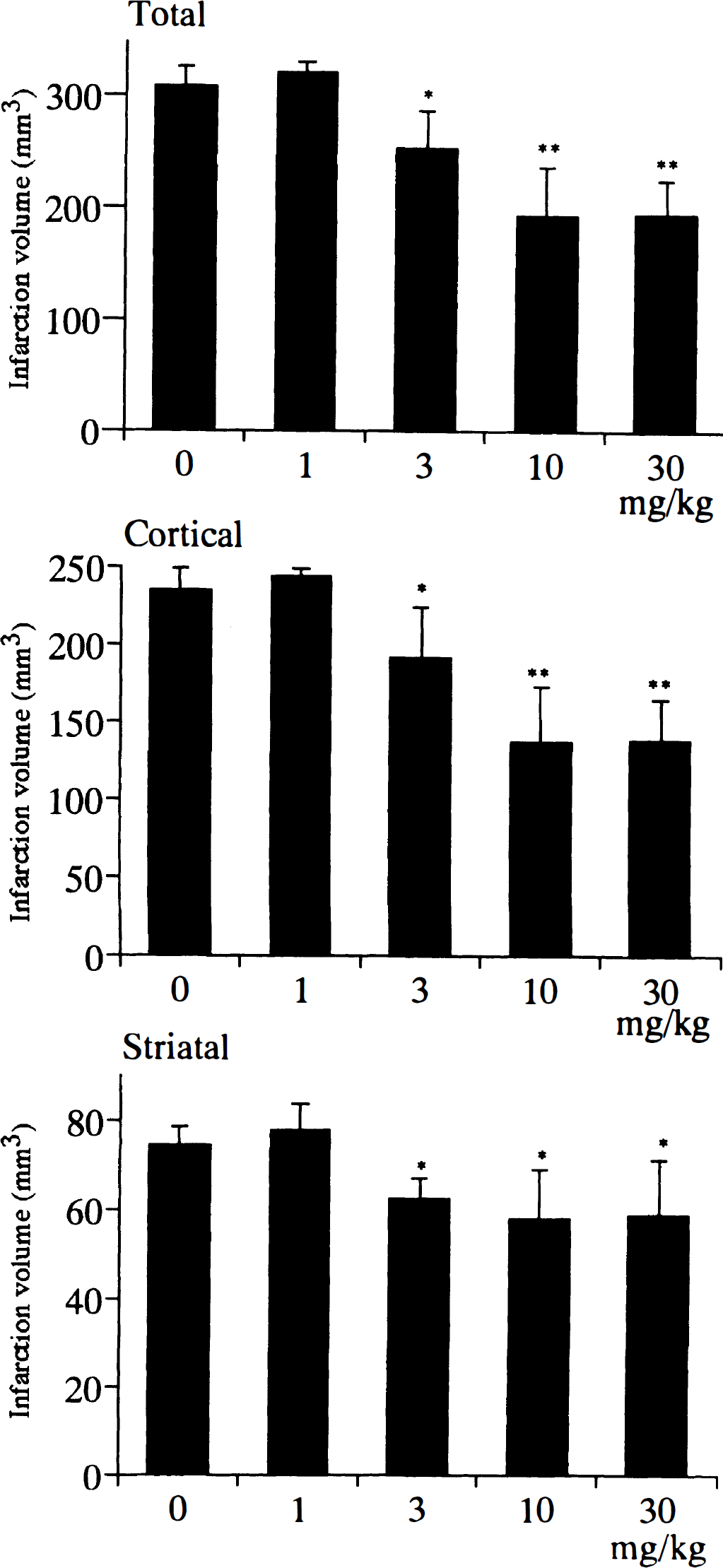
Total, cortical, and striatal infarction volumes 24 hours after intraluminal, permanent right middle cerebral artery (MCA) occlusion in animals that received 0 (vehicle), 1, 3, 10, and 30 mg/kg of intraventricular administration of 3-ABA 30 minutes before ischemic insult. The 3-ABA reduced total, cortical, and striatal infarction volume dose dependently. The maximum reduction is obtained at a dose of 10 mg/kg. Data are presented as means ± SD, and *P < 0.05, **P < 0.01 when compared with vehicle.
