
Abstract
Contrary to previous dogmas, it is now well established that brain cells can produce cytokines and chemokines, and can express adhesion molecules that enable an in situ inflammatory reaction. The accumulation of neutrophils early after brain injury is believed to contribute to the degree of brain tissue loss. Support for this hypothesis has been drawn from many studies where neutrophil-depletion blockade of endothelial-leukocyte interactions has been achieved by various techniques. The inflammation reaction is an attractive pharmacologic opportunity, considering its rapid initiation and progression over many hours after stroke and its contribution to evolution of tissue injury. While the expression of inflammatory cytokines that may contribute to ischemic injury has been repeatedly demonstrated, cytokines may also provide “neuroprotection” in certain conditions by promoting growth, repair, and ultimately, enhanced functional recovery. Significant additional basic work is required to understand the dynamic, complex, and time-dependent destructive and protective processes associated with inflammation mediators produced after brain injury. The realization that brain ischemia and trauma elicit robust inflammation in the brain provides fertile ground for discovery of novel therapeutic agents for stroke and neurotrauma. Inhibition of the mitogen-activated protein kinase (MAPK) cascade via cytokine suppressive anti-inflammatory drugs, which block p38 MAPK and hence the production of interleukin-1 and tumor necrosis factor-α, are most promising new opportunities. However, spatial and temporal considerations need to be exercised to elucidate the best opportunities for selective inhibitors for specific inflammatory mediators.
Keywords
Stroke is the third largest cause of death and the leading cause of long-term disability in the United States. No medical treatment is approved for the treatment of stroke beyond tissue plasminogen activator, a thrombolytic agent restricted to administration within 3 hours after stroke. In fact, only few patients (5% to 8%) qualify for treatment within this short time from stroke onset. Aspirin, other antiplatelets, and anticoagulants (where embolic phenomena are documented) are used as preventative therapy. Overall, stroke represents a large, unmet medical need. Estimates indicate an incidence of approximately 750,000 strokes per year in the United States, with prevalence of about 4 million survivors, who are at high risk of a secondary cardiovascular event. The stroke therapies market is estimated at $1 billion with an annual health care cost of $30 to $50 billion (this includes half of all patients hospitalized for acute neurologic disease). Increased prevalence of stroke is expected by the year 2050 with more than 1 million strokes per year expected in the United States. (Stephenson, 1998; Fisher and Bogousslavsky, 1998; Pancioli et al., 1998).
Stroke is most commonly the outcome of an obstruction of blood flow in a major cerebral vessel (usually the middle cerebral artery), which, if not resolved within a short period of time, will lead to a core of severely ischemic brain tissue that may not be salvaged. However, the ultimate size of the brain infarct also depends on the penumbra, a zone of tissue around the core of the infarct where neuronal electrophysiology is not compromised and blood flow is still maintained above a neuronal disabling level (i.e., the critical 20% to 25% of normal blood flow). If blood flow in this penumbral zone further decreases below this critical level and/or energy requirements are exceeded, the infarct zone will inevitably expand. Thus, blood flow levels are the driving force in determining the size of the ultimate infarct by providing the conditions essential for maintenance of cellular energy hemostasis. Figure 1 provides a schematic diagram outlining some of the known events involved in neuronal death after ischemic stroke. Decreased blood flow leads to a reduction in phosphocreatinine and ATP (especially the latter), and if ischemia is prolonged, the energy depletion will be sufficient to lead to severe impairment of cellular function by disruption of ATP-dependent processes.
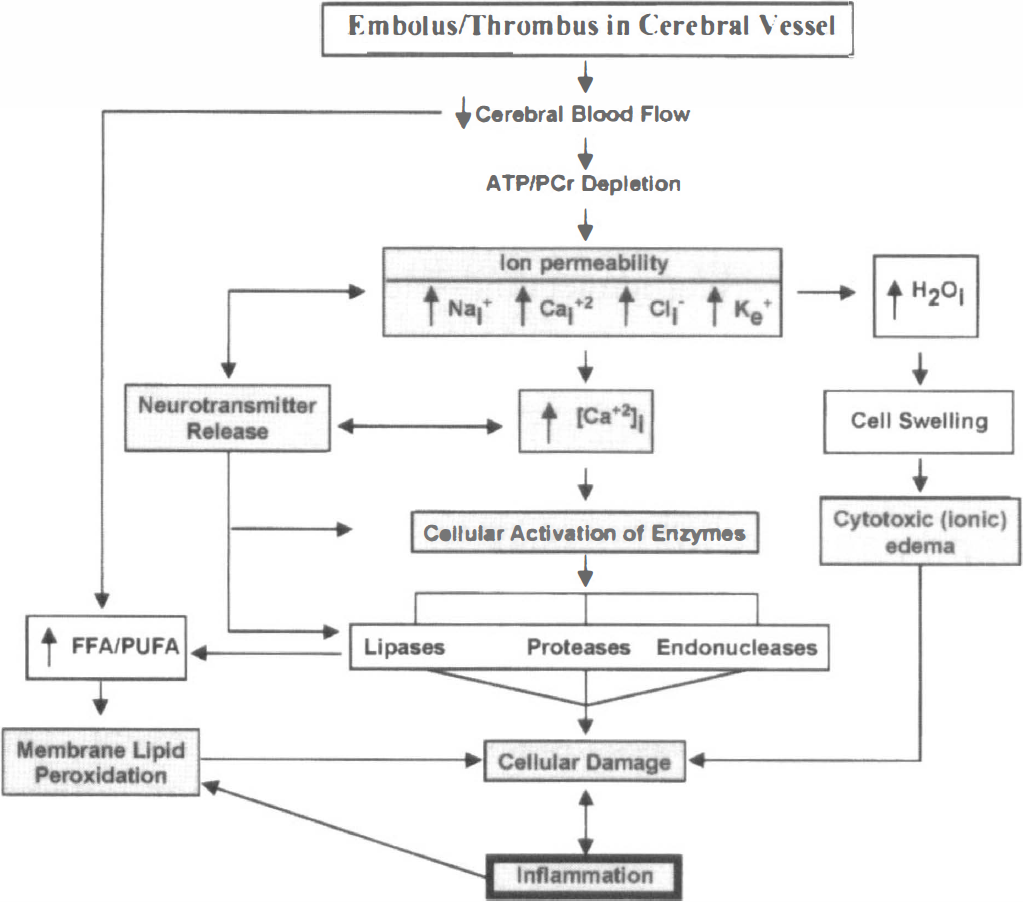
Schematic diagram illustrating major changes (shaded boxes) that occur in thromboembolic ischemic stroke. Ischemic stroke reduces energy availability and therefore membrane ionic primes fail rapidly. The increase in extracellular potassium can reach levels sufficient to release excitotoxic neurotransmitters (e.g., glutamate and aspartate) to stimulate sodium/calcium channels coupled to glutamate receptors that can facilitate developing cytotoxic edema. The significant influx of calcium through calcium channels increase free cytosolic calcium that causes mitochondrial calcium overload, cessation of already compromised ATP production, and extensive breakdown of cellular phospholipids, proteins, and nucleic acids owing to Ca2+ activation of phospholipases, proteases, and endonucleases. Free radicals are produced in the process and contribute to membrane lipid peroxidation, protein and nuclear DNA toxic changes, and cellular injury (i.e., necrosis and/or apoptosis).
