
Abstract
Poly(ADP-ribose)polymerase (PARP, EC 2.4.2.30), an abundant nuclear protein activated by DNA nicks, mediates cell death in vitro by nicotinamide adenine dinucleotide (NAD) depletion after exposure to nitric oxide. The authors examined whether genetic deletion of PARP (PARP null mice) or its pharmacologic inhibition by 3-aminobenzamide (3-AB) attenuates tissue injury after transient cerebral ischemia. Twenty-two hours after reperfusion following 2 hours of filamentous middle cerebral artery occlusion, ischemic injury was decreased in PARP−/− and PARP+/− mice compared with PARP+/+ litter mates, and also was attenuated in 129/SV wild-type mice after 3-AB treatment compared with controls. Infarct sparing was accompanied by functional recovery in PARP−/− and 3-AB–treated mice. Increased poly(ADP-ribose) immunostaining observed in ischemic cell nuclei 5 minutes after reperfusion was reduced by 3-AB treatment. Levels of NAD—the substrate of PARP—were reduced 2 hours after reperfusion and were 35% of contralateral levels at 24 hours. The decreases were attenuated in PARP−/− mice and in 3-AB–treated animals. Poly(ADP-ribose)polymerase cleavage by caspase-3 (CPP-32) has been proposed as an important step in apoptotic cell death. Markers of apoptosis, such as oligonucleosomal DNA damage, total DNA fragmentation, and the density of terminal deoxynucleotidyl transferase dUTP nick-end–labelled (TUNEL +) cells, however, did not differ in ischemic brain tissue of PARP−/− mice or in 3-AB–treated animals versus controls, although there were differences in the number of TUNEL-stained cells reflecting the decrease in infarct size. Thus, ischemic brain injury activates PARP and contributes to cell death most likely by NAD depletion and energy failure, although the authors have not excluded a role for PARP in apoptotic cell death at earlier or later stages in ischemic cell death. Inhibitors of PARP activation could provide a potential therapy in acute stroke.
Poly(ADP-ribose)polymerase (PARP, EC 2.4.2.30), an abundant nuclear protein present in all nucleated cells, is believed to be involved in DNA repair and participates in cell proliferation, differentiation, and transformation. Poly(ADP-ribose)polymerase catalyzes the covalent attachment of ADP-ribose subunits from its substrate, nicotinamide adenine dinucleotide (NAD), to numerous nuclear proteins, including PARP itself. Formation of poly(ADP-ribose) is a unique post-translational modification that can be induced by DNA strand breaks after exposure to nitric oxide (NO) or oxygen-free radicals (de Murcia et al., 1992, 1994; Lindahl et al., 1995; Shall, 1995).
The “PARP suicide hypothesis” states that excessive poly(ADP-ribose) formation due to PARP activation by DNA strand breaks and subsequent depletion of its substrate NAD might contribute to cell death through an energy failure mechanism. This may explain the neurotoxic properties of NO (Zhang et al., 1994) and why pancreatic islet cells die after exposure to reactive oxygen species (ROI) in vitro (Radons et al., 1994). In fact, inhibitors of PARP, such as 3-aminobenzamide (3-AB), can partially prevent NO and ROI cytotoxicity in neuronal and pancreatic cells. Nitric oxide and ROI formed during ischemia/reperfusion are known DNA-damaging agents and are potent mediators of cell death (Beckman et al., 1990).
Recently, PARP was recognized as a substrate of activated caspase-3 (CPP-32), a mammalian homologue of the C. elegans ced-3 death gene, during apoptosis (Tewari et al., 1995; Nicholson et al., 1995). The role of PARP in apoptosis, however, remains unclear and somewhat controversial. On the one hand, cleaved PARP appears to facilitate apoptosis, possibly by interrupting DNA binding and repair at an earlier step than internucleosomal DNA fragmentation (Patel et al., 1996). Cleavage decreases PARP enzymatic activity and could facilitate DNA laddering by upregulating Ca2+/Mg2+-dependent endonuclease as a consequence of reduced poly(ADP-ribosyl)ation (Yoshihara et al., 1975, Nicholson et al., 1995). However, inhibition of poly(ADP-ribosyl)ation of histone H1 by 3-AB abolishes internucleosomal DNA fragmentation in vitro (Yoon et al., 1996). Treatment with enzymatic inhibitors of PARP reportedly blocked or augmented apoptosis, depending on the paradigm (Kaufmann et al., 1994).
In the present study, we analyzed the function of PARP in cerebral ischemia in vivo by studying the effects of transient focal cerebral ischemia in PARP null mice and after 3-AB treatment, a widely used, nonselective PARP inhibitor. Poly(ADP-ribose)polymerase null mice are phenotypically normal and develop without obvious behavioral defects. Although DNA repair is efficient after ultraviolet or alkylating agents, primary fibroblasts proliferate more slowly. Moreover, the mice are susceptible to epidermal hyperplasia (Wang et al., 1995). Heller et al. (1995) demonstrated that 3-AB prevents ROI and NO cytotoxicity in PARP+/+ but not in PARP−/− islet cells in vitro, identifying PARP as the target of 3-AB-′s cytoprotective effects.
We show in the present study that PARP deletion or inhibition of PARP enzyme activity promotes resistance to ischemic brain injury after middle cerebral artery occlusion (MCAO). We further measured poly(ADP-ribose) formation and NAD levels in ischemic tissue as well as markers of apoptosis in ischemic tissue to decipher the mechanism by which activation of PARP mediates ischemic brain injury.
METHODS
Physiology
Regional cerebral blood flow was measured by laser Doppler flowmetry (PF2B, Perimed, Stockholm, Sweden), along with arterial blood pressure and heart rate, as described (Hara et al., 1996, 1997a,b). Arterial blood samples (50 μL) were analyzed for pH, arterial oxygen pressure (PaO2) and partial pressure of carbon dioxide (PaCO2) using a blood gas/pH analyzer (Corning 178, Ciba-Corning Diagnostics, Medford, MA, U.S.A.). Core temperature was maintained at approximately 37°C with a thermostat (FHC, Brunswick, ME, U.S.A.) and a heating lamp (Skytron, Daiichi Shomei, Tokyo, Japan) until 1 hour after reperfusion and during the treatment and monitoring period.