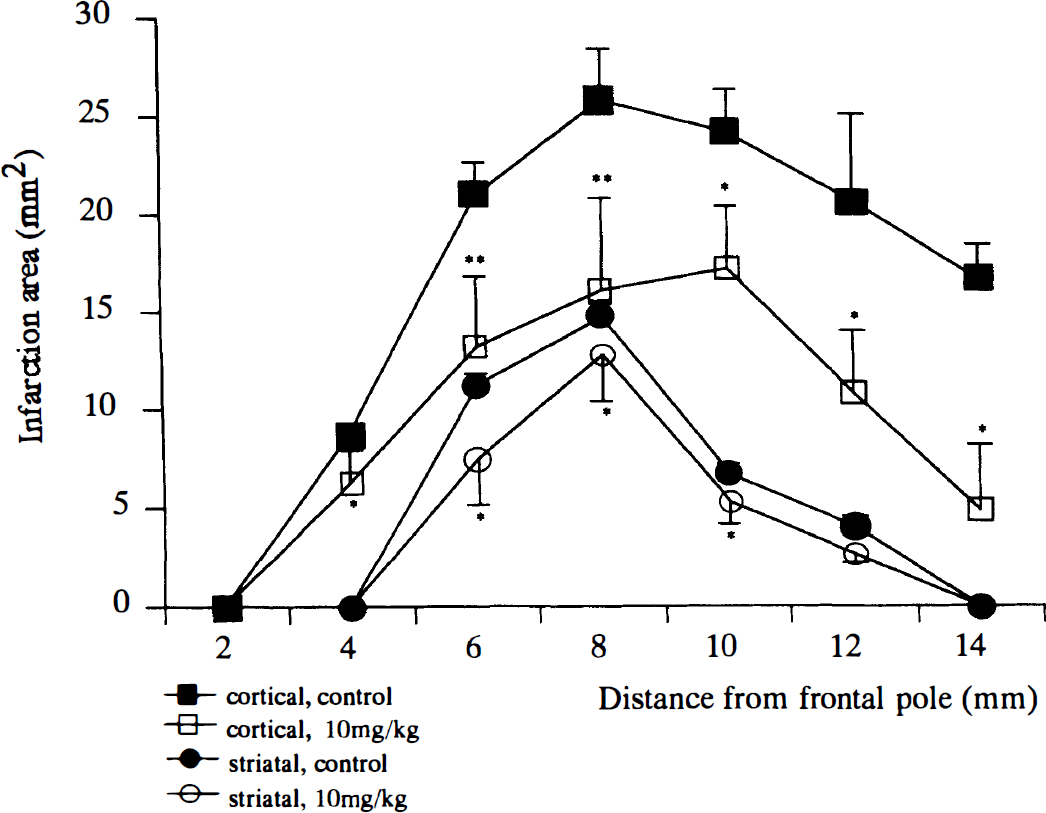
Cortical (square) and striatal (circle) infarction areas at each coronal section (2, 4, 6, 8, 10, 12, and 14 mm from frontal pole) in animals that received vehicle (control; closed) and 10 mg/kg of intraventricular administration of 3-ABA (open) 30 minutes before ischemic insult. Data are presented as means ± SD, and *P < 0.05, **P < 0.01 when compared with vehicle.