The disruption of ionic gradients across excitable (neuronal) and nonexcitable (glial) membranes owing to loss of ATP is characterized by efflux of K+ from the cells, cellular depolarization, and influx of Na+, Cl−1, and Ca2+ into the cells (Siesjö, 1992a, b ). Of these events, the increase in extracellular K+, along with a decrease in pH, precedes the other ionic changes. In this phase, ATP stores are rapidly depleted and ultimately result in marked changes in ion conductance. It is postulated that the increase in K+ can reach levels sufficient to release neurotransmitters such as glutamate, which in turn will stimulate Na+/Ca2+ channels coupled to the NMDA receptor; these events will further lead to Na+, Cl−, and H2O accumulation, cell swelling, and cytotoxic edema. In parallel, extracellular Ca2+ enters into cells through both voltage-operated and receptor-operated calcium channels, leading to elevated free cytosolic Ca2+, which causes mitochondrial calcium overload and further compromise of ATP production. Transient extracellular K+- induced depolarizations can also contribute to the expansion of the neuronal lesion. Peri-infarct depolarization produces disruption of ionic gradients and transmitter release, with the associated accumulation of free Ca2+ in the cells leading to rapid and extensive breakdown of phospholipids, proteins, and nucleic acids by activation of calcium-dependent phospholipases, proteases, and endonucleases. Depolarization causes an increase in intracellular calcium and an increase in extracellular glutamate. Glutamate, an excitatory neurotransmitter implicated in ischemic neuronal damage, results in excitotoxicity in which excessive extracellular glutamate kills neurons through an increase in intracellular calcium. Glutamate-mediated excitotoxicity is thought to occur because of excessive activation of AMPA and NMDA synaptic glutamate receptors. The NMDA receptor channel conducts Na+ and calcium as do some AMPA receptor channels. AMPA receptors also control the initial membrane depolarization caused by glutamate, and affect the opening of the NMDA receptors. AMPA and NMDA receptors are two targets currently being pursued pharmacologically.
Accumulation of products such as free fatty acids (especially the unsaturated) that are metabolized to toxic lipid peroxides (via lipid peroxidation) further contribute to structural and functional perturbations of the membrane and cell function. Free radicals (a group of highly reactive oxygen species) are generated during ischemia and cause considerable damage to lipids, DNA, and proteins, and contribute to the process of neuronal death. Free radicals also contribute to the breakdown of the blood-brain barier and brain edema. Levels of free radical scavenging enzymes (e.g., superoxide dismutase) decrease during ischemia and nitric oxide levels are elevated. Nitric oxide generated primarily by neuronal and inducible nitric oxide synthases promote neuronal damage after ischemia. Activation of cytosolic proteases by the increased intracellular Ca2+ can result in the direct disassembly of the cellular microtubule system and proteolytic degradation of structural and functional protein. A specific event associated with protease activation, the conversion of xanthine dehydrogenase to xanthine oxidase, bears directly on cellular formation of toxic structures (Barone, et. al., 1998; Koroshetz and Moskowitz, 1996; Ryan et al., 1997; Albers, 1997; Wahlgren, 1997).
In summary, reduction in cerebral blood flow depletes energy to sufficiently low levels that perturb cellular ionic homeostasis. A key consequence is an increase in intracellular calcium. This has been the main, consistent event focused on by scientists during the past three decades of stroke research. Increased intracellular calcium is responsible for the release of neurotransmitters and the activation of many enzymes. Glutamate has been focused on as the excitotoxic neurotransmitter released by the ischemia-induced depolarization. The consequences of endonucleases activation may include apoptosis (programmed cell death) (Li et al., 1998b; Morrison et al., 1998; MacManus and Linnik, 1997) as DNA damage is well documented in brain ischemia.
PREVIOUS THERAPEUTIC APPROACHES HAVE BEEN UNSUCCESSFUL
Basic research conducted in the areas cited above yielded numerous pharmacologic agents such as calcium channel blockers and a variety of glutamate receptor antagonists (i.e., targeting various glutamate and glutamate-associated receptors). Approaches targeting oxygen free radicals have also been pursued. Although development of compounds based on all these mechanisms continues, no calcium channel blocker, oxygen radical scavenger, or glutamate receptor antagonists successfully completed phase III clinical development.
One reason for the difficulties in demonstrating therapeutic benefits with these compounds is that changes associated with these targets occur very soon after stroke (Fig. 2). Calcium changes happen within seconds to minutes after the ischemia insult. In fact, calcium levels maximize in about 2 minutes. Glutamate release has been demonstrated to be at maximum after 20 to 40 minutes, and most of the damage associated with its excitoxicity may occur within the first 60 minutes. Oxygen free radicals are very short-lived. Thus by the time the patient is available for treatment in the emergency room these mechanisms of damage might be largely exhausted.