Ischemia Model
Adult male 129/SV mice (18–20 g, Taconic farm, German-town, NY, U.S.A.) were anesthetized for induction with 1.5% halothane and maintained in 1.0% halothane in 70% nitrous oxide (N2O) and 30% oxygen (O2) using a Fluotec 3 vaporizer (Colonial Medical, Amherst, NH, U.S.A.). PARP−/− mice and PARP+/− and +/+ litter mates (generated and genotyped by Dr. Wang at the Institute of Molecular Pathology, Vienna, as described in Wang et al., 1995) were anesthetized with 0.1 mL chloral hydrate (7% w/w in phosphate-buffered saline [PBS] given intraperitoneally). Ischemia was induced by occlusion with a 8-0 nylon monofilament coated with resin/hardener mixture (Xantopren and Elastomer Activator, Bayer Dental, Osaka, Japan) as described previously (Hara et al., 1996). For filament withdrawal after 2 hours of ischemia, the animals were reanesthetized briefly with halothane. Twenty-two hours after reperfusion, animals were killed and the brains were frozen immediately in 2-methylbutane on dry ice for cryostat sections or directly divided into five coronal 2-mm sections using a mouse brain matrix (RBM-200C, Activational Systems, Ann Arbor, MI, U.S.A.). Infarction volume was quantitated with an image analysis system (M4, St. Catharines, Ontario, Canada) on 2% 2,3,5 triphenyltetrazolium chloride-stained 2-mm slices (3-AB experiments) or hematoxylin & eosin-stained cryostat sections (20 μm; PARP−/−, +/−, +/+ experiments) and calculated by summing the volumes of each section determined directly (Huang et al., 1994) or indirectly using the following formula: contralateral hemisphere (mm3) – undamaged ipsilateral hemisphere (mm3) (Swanson et al., 1990). 3-Aminobenzamide (Sigma Chemical Co., St. Louis, MO, U.S.A.; IC50 on PARP activity = 33 μmol/L, 97% PARP inhibition at 5mmol/L) was dissolved in PBS (Poly Scientific, Bay Shore, NY, U.S.A.) at a concentration of 27.2 mg/mL (0.2 mol/L). Two μL of this solution (0.4 μmol) were injected intracerebroventricularly (bregma −0.9 mm lateral, −0.1 mm posterior, −3.1 mm deep) 10 minutes before ischemia using a Hamilton injection syringe (Fisher Scientific, Pittsburgh, PA, U.S.A.).
Neurological Deficits
Mice were tested for Neurological deficits and scored as described by Bederson et al. (1986) with the following minor modifications (Hara et al., 1996): 0 = no observable Neurological deficit (normal); 1 = failure to extent right forepaw (mild); 2 = circling to the contralateral side (moderate); and 3 = loss of walking or righting reflex (severe). Animals were rated by the operator and by a second rater blinded to the treatment protocol and group identity. Assessments were made after 30 minutes and after 24 hours.
Poly(ADP-ribose) Immunohistochemistry
Rabbit polyclonal antisera directed against purified poly-(ADP-ribose) was provided by Dr. Kunihiro Ueda (Kyoto University). The antibody is specific for poly(ADP-ribose), and immunoreactivity reflects both the number of ADP ribosylation sites and poly(ADP-ribose) chain length (Ikai et al., 1980). Mice were anesthetized with sodium pentobarbital (100 mg/kg intraperitoneally) and transcardially perfused with 4% paraformaldehyde. After immediate removal, brains were stored in the same buffer containing 20% sucrose, and free-floating sections (40 μm) were prepared on a freezing microtome. Immunohistochemistry was performed according to a three-stage avidin-biotin method with biotinylated goat antirabbit immunoglobulin G as secondary antibody and 3′-3′-diaminobenzidine as chromogene. The 3′-3′-diaminobenzidine reaction was intensified with 0.04% nickel chloride. The poly(ADP-ribose) polyclonal antibody was used at a concentration of 1:100. Negative controls were performed by omission of either the primary or secondary antiserum.
Nicotinamide Adenine Dinucleotide Determination
For in vitro measurements, animals were killed and the brains were removed immediately and cut into five coronal 2-mm slices. Tissue was obtained from within the ischemic zone in the third and fourth coronal section (4–8 mm) and snap-frozen. The samples were weighed, and the tissue was disrupted by freezing and thawing followed by homogenization in 4 × volumes PBS. The homogenate was centrifuged for 20 minutes at 15,000 × g at 4°C, and the supernatant was stored at −70°C until further use (Heller et al., 1995). Cellular NAD was determined by an enzymatic cycling method using alcohol dehydrogenase (Boehringer Mannheim, Mannheim, Germany) from Saccharomyces cerevisiae, as described previously (Nisslbaum and Green, 1969). Levels in ischemic tissue are expressed as percent of contralateral side. Contralateral values were stable at 0.00744 ± 0.00135 units/minute per milligram of tissue and did not differ between groups (PARP+/+, PARP−/−, 3-AB, PBS). For validation, NAD was added to homogenate prepared from ischemic and control tissue. Measurements increased linearly by addition of external NAD to the homogenate.
For qualitative in situ analysis, we adapted the method for tissue sections. The reaction mixture needed for enzymatic cycling was added to a 2% low-melt agarose solution at 45°C. Alcohol dehydrogenase was used at a final concentration of 50 units/mL, all other components were used at the same final concentration as in the in vitro assay. Glass slides were covered with 200 μL of the warm mixture in a homogeneous layer (approximately 1 mm thick) and kept horizontally on ice. Within 30 minutes, 12-μm cryostat brain sections were cut and placed onto the agarose-covered slide, immediately incubated at 37°C for 15 to 30 minutes in a humid chamber, and analyzed with the M4 image analysis system.
Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick End-Labelling (TUNEL)
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labelling was performed according to the method of Gavrieli et al. (1992) with minor modifications according to Wood et al. (1993). Terminal deoxynucleotidyl transferase (TdT) and biotinylated dUTP were obtained from Boehringer Mannheim (Mannheim, Germany). The biotinylated dUTP was visualized by the avidin-biotin method with 3'-3'-diaminobenzidine as chromogene. For negative controls, either TdT or biotinylated dUTP was omitted. For positive controls, the sections were treated with DNase I.
DNA Analysis
For DNA analysis, samples were prepared from the ischemic area and contralateral hemisphere at 24 hours. The brain was cut with a brain matrix, and the second (2–4 mm) slice was placed on a chilled glass plate. Tissue preparation was performed under a dissecting microscope, and only tissue from within the pale ischemic area was isolated for DNA preparation; corresponding tissue was taken from the contralateral side.