DISCUSSION
Poly(ADP-ribose) polymerase is activated by DNA breakage, resulting in the addition of up to 100 ADP-ribose groups to acceptors such as histone and PARP itself. After limited damages of DNA, poly(ADP-ribosyl)ation plays a critical role in DNA repair (de Marcia and de Marcia, 1994). However, when massive damage of DNA occurs, the associated extensive activation of PARP is thought to lead to depletion of NAD, which is the donor of the ADP-ribose group. Also, ATP is depleted in efforts to resynthesize NAD, resulting in cell death. Zhang and associates (1994) showed that NO activated PARP and that the inhibition of PARP rescued from NO- and N-methyl-D-aspartate-mediated neurotoxicity in a cortical culture. Cosi and colleagues (1994) showed by immunohistochemical study that glutamate induced poly(ADP-ribosyl)ation in cerebellar granule cells. They also showed that inhibitors of PARP reduced glutamate neurotoxicity. Also, in other cell lines, the inhibition of PARP has been shown to induce protective effects on cytotoxicity by several stimuli, including NO (Burkart et al., 1995; Heller et al., 1995; Kuo et al., 1996; Nosseri et al., 1992; Radons et al., 1994; Zingarelli et al., 1996). These results strongly suggest the involvement of poly(ADP-ribosyl)ation in the process of cell death. Another mechanism of cell death by poly(ADP-ribosyl)ation other than energy failure was proposed by Nosseri and coworkers (1992). They speculated that poly(ADP-ribosyl)ation may represent a quantity of DNA damages, and some intracellular signal transduction mechanisms for cell death may exist after poly(ADP-ribosyl)ation.
The current study showed that poly(ADP-ribosyl)ation was detected 1 hour and peaked 2 hours after focal brain ischemia model, where infarction developed several hours later. These findings indicate that PARP is activated in vivo in the early phase of the development of infarction. Because PARP recognizes the site of DNA single-strand breaks but not the site of double-strand breaks, poly(ADP-ribosyl)ation in the focal cerebral ischemia may reflect early damages of single-strand DNA in ischemic regions.
When we used the antibody by Ikai and colleagues, poly(ADP-ribose) immunoreactivity was mainly located in the nucleus of neurons, where DNA damages and repairs should occur. A few neurons in severe ischemic regions showed weak poly(ADP-ribose) immunoreactivity in the cytoplasm, along with strong poly(ADP-ribose) immunoreactivity in the nucleus. The commercially available polyclonal antibody (Trevigen) produced diffuse, positive staining both in the nucleus and cytoplasm. Further study is needed to clarify whether activation of PARP and poly(ADP-ribosyl)ation will occur in the cytoplasm, as well as the locus of poly(ADP-ribosyl)ation in the cytoplasm.
The current study also demonstrates that 3-ABA, which is a PARP inhibitor, reduced mortality rate and also reduced infarction volume dose dependently, along with the reduction of poly(ADP-ribosyl)ation in permanent MCA occlusion model in rats. The maximum reduction in infarction volume was obtained by 40% at a dose of 10 mg/kg. In this dosage, poly(ADP-ribosyl)ation did not disappear completely. During the preparation of this manuscript, Eliasson et al. (1997) showed that genetic disruption of PARP provides profound protection against glutamate-NO-mediated ischemic insults in vitro and major decreases in infarction volume after reversible MCA occlusion. They have demonstrated 80% reduction in infarction volume in PARP knockout mice. The differences in the neuroprotective effect of PARP inhibition in these two experiments may reflect the difference in the ischemia model(permanent versus transient) and the drug delivery and bioavailability.
It has been recently shown that NO and glutamate are able to activate PARP through damaging DNA in neuronal cell culture (Cosi et al., 1994; Zhang et al., 1994, 1995). We have shown in the current study that focal brain ischemia induced poly(ADP-ribosyl)ation, and that 7-NI, a specific nNOS inhibitor, reduced poly(ADP-ribosyl)ation in vivo. Because 7-NI could not block poly(ADP-ribosyl)ation completely, PARP may be activated through NO, as well as other stimuli, during focal brain ischemia.
The current study demonstrates the strong involvement of poly(ADP-ribosyl)ation in the development of cerebral infarction in focal ischemia model, and implies a new therapeutic modality for ischemic brain injury through the inhibition of poly(ADP-ribosyl)ation.