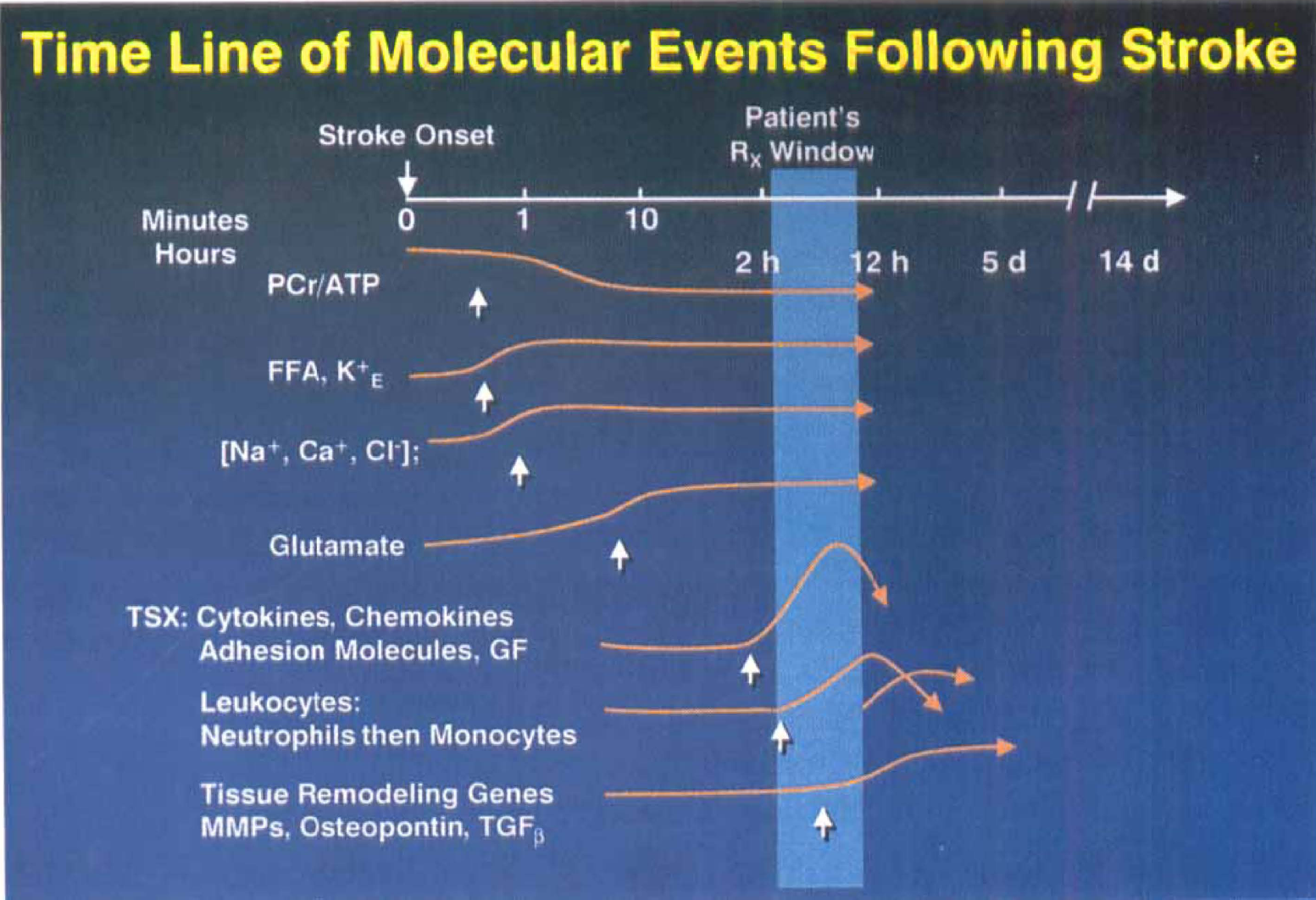
Relative (log scale) time frames for events that occur after ischemic stroke. Vertical arrows indicate onset of individual changes. Early depletion in energy-rich phosphates has immediate effects on membrane transport processes resulting in increased extracellular potassium, increased intracellular sodium, and calcium and glutamate release. All these events are over with within 1 to 2 hours. The inflammatory response to tissue injury occurs after these initial changes but contributes to the ongoing evolution of tissue injury. Immediate early response genes are expressed within 2 hours, and then inflammatory cytokines (e.g., interleukin (IL)-1β and tumor necrosis factor (TNF)-α), chemokines and adhesion molecules (e.g., ICAM-1) are expressed and drive the inflammatory response. Leukocytes accumulate within vessels and tissues (e.g., initially neutrophils and then later monocytes/macrophages). Tissue remodeling factors (e.g., transforming growth factor-β) are expressed later and are associated with resolution/healing of infarcted tissue. The shaded area indicates the time window post-stroke where one might expect acute intervention to be successful. After 3 hours, tissue plasminogen activator (the only approved therapy for strokes) cannot be administered for thrombolysis because of increased hemorrhage liability. Typically, patients are available for therapeutic intervention beyond 3 hours after stroke onset.
THE BRAIN INFLAMMATORY RESPONSE TO INJURY: A THERAPEUTIC OPPORTUNITY
Direct trauma, deprivation of oxygen and nutrients (ischemia), neurotoxicity, viral infection, or immunological challenge produce a well-defined response of gliosis (Perry and Gordon, 1991). The activation, proliferation, and hypertrophy of cells derived from the mononuclear phagocytic system (e.g., macrophages and microglia) are the hallmark of this reaction. Originally, this response was thought to mediate repair by restoring blood supply, reestablishing the integrity of the blood-brain barrier, and promoting general homeostasis at the site of injury (Norton et al., 1992; O'Callaghan, 1991; Lindsay, 1986; Eng, 1988; Giulian et al., 1988).