For quantitation of DNA damage, a terminal deoxynucleotidyl transferase-dependent isotopic end-labelling method was used (Tilly and Hsueh, 1993) with minor modifications as described previously (Hara et al., 1997b). Three micrograms of DNA were used in the labelling procedure together with 35 ng of a 100-base pair DNA fragment as the internal standard. The DNA was electrophoresed on a 2.0% agarose gel (agarose 3:1, Amresco, Solon, OH, U.S.A.), autoradiographed together with a radioactive standard, and analyzed with the M4 image analysis system. DNA less than 10 kilobase was used as an index of total DNA fragmentation. To measure oligonucleosomal damage more specifically, laddered DNA < 1000 base was measured by summing the areas of each peak (areas under the curve) minus baseline densitometry readings. The method of quantitation was validated using an artificial “smear-ladder” system in which smeared DNA was obtained by extracting DNA from a decapitated mouse brain incubated at 37°C for 48 hours (MacManus et al., 1996). In a total amount of 1 μg of DNA, 0.02, 0.04, 0.08, and 0.12 μg of a commercially available 200-bp ladder (Invitrogen, Carlsbad, CA, U.S.A.) was added to a constant amount of smeared DNA. In another experiment, increasing amounts of smeared DNA (0.3, 0.5, 0.7, 0.9 μg) were added to a constant amount of ladder (0.1 μg). The readings for “total DNA fragmentation” and “oligonucleosomal damage” were related linearly to the amount of total (smeared) and laddered DNA (error < 10%).
Statistical Analysis
Data are presented as mean ± standard deviation. Statistical comparisons were made by two-tailed Student's t-test (infarct size, DNA quantitation) or by two-way analysis of variance followed by Student's t test (NAD determination) or one-way analysis of variance followed by Tukey test (physiology) or Dunnett's test (regional cerebral blood flow). For Neurological deficits, the Mann–Whitney rank sum test was applied to compare two groups, and Kruskal–Wallis one-way analysis of variance on ranks followed by Dunnett's test was used for three groups. Analysis was made using the software EXCEL (Microsoft, Redmond, WA, U.S.A.) or SigmaStat (Jandel, Corporation, San Rafael, CA, U.S.A.). P < 0.05 was considered statistically significant.
RESULTS
Physiologic Data
No significant differences in mean arterial blood pressure, heart rate, RCBF, or arterial blood gases were detected between 3-AB– and vehicle-treated 129/SV mice (n = 5 per group; Table 1). Regional cerebral blood flow decreased to less than 20% of baseline immediately after MCAO and sustained during 2 hours of ischemia. After reperfusion, RCBF increased to 90% to 100% of baseline within 5 minutes.
rCBF, mean arterial blood pressure, and physiological parameters before, during, and after 2 hours of MCA occlusion and 3-aminobenzamide treatment
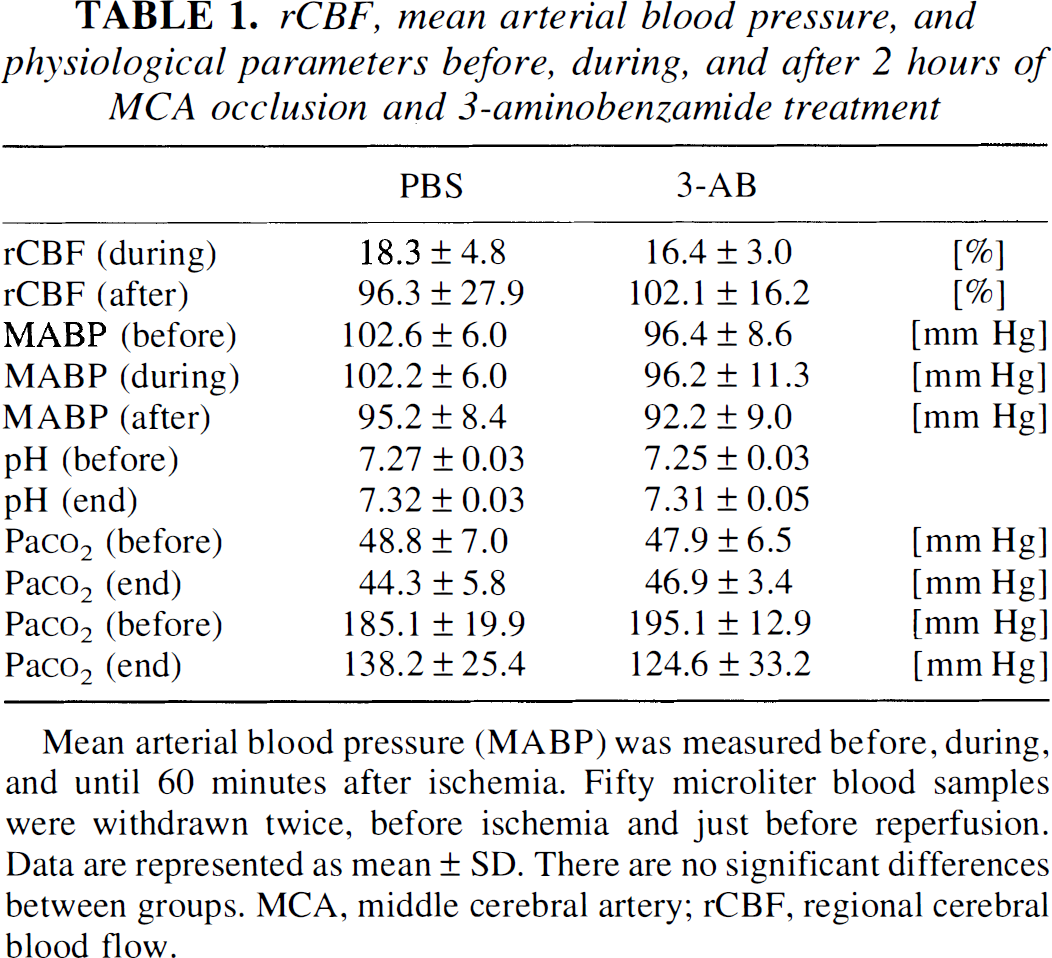
Mean arterial blood pressure (MABP) was measured before, during, and until 60 minutes after ischemia. Fifty microliter blood samples were withdrawn twice, before ischemia and just before reperfusion. Data are represented as mean ± SD. There are no significant differences between groups. MCA, middle cerebral artery; rCBF, regional cerebral blood flow.