Since cytokines activate glial cells (Eng, 1988), which then produce cytokines (Giulian et al., 1986; Lieberman et al., 1989, 1992; Sawada et al., 1989), a close relationship appears to exist between inflammation, cytokine production, and gliosis. Indeed, gliosis can be induced by tumor necrosis factor-α (TNF-α), interleukin (IL)-1β, and interferon α (Balasingam et al., 1994; Selmaj et al., 1990; Sawada et al., 1989). Further implications for the potential central involvement of cytokines in brain inflammation have been derived from studies on the localization of TNF-α and IL-1β after surgical injury to the hippocampus in rats (Tchelingerian et al., 1993), in multiple sclerotic plaques in human brain (Hofman et al., 1989), and in human head injury (Griffin et al., 1994).
Several cell types within the brain are able to secrete cytokines: microglia, astrocytes, endothelial cells, and neurons. In addition, there is also evidence to support the involvement of peripherally derived cytokines in brain inflammation. The blood-brain barrier is permeable to cells of the immune lineage (Tsuchihashi et al., 1981; Giulian et al., 1989). Peripherally derived mononuclear phagocytes, T-lymphocytes, natural killer cells, and polymorphonuclear leukocytes (PMNs), which produce and secrete cytokines, can all contribute to central nervous system (CNS) inflammation and gliosis. In support of this, irradiation of the bone marrow or treatment in vivo with colchicine, attenuate gliosis, wound repair, neovascularization, and generalized inflammation (Giulian et al., 1989).
The inflammatory response to brain injury after focal stroke has been studied systematically by several investigators. The early accumulation of neutrophils in ischemic brain damage has been clearly shown based on histopathological (Garcia and Kamijyo, 1974; Hallenbeck et al., 1986; Chen et al., 1992; Dereski et al., 1992; Clark et al., 1993, 1994; Zhang et al., 1994a; Ritter et al., 1998), biochemical (Barone et al., 1991, 1992b, 1995), and 111ln-labeled leukocyte studies (Pozzilli et al., 1985; Dutka et al., 1989). Ameboid microglia, a form of activated microglia, can be identified within 2 hours of ischemia. Unlike normal brain microvessels that are clear of inflammatory cell, brain microvessels from ischemic zones are filled with leukocytes and a significant zone of edema surrounds them (see Fig. 3, rat focal stroke). Many of the leukocytes, primarily neutrophils, found in vessels within ischemic tissue are adherent to the endothelium, a situation not normally observed in normal brain microvessels. Some of these neutrophils migrate outside the vascular walls into the focal ischemic cortex. Brain injury is associated with the expression of inflammatory mediators, e.g., inflammatory cytokines (IL-1 and TNF-α) and chemokine (IL-8 for neutrophils, MCP-1 for monocytes, RANTES, LTB4, IP-10), while up-regulation of adhesion receptors (ICAM-1, selectins) support leukocyte adherence to the endothelium (Feuerstein et al., 1998). Tumor necrosis factor-α and IL-1β predispose or “prime” endothelium for cellular adherence. Additionally, adhesion molecules such as CD11/CD18 integrins are also thought to be pivotal in this inflammatory process (Pober and Cotran, 1990). The importance of leukocyte infiltration in the pathogenesis of brain injury has been reviewed previously (Barone, 1998; Feuerstein et al., 1998; Kochanek and Hallenbeck, 1992; Kogure et al., 1996; Stoll et al., 1998). This inflammatory reaction not only contributes to lipid-membrane peroxidation, but also exacerbates the degree of tissue injury caused by the rheologic effects of “sticky” leukocytes in the blood vessels (i.e., an interference with normal microvascular perfusion attributable to vascular plugging in an already compromised ischemic tissue bed), and also caused by the release of cytotoxic products from these activated leukocytes (i.e., by generation and release of oxygen radicals and cytotoxic products that are cytodestructive to the already compromised tissue) (Kochanek and Hallenbeck, 1992; Kogure et al., 1996; Hallenbeck and Dutka, 1990; Del Zoppo et al., 1991; Grau et al., 1992; Zhang et al., 1994b; Schmid-Schonbein, 1987). The exact nature of the signaling mechanisms in brain inflammation still remains to be elucidated but undoubtedly involves TNF-α and IL-1β, chemotactic cytokines (e.g., chemokines such as IL-8), as well as the expression of adhesion molecules and proteinases that together promote both recruited cell adherence and infiltration, and enhanced permeability of brain endothelium. For example, for a neutrophil to adhere to the endothelium and then migrate unidirectionally into the tissue, adhesion molecules have to be up-regulated. To upregulate adhesion molecule expression, injury must up-regulate specific cytokines, such as TNF-α and IL-1β. This has to happen very rapidly, and proteins also have to be rapidly translated. In addition, when those cytokines are expressed and translated, they have to upregulate adhesion molecules and produce chemokines that will induce chemoattraction to drive neutrophils into the tissue.