Ischemic Infarction in Poly(ADP-Ribose)Polymerase Null Mice
Twenty-two hours after reperfusion, direct infarct volume was significantly smaller in PARP null mice (n = 8) and PARP heterozygotes (n = 8) compared with wild-type litter mates (n = 8; Fig. 1A). Infarction area was significantly smaller in all five coronal sections in PARP null mice and in section 5 in PARP heterozygote mice compared with wild-type litter mates (Fig. 1B). Statistical significance also was achieved with an indirect method to determine infarction (71.7 ± 20.4 mm3 [PARP−/−], 76.5 ± 20.1 mm3 [PARP+/−], 110.3 ± 7.4 mm3 [PARP+/+], P < 0.01). All animals exhibited a Neurological score of 2 or higher 30 minutes after the onset of ischemia and after animals recovered from anesthesia. At 24 hours, the deficit was significantly smaller in PARP null mice compared with wild-type mice (0.7 ± 0.9 vs. 1.6 ± 0.8, P < 0.01). The deficit was less but did not reach statistical significance in the PARP heterozygote mice (1.0 ± 0.8 vs. 1.6 ± 0.8).
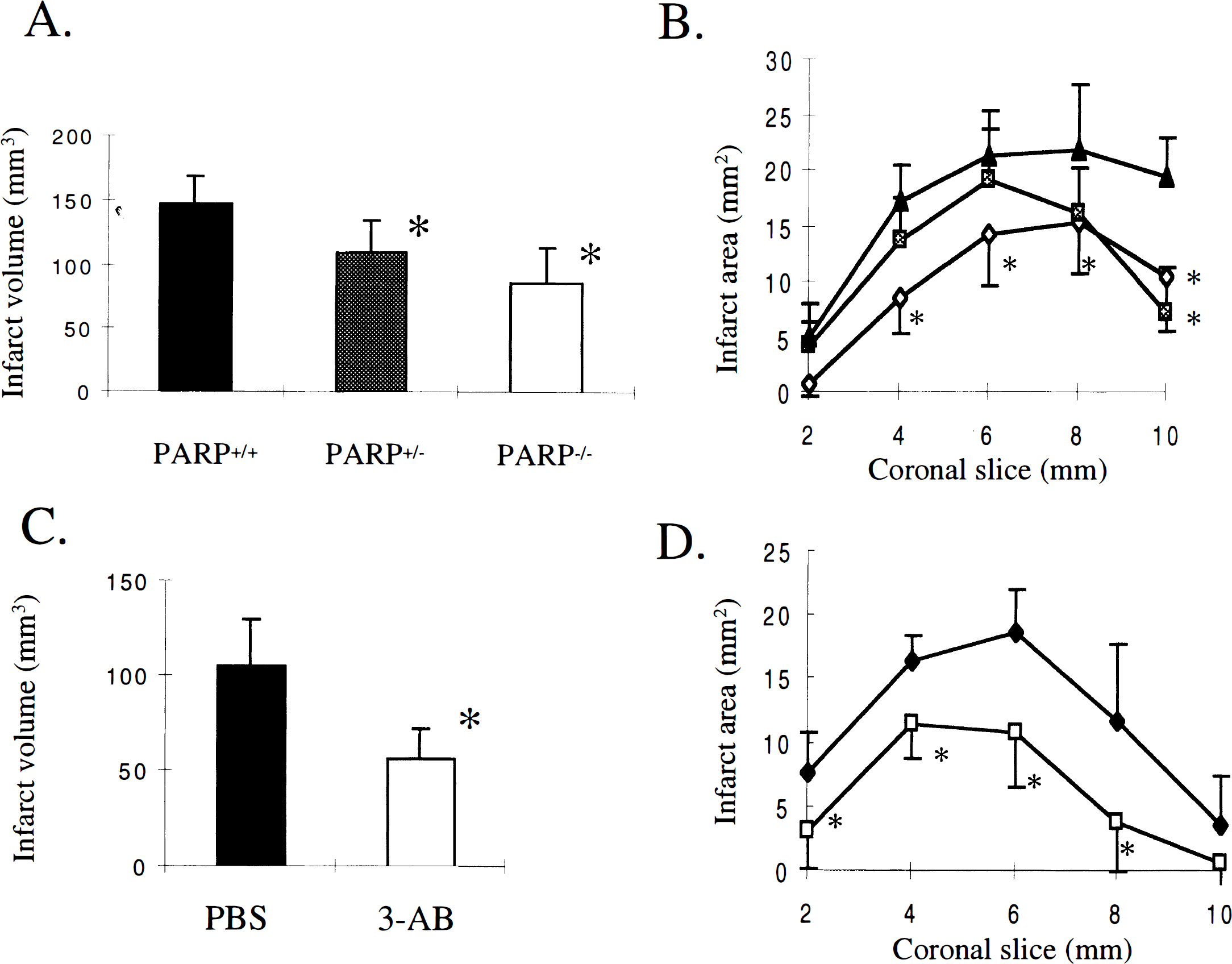
Infarct size was smaller in Poly(ADP-ribose)polymerase (PARP)−/− and 3-aminobenzamide (3-AB)-treated animals compared with their respective controls 22 hours after 2-hour middle cerebral artery occlusion. Brain infarct volume
Infarction Volume in 3-Aminobenzamide–Treated Animals
3-aminobenzamide pretreatment (n = 8) significantly reduced direct infarct volume and area (sections 1–4) in 129/SV mice compared with PBS-injected control mice (n = 7) 22 hours after reperfusion (Figs. 1C and 1D). Also, infarct volume determined indirectly was significantly smaller (50.6 ± 12.0 mm3 vs. 94.3 ± 19.2 mm3, P < 0.01). Neurological deficits were significantly improved in 3-AB–treated animals (1.3 ± 0.5 vs. 2.3 ± 0.5, P < 0.05).
Poly(ADP-Ribose) Formation
To analyze PARP activation after ischemia, poly-(ADP-ribose) immunohistochemistry was performed. The number of poly(ADP-ribose) positive cells that could be identified clearly by specific nuclear staining increased two- to threefold in the ischemic tissue compared with the contralateral side 5 minutes after reperfusion following 2-hour ischemia. Also, the intensity of staining per cell was increased. Positive cells frequently showed swelling and nuclear disruption as early signs of cell damage. Poly(ADP-ribose) formation after MCAO and reperfusion was inhibited strongly by 3-AB (Fig. 2); staining was not detected in PARP null mice. After longer periods of reperfusion (3 or 6 hours) after 2-hour ischemia, or after 1-hour ischemia without reperfusion, increased poly(ADP-ribose) formation was not observed (not shown).