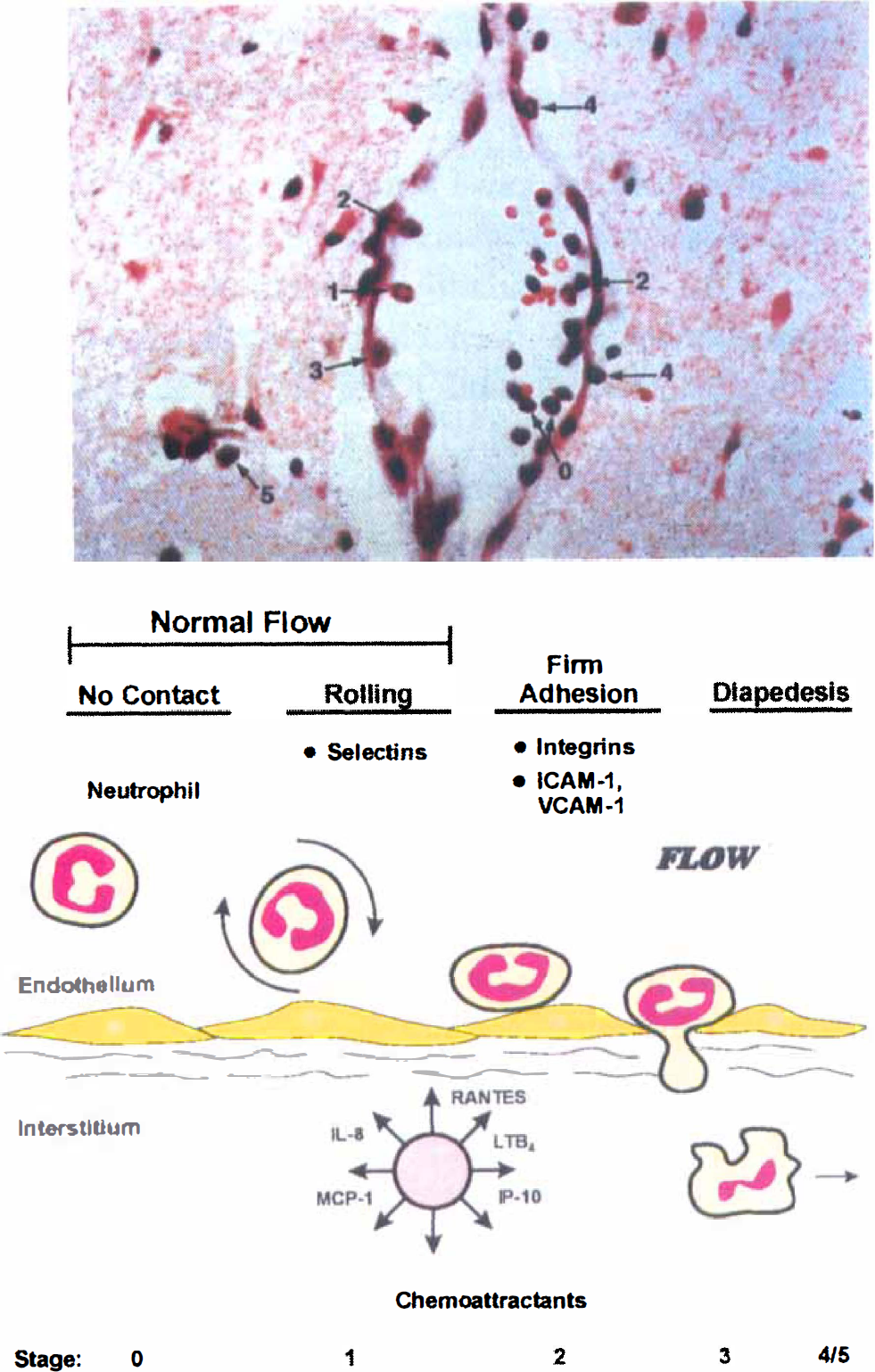
Histologic and schematic representations of changes in leukocyte behavior in the brain microvessels after focal ischemia. Shortly (within 1 to 6 hours) after experimental stroke, many of the leukocytes, primarily neutrophils, in the ischemic tissue vessels are adherent to the post-capillary venuole and capillary walls. This can modify and exacerbate the decreased blood flow occurring in the already ischemic brain. Then, these neutrophils can find their way outside the vascular walls into the focal ischemic cortex over the next 6 to 24 hours. Macrophages move into the brain later (i.e., over 1 to 5 days) and significantly accumulate in the infarcted brain. These changes in leukocyte behavior are mediated by increased brain inflammatory cytokine, adhesion molecule(s), and chemokine expression in the ischemic/injured brain.
In the remainder of this discussion, the role of TNF-α and IL-1β in traumatic and ischemic brain injury and the cooperative actions of chemokines and adhesion molecules in this process will be highlighted. Furthermore, the cascade of new gene expression that unfolds after focal stroke, which are key to the inflammatory reaction, will be explained, and the complex nature of protective and destructive gene products on histologic and functional outcomes after stroke will be addressed. Finally, novel approaches to block cytokines and brain inflammation will be presented.
ROLES OF TNF-α IN TRAUMATIC BRAIN TRAUMA AND STROKE
Tumor necrosis factor-α is a pleotrophic cytokine released by many cell types on diverse stimulation. Tumor necrosis factor-α exerts a multiple array of biological activities including stimulation of acute phase protein secretion and vascular permeability (Tracey and Cerami, 1993). Tumor necrosis factor-α and its receptors are present in the CNS (Smith and Baglioni, 1992). In addition, several clinical studies have shown a distinct relationship between elevated levels of cytokines, including TNF-α, neurodegenerative disorders, and brain injury (for reviews, see Feuerstein et al., 1998; Barone, 1999).
Elevated TNF-α has been repeatedly shown in various experimental models of brain injury. Systemic kainic acid administration induces TNF-α mRNA levels in cerebral cortex, hippocampus and hypothalamus 2 to 4 hours later (Minami et al., 1991). Systemic or intracerebroventricular administration of lipopdysaccharide endotoxin has also been shown to increase brain TNF-α levels as determined by bioassay (Siren et al., 1992), while injection of the excitotoxin ibotenic acid into the rat posterior hypothalamic region increased hippocampal TNF-α 2 weeks later (Alvarez et al., 1994). In a model of closed head injury, Shohami et al. (1994) reported an early increase in TNF-α peptide at the site of the focal insult. Also, in rat traumatic head injury, TNF-α mRNA and protein levels are rapidly elevated (Fan et al., 1996; Taupin et al., 1993). Furthermore, in mice challenged with particles of charcoal injected into the hippocampus, an increase in striatal levels of TNF-α mRNA was observed (Tchelingerian et al., 1993). Elevated serum TNF-α was also observed after severe head injury in humans (Goodman et al., 1990).
Elevated expression of TNF-α mRNA and protein occurs shortly (1 to 3 hours) after middle cerebral artery occlusion (MCAO) in rats (Liu et al., 1994; Wang et al., 1994b). Ischemic cortex levels of TNF-α mRNA were elevated as early as 1 hour after occlusion (i.e., before significant influx PMN), peaked at 12 hours, and persisted for approximately 5 days. The early expression of TNF-α mRNA preceding leukocyte infiltration suggests that TNF-α may be involved in this response. Double-label immunofluorescence studies localized the de novo synthesized TNF-α to neurons but not astroglia. At 5 days after the ischemic insult, neuronally associated TNF-α was diminished, and TNF-α immunoreactivity was localized in the inflammatory cells. The significance of TNF-α expression in the brain was studied by microinjection of TNF-α into the rat cortex; TNF-α-induced leukocyte adhesion to the capillary endothelium, but no evidence for neurotoxicity at the site of injection was found. Buttini et al. (1996) identified a rapid upregulation of TNF-α mRNA and protein in activated microglia and macrophages after focal stroke, again suggesting that TNF-α is part of an intrinsic inflammatory reaction of the brain following ischemia. Tumor necrosis factor-α may exert a primary effect on microvascular inflammatory response as reflected by TNF-α–induced neutrophil adhesion to brain capillary endothelium (Liu et al., 1994). Furthermore, intracerebroventricular injection of TNF-α 24 hours before MCAO exacerbates the ischemia-induced tissue injury (Barone et al., 1997a). This effect was reversed by ventricular administration of anti-TNF-α monoclonal antibody in the contralateral ventricle. Further evidence for the involvement of TNF-α in stroke-induced injury is supported by findings that spontaneously hypertensive rats that are stroke prone have higher levels of TNF-α production in the brain as compared with normotensive rats (Siren et al., 1992). These data suggest that TNF-α may prime the brain for subsequent damage by activating capillary endothelium to a pro-adhesive state, possibly through the up-regulation of surface endothelial adhesion molecules (see below).
INTERLEUKIN-1β IN BRAIN TRAUMA AND STROKE
Accumulating evidence shows that IL-1β can be produced in the CNS from various cellular elements including microglia, astrocytes, neurons, and endothelium. Like TNF-α, IL-1β has many pro-inflammatory properties, and receptors for this cytokine have been shown in the CNS (Rothwell, 1991; Dinarello, 1988; Takao et al., 1990). Increase in IL-1β mRNA expression has been shown to occur after several types of injury to the brain including kainate excitotoxicity (Minami et al., 1991) and lipopdysaccharide endotoxin (Ban et al., 1992; Buttini and Boddeki, 1995). Furthermore, mechanical damage after implantation of a microdialysis probe has been shown to induce expression of IL-1β (Woodroofe et al., 1991). After fluid percussion brain trauma in the rat, a rapid increase in IL-1β mRNA expression has been reported (Fan et al., 1995). Microglial IL-1α expression has been observed in human head injury (Griffin et al., 1994). Interleukin-β mRNA expression has been shown to increase after transient brain ischemia in the rat (Minami et al., 1992; Yabuuchi et al., 1994). The exacerbation of ischemic brain injury caused by exogenous IL-1β administered into the brain has been observed (Yamasaki et al., 1995; Loddick and Rothwell, 1996). A rapid (3 to 6 hours after ischemia) increase in IL-1β mRNA after MCAO peaked at 12 hours but returned to basal values at 5 days (Liu et al., 1993). Early IL-1β expression after focal stroke has also been shown using in situ hybridization (Buttini et al., 1994). The recent development of tools such as specific antibodies to rat IL-1β has permitted the identification (by immunohistochemistry) of IL-1β peptide in cerebral vessels, microglia and macrophages after focal stroke (Davies et al., 1999; Zhang et al., 1998b).
Interleukin-1 receptor antagonist (IL-1ra), a 23 to 25 Kda glycosylated protein, is a naturally occurring inhibitor of IL-1 activity that competes with IL-1 for occupancy of IL-1RI without inducing a signal of its own. Interleukin-1 receptor antagonist is produced by many different cellular sources including monocytes/macrophages, endothelial cells, fibroblasts, neurons, and glial cells (Dinarello, 1996). The expression of IL-1ra and IL-1R mRNA after focal stroke was also reported (Wang et al., 1997). The level of IL-1a mRNA was markedly increased in the ischemic cortex at 6 hours, then reached a significantly elevated level from 12 hours to 5 days after MCAO. The presence of IL-1ra in the normal brain and the upregulation of IL-1ra mRNA after ischemic injury suggest that IL-1ra may serve as a defense system to attenuate the IL-1-mediated brain injury. It is interesting to observe that the temporal induction profile of IL-1ra after MCAO virtually parallels that of IL-1β as shown previously (Liu et al., 1993a), except that IL-1ra mRNA exhibited prolonged elevation beyond that of IL-1β. Thus, the balance between the levels of IL-1β and IL-1ra expressed after ischemia may be more critical to the degree of tissue injury than IL-1 levels per se.
The mediators responsible for IL-1ra induction after focal stroke are not known. However, previous studies indicate that some cytokines such as IL-1, TNF, IL-6, and transforming growth factor-β are inducers of IL-1ra (Dinarello, 1996). Ischemia-induced expression of IL-1ra mRNA could originate from monocytes/macrophages, endothelial cells, fibroblasts, neurons, and glial cells as observed previously under normal conditions (Dinarello, 1996). The same cellular sources may be responsible for IL-1β and IL-1ra production based on the close temporal, and perhaps functional coupling of these two genes after focal stroke.
CHANGES IN GENE EXPRESSION
As pointed out previously, focal ischemia is a very powerful stimulus to elicit genomic responses in the brain in the form of multiple gene expression. Tumor necrosis factor-α or IL-1β certainly are not the only inflammatory genes that are expressed in focal cerebral ischemia. Focal ischemia is a powerful stimulus that results in comprehensive genomic reformation of the brain. Figure 4 summarizes a large amount of experimental data and illustrates that in response to ischemia, numerous pro-inflammatory genes are up-regulated. Transcription factors (immediate early genes are the first “wave” as shown by c-fos and others (c-jun, zif268, Jun-B), but their expression is transient (Hsu et al., 1993; Nowak et al., 1990; Uemura et al., 1991; Wang et al., 1995c; Welsh et al., 1992). A second wave consists of the heat shock proteins. Heat shock protein mRNA is usually expressed within 1 to 2 hours and then down-regulated by 1 to 2 days (Nowak et al., 1990; Welsh et al., 1992).