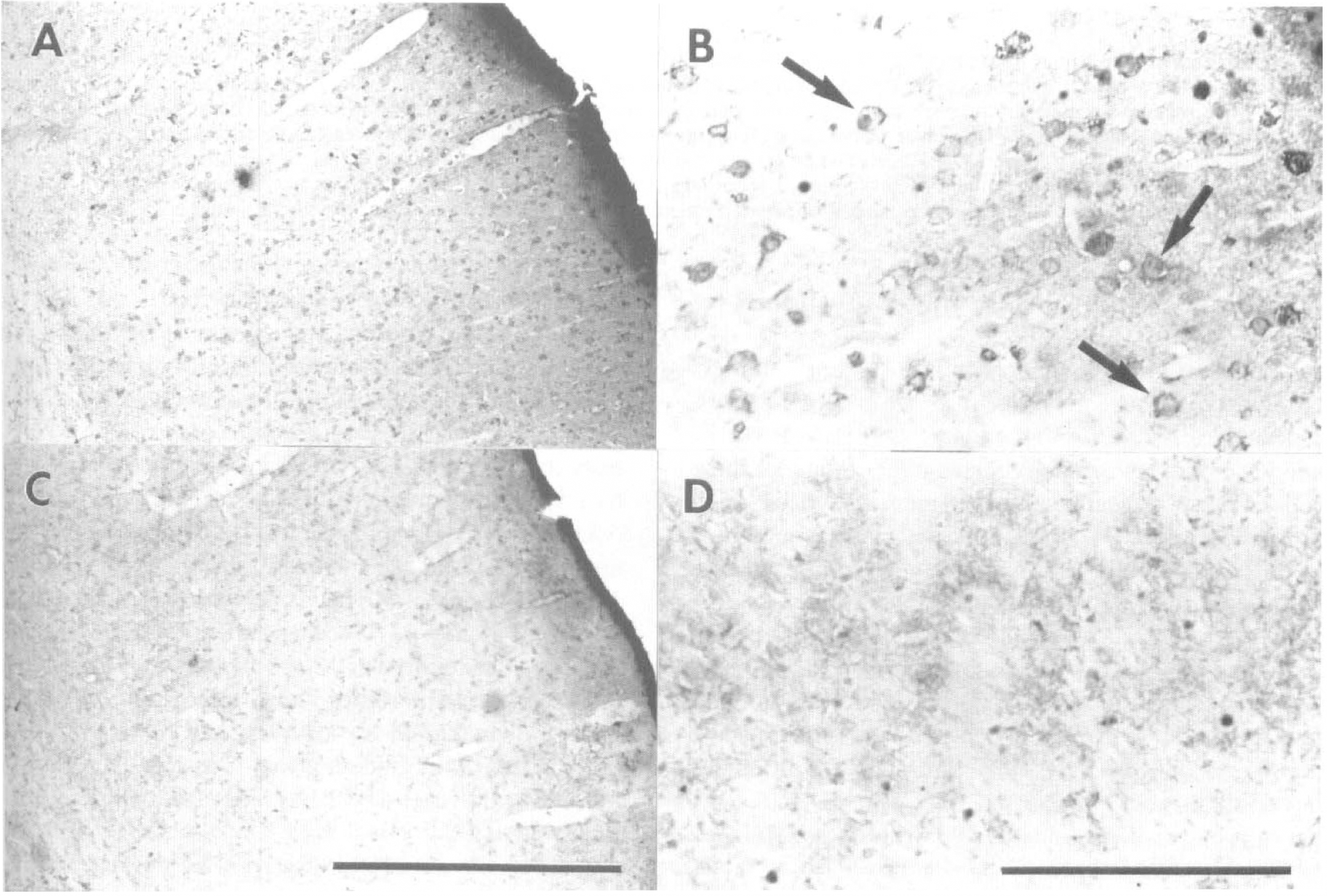
Poly(ADP-ribose) immunoreactivity was increased in ischemic tissue 5 minutes after reperfusion following 2-hour middle cerebral artery occlusion. Poly(ADP-ribose) was visualized using a polyclonal poly(ADP-ribose) antibody.
Nicotinamide Adenine Dinucleotide Determination
Brain levels of NAD did not differ under basal conditions between PARP null mice and wild-type litter mates and 3-AB versus vehicle-treated controls as assessed both in vitro and in vivo. Levels of NAD decreased to approximately 35% within the ischemic territory at 24 hours in vehicle-treated and PARP+/+ mice. Higher NAD levels were measured in ischemic territory of PARP null mice compared with wild-type (n = 5 in duplicate per group), and in 3-AB–treated mice compared with vehicle controls (n = 4 in duplicate per group) after 24 hours (Fig. 3B and D).
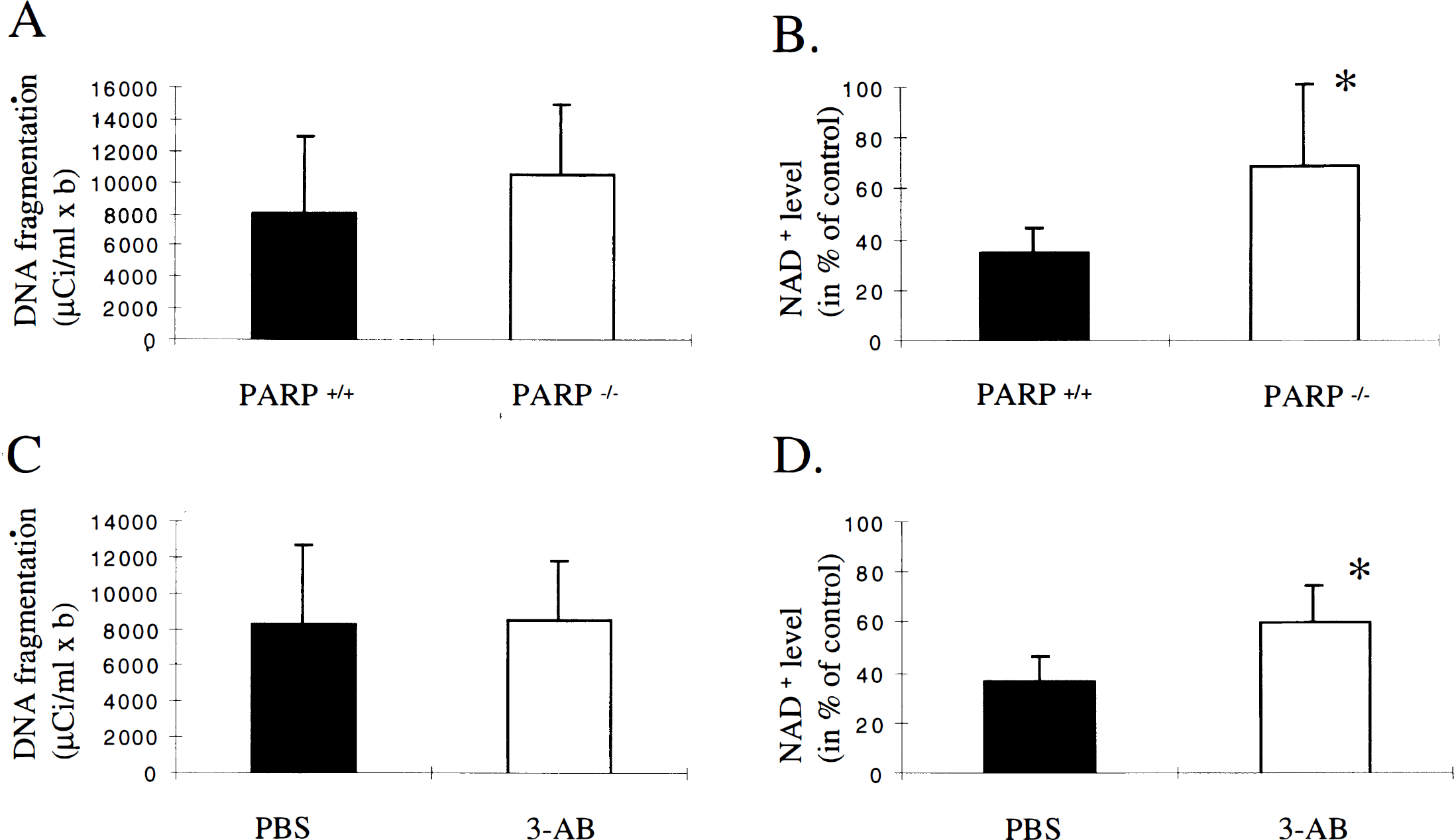
Ischemic brain tissue from poly(ADP-ribose)polymerase (PARP)−/− mice and 3-aminobenzamide (3-AB)-treated mice exhibited the same amount of DNA fragmentation
Decreased NAD staining was observed in the middle cerebral artery territory as early as 2 hours after recirculation and was further decreased at 4 and 6 hours (Fig. 4). The decrease was attenuated after 3-AB treatment (Fig. 4). The decreased NAD staining preceded gross morphologic damage on hematoxylin and eosin stained sections (not shown).
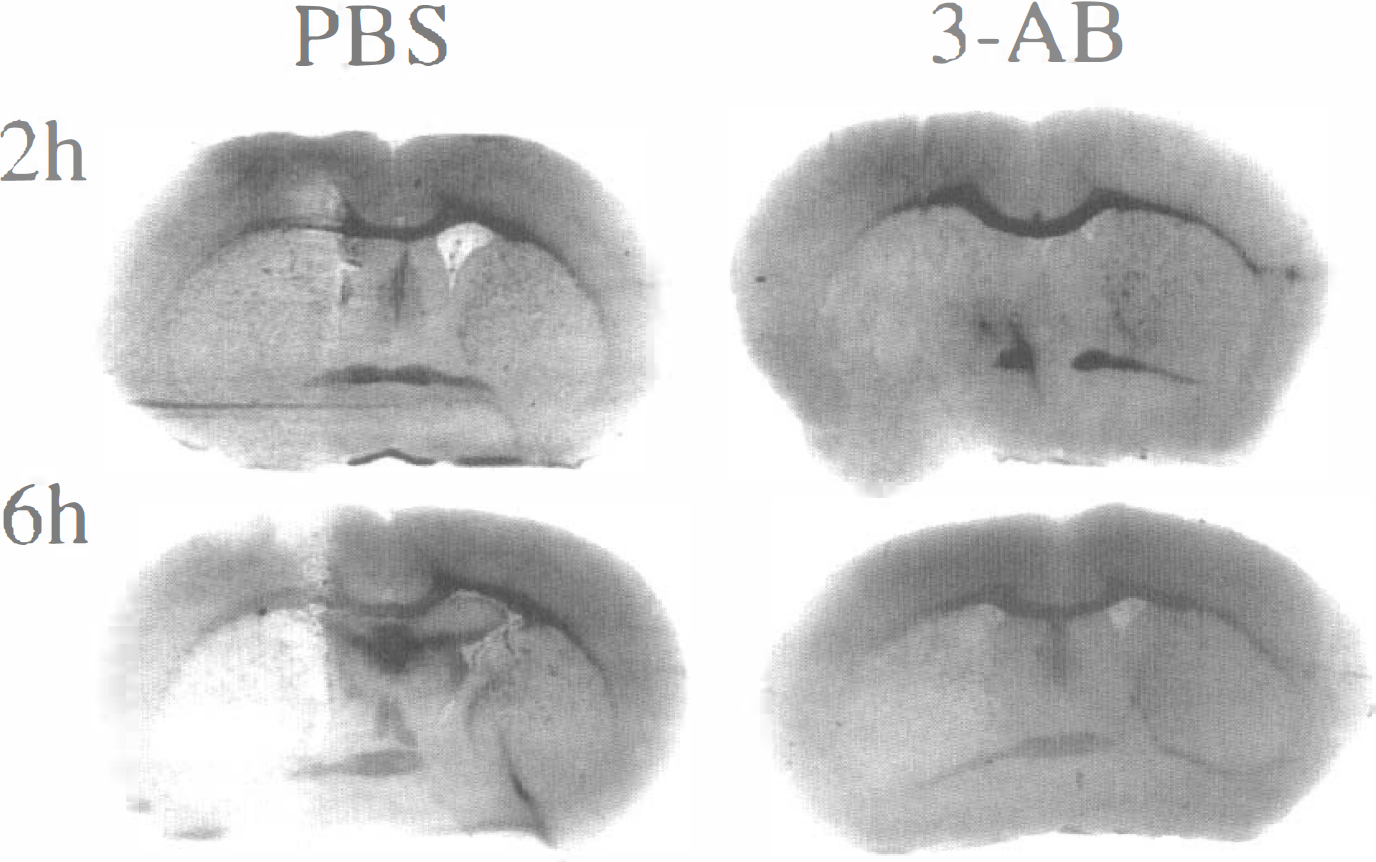
The decreases in nicotinamide adenine dinucleotide (NAD) staining after middle cerebral artery occlusion (MCAO) were attenuated after 3-aminobenzamide (3-AB) pretreatment. Nicotinamide adenine dinucleotide histochemistry was performed in coronal brain slices from phosphate-buffered saline-treated (left) and 3-AB–treated animals (right) 2 and 6 hours after 2-hour MCAO. Histochemical staining for NAD was performed with an enzymatic cycling method (Nisslbaum et al., 1969) modified for cryostat sections (12 μm). Nicotinamide adenine dinucleotide staining (dark color) is detected throughout the brain but is depleted in the territory of the left middle cerebral artery at 2 hours and more pronounced at 6 hours in vehicle-treated animals (left). Nicotinamide adenine dinucleotide depletion was less severe in 3-AB–treated animals (right).