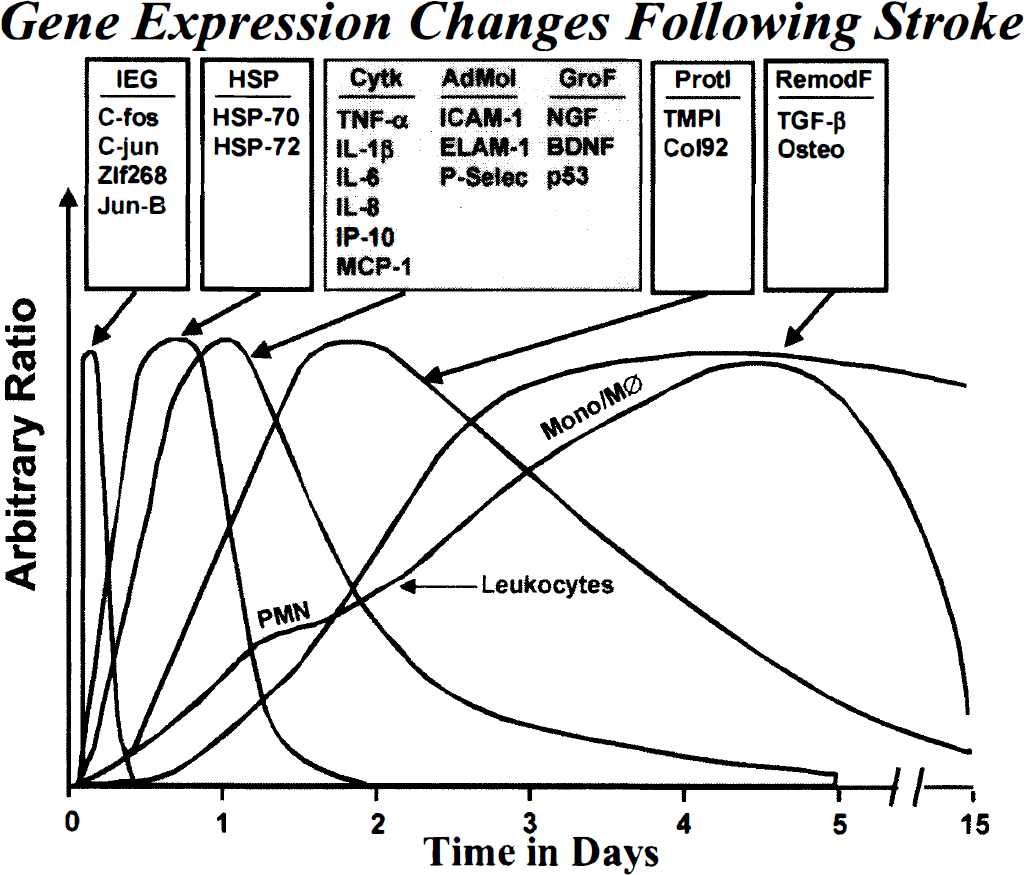
Time expression of gene expression after focal ischemia in rat cortex induced by middle cerebral artery occlusion. Five waves of ischemic cortex gene expression include a broad range of up-regulated transcription factors (wave 1), heat-shock proteins (wave 2), pro-inflammatory mediators including cytokines, chemokines, and adhesion molecules, and including neurotrophic (e.g., neuroprotective) growth factor and oncogene expression (wave 3; stroke), proteinase and proteinase inhibitor gene expression (wave 4), and delayed remodeling proteins involved in resolution of the tissue injury (wave 5). A leukocyte wave, including early polymorphonuclear ([PMN] neutrophil) infiltration and later mononuclear/macrophage infiltration (mono/MØ), is also included for comparison.
Of great interest is the third wave (shaded in Fig. 4) that is largely comprised of increased cytokine gene expression as described earlier for TNF-α and IL-1β (Liu et al., 1993a, 1994; Wang et al., 1994b), and including IL-6 (Wang et al., 1995c) and IL-1 ra (Wang et al., 1997). Chemokines such as IL-8 (Liu et al., 1993a), IP-10 (Wang et al., 1998b), and MCP-1 (Wang et al., 1995b; Kim et al., 1995) are also increased, and are likely to play a role in neutrophil and mononuclear cell infiltration. The presence of recruited leukocytes at the site of inflammation is critically dependent on the coordinated expression of adhesion molecules (ligands and receptors) on inflammatory cells and the activated capillary endothelium, respectively. These timed responses facilitate the efficient “docking” of activated immune cells to their respective receptors which is the primary step in the process of transendothelial migration and diapedesis. Some of the candidate molecules include ICAM-1, ELAM-1, and P-selectin on the endothelial side, and CD11/CD18, MAC-1, and LFA-1 on the leukocyte side (Beerhuizen and Van Furth, 1993; Granger and Kvietys, 1993). After MCAO in rats, ICAM-1 (Wang et al., 1994) and ELAM-1 (Wang et al., 1995) mRNA expression is increased within 1 to 3 hours. ICAM-1 protein was localized to the capillary endothelium in the infarcted cortex. After stroke in baboons (Okada et al., 1994), P-selectin and ICAM-1 were shown to be up-regulated on the endothelium of postcapillary microvessels in the ischemic penumbra. The third wave also includes increased expression of the endothelial cell adhesion molecules such as ICAM-1, ELAM-1, and P-selectin (Wang et al., 1994, 1995; Okada et al., 1994; Zhang et al., 1995, 1996, 1998a) that are important in establishing endothelial adhesion to the leukocyte before infiltration. In addition, growth factors (Hsu et al., 1993) (e.g., nerve growth factor, brain-derived nerve growth factor) that might be expected to play neuroprotective role(s) after stroke, and the tumor suppressor gene p53 (Li et al., 1994) also increase in this time frame. This inflammatory wave is believed to play a key role in leukocyte infiltration (initially neutrophils; PMN, that are followed by monocytes/macrophages; Mono/MØ as indicated in Fig. 4 (Clark et al., 1993, 1994; Barone et al., 1991, 1992b, 1995). The third wave commences about one hour after stroke, peaks at about 12 hours, and subsides 2 to 5 days later. In this report, it is noteworthy that sensitivity of detection methods play an important role in defining these time frames; upon improved methodology, it is possible that earlier transcriptional event might be monitored.