DNA Damage and Oligonucleosomal Fragmentation (Laddering)
TUNEL(+) cells were present in great numbers throughout the ischemic tissue in all animals at 24 hours, especially in the peri-infarct zone (n = 3 for PARP−/− and +/+, 3-AB– and vehicle-treated mice). Many of these cells showed typical features of apoptosis. The lesion areas containing TUNEL(+) cells were smaller in PARP−/− mice and 3-AB–treated animals compared with their controls, matching smaller infarction areas as determined by hematoxylin & eosin staining on adjacent sections (data not shown). Estimates of cell density within the ischemic area were made in animals from all four groups at the level of the anterior commissure and 2 mm posterior to the anterior commissure. No difference in cell density within the ischemic area was found between groups when evaluated by two observers blinded to the treatment groups.
Brain tissue from within the ischemic zone from PARP null mice, 3-AB–treated animals, and controls exhibited DNA laddering on agarose gels (Fig. 5). Total DNA fragmentation did not differ between PARP null mice (n = 5) and wild-type litter mates (n = 5) after 24 hours. Moreover, 3-AB–treated mice (n = 6) did not differ from vehicle-treated controls (n = 5) after 24 hours (Figs. 4A and 4C). The amount of laddered DNA (oligonucleosomal damage) did not differ between PARP−/− and +/+ at 24 hours (20 ± 6 vs. 22 ± 16 μCi/mL × b, n = 5) and 3-AB–treated mice compared with controls at 24 hours (30 ± 11 vs. 24 ± 14 μCi/mL × b, n = 6 or 5).
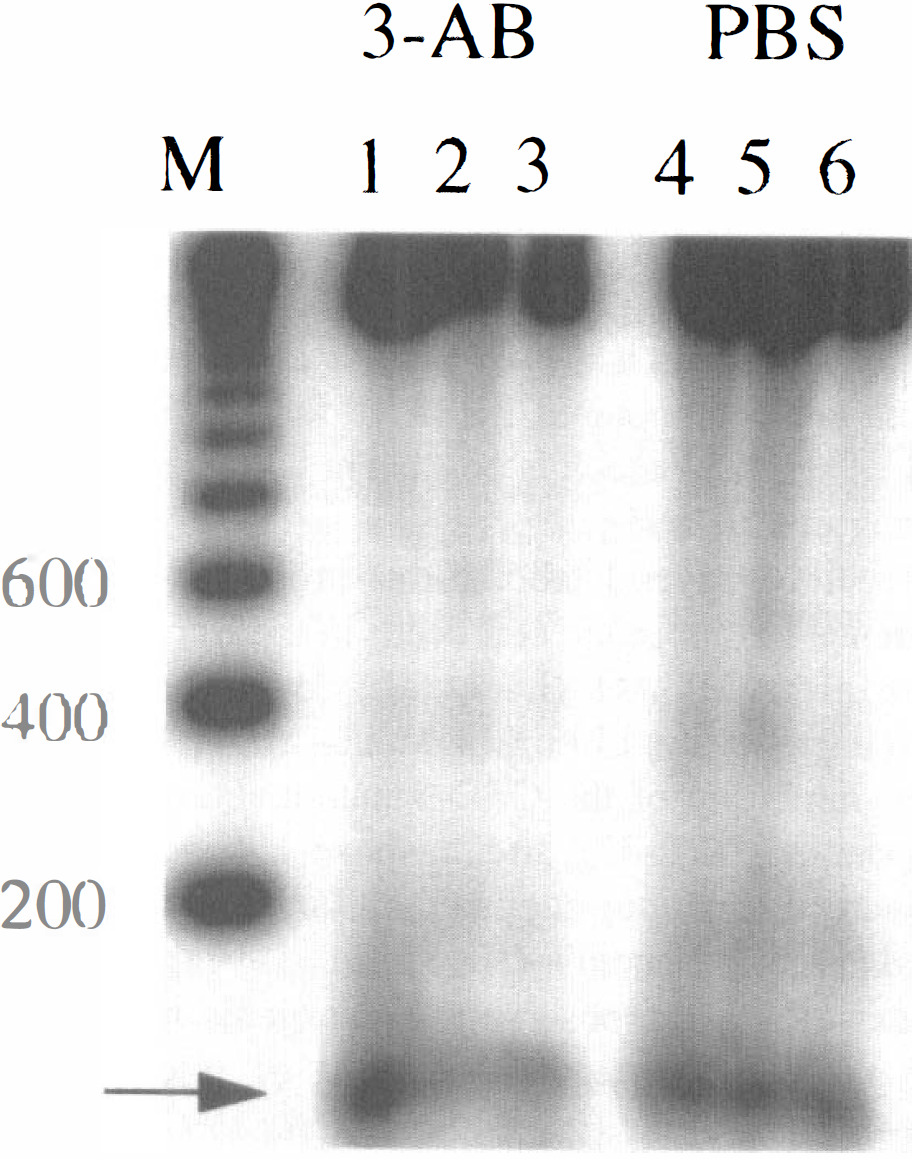
Ischemic brain tissue from 3-aminobenzamide (3-AB)- and phosphate-buffered saline (PBS)-treated animals exhibited DNA laddering on agarose gel autoradiography 22 hours after middle cerebral artery occlusion. DNA was isolated from ischemic tissue and a [32P]ddATP-end-labelling method with terminal transferase was performed. M: 200-bp standard ladder; Lane 1, 2, 3: individual 3-AB–treated animals; lane 4, 5, 6: individual PBS-treated animals; arrow indicates 100 bp internal standard. Quantitative analysis showed no significant difference between groups.
DNA fragmentation and DNA laddering also did not differ in 3-AB–treated animals compared with controls at 12 and 72 hours (n = 3 each). DNA fragmentation at 12 hours was 4250 ± 1770 versus 4110 ± 1310 μCi/mL × b and DNA laddering was 13 ± 3 versus 16 ± 12 μCi/mL × b (3-AB vs. PBS, respectively). DNA fragmentation at 72 hours was 23,700 ± 6450 versus 20,740 ± 9210 μCi/mL × b, and DNA laddering was 61 ± 26 versus 53 ± 27 μCi/mL × b (3-AB vs. PBS).
DISCUSSION
The present study provides both pharmacologic and genetic evidence that poly(ADP ribosyl)ation contributes to ischemic cell death, and that PARP inhibition protects tissue within the ischemic territory. Mice deficient in PARP were resistant to brain injury after transient focal cerebral ischemia. Pharmacologic inhibition by 3-AB also conferred resistance to injury. Infarct sparing in both experiments was accompanied by improved Neurological scores, suggesting that reduced PARP activity preserves Neurological function. The differences between 3-AB and vehicle treated groups could not be explained by obvious effects on blood pressure or blood gases. These results recently have been confirmed by independent studies on PARP−/− mice (Eliasson et al., 1997, in press).
Poly(ADP-ribose) immunoreactivity increased in ischemic tissue but only at early time points after reperfusion (5 minutes). The increase in immunoreactivity in vehicle-treated animals did not sustain over time, possibly because of its short half-life (de Murcia and Ménisser-de Murcia, 1994). As expected, PARP−/− mice are deficient in poly(ADP-ribose) and remain so after MCAO. 3-aminobenzamide–treated mice also showed decreased poly(ADP-ribose) formation, which is in agreement with Heller et al. (1995). Increases were less dramatic without reperfusion, as was protection by 3-AB treatment in a model of permanent ischemia (not shown). Thus, the extent of neuroprotection after PARP inhibition/deletion may depend on the injury paradigm and the production of ROI and NO on reperfusion.
Poly(ADP-ribose)polymerase activation and subsequent depletion of NAD seem to be sequential events after the onset of reperfusion. Nicotinamide adenine dinucleotide levels decreased shortly after reperfusion in wild-type animals. Both 3-AB–treated mice and PARP knockout mice exhibited higher levels within the middle cerebral artery territory, suggesting that the mechanism of protection may relate to energy sparing. Our measurements did not reflect the redox state of the tissue because we measured total NAD and not NADH/NAD+. Resynthesis of NAD contributes to energy depletion because four molecules of adenosine triphosphate are needed to generate one molecule of NAD (Zhang et al., 1994). Under global ischemic conditions (Siesjö et al., 1973; Pulsinelli and Duffy, 1983; Nowak et al., 1985), levels of adenosine triphosphate and adenosine, and indices of mitochondrial function decrease minutes after global ischemia and return toward normal levels after 10 to 20 minutes, but not after longer ischemic periods (Phillis et al., 1996).
Current data suggest that oxidative and deaminative DNA damage develops early during reperfusion, and that this is distinct from DNA laddering/oligonucleosomal damage. DNA damage is mediated in part by NO, ROI, and possibly other metabolites, causing base modifications, and single- and double-strand breaks (Tamir et al., 1996). Liu and colleagues (1996) provided evidence for free radical-mediated DNA damage during transient forebrain ischemia, e.g., 8-hydroxy-2′-deoxyguanosine formation and DNA strand breaks increase during reperfusion. We observed random DNA fragmentation early after reperfusion whereas DNA laddering indicative of apoptosis was first evident 6 hours after 2 hours of focal cerebral ischemia and increased thereafter (Fink et al., 1997). Because DNA damage as judged by total DNA fragmentation, did not differ between groups in the present study, this might indicate that DNA repair does not occur early after ischemia or that DNA repair mechanisms are not impaired in PARP knockout and 3-AB–treated animals, which is consistent with previous results (Wang et al., 1995).
Recently, several laboratories provided experimental evidence for apoptotic cell death in the ischemic border-zone after MCAO (Li et al., 1995a,b; Charriaut-Marlangue et al., 1996; MacManus et al., 1996). Moreover, inhibition of the CED-3 mammalian homologues, caspase-3 (CPP-32) and caspase-1 (interleukin-1β–converting enzyme) attenuates infarction after transient focal cerebral ischemia (Hara et al., 1997a), and smaller infarcts also were observed in transgenic mice expressing a dominant negative mutant of interleukin-1β–converting enzyme (Friedlander et al., 1997; Hara et al., 1997b). Interestingly, PARP is a substrate for caspase-3, exhibiting only residual activity after cleavage. However, the functional role of PARP cleavage remains unclear. Because we did not observe differences in TUNEL cell density or DNA laddering within ischemic tissue in PARP−/− and +/+ mice or 3-AB–treated animals compared with controls, our results suggest that if PARP deletion/inhibition blocks apoptosis in ischemia, it is not as robust as after administering peptide inhibitors of the interleukin-1β–converting enzyme family (Endres et al., 1997). The decrease in the number of TUNEL-stained cells in the knockout/treatment group probably only reflects infarct sparing because the density of positive cells did not differ between groups. These findings are consistent with studies by Liu et al. (1997), demonstrating that DNA fragmentation does not require the cleavage of PARP, and the fact that PARP knockout animals show no detectable defect in apoptosis during development (Wang et al., 1995). Of course, caspase-3 cleaves numerous substrates, one or more of which might mediate apoptosis in ischemia by events unrelated to PARP cleavage.
Thus, PARP activation contributes to ischemic cell death in brain, and the mechanism—although still not completely understood—may relate primarily to the consequences of depleting NAD levels. When given early, PARP inhibitors may be promising drugs for the treatment of acute stroke in humans.