A fourth wave of new gene expression may well be associated with the inflammatory reaction to ischemia. This fourth wave includes proteolytic enzymes (metalloproteinases) implicated in remodeling of the extracellular matrix (Rosenberg et al., 1996; Romanic et al., 1998), and their endogenous protease inhibitors, TMPI, (Wang et al., 1998), after focal stroke. The expression of these genes in stroke appears to be related to the influx of inflammatory cells and is associated with secondary brain injury and repair processes after stroke. The fifth wave of de novo transcription includes mediators such as transforming growth factor-β (Wang et al., 1995) and osteopontin (Wang et al., 1998; Ellison et al., 1998) which appear to be important in tissue remodeling, including formation of a new barrier to protect the normal, viable brain tissue. The relationship of the diverse gene expression to the progression of brain injury and the healing process (including plasticity that enables recovery of function long term after stroke) is depicted in Fig. 5.
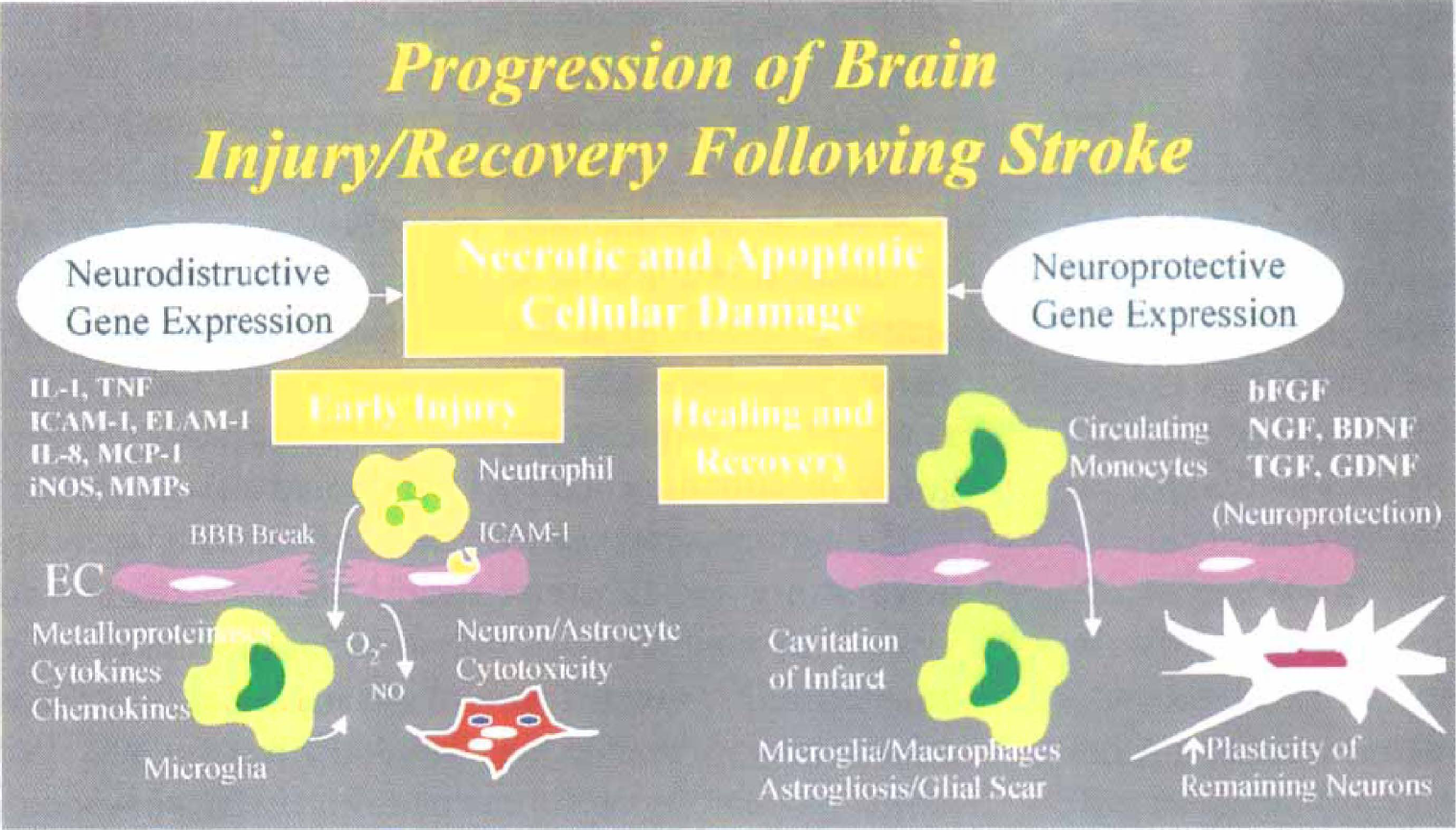
After initiation of brain injury (as depicted in Fig. 1 and outlined in the text), various neurodestructive and neuroprotective genes are expressed. Neurodestructive gene expression (e.g., primarily those inflammatory cytokines, adhesion molecules, chemokines, and inflammatory proteins such as inducible NOS and metalloproteinases) can drive brain inflammation and necrotic/apoptotic cell death. Neuroprotective gene expression includes neurotrophic and growth factors from circulating mononuclear cells that have infiltrated into damaged tissue. In addition, resident-activated glial cells can also protect cells/tissue by facilitating repair and remodeling, and/or increasing neuronal plasticity and thereby facilitating the recovery of function for the remaining viable neurons/tissue.
BRAIN INFLAMMATION: A DUAL-EDGED SWORD
The direct injection of minute amounts of TNF-α into the brain results in leukocyte adhesion to microvessel endothelium and infiltration into the tissue; however, no direct neurotoxicity to neurons at the site of injection was reported (Liu et al., 1994). Others have also shown that TNF-α is not directly toxic to neurons (Garcia et al., 1992; Piani et al., 1992) and some investigators even suggest a protective effect of TNF-α on neurons (Barger et al., 1995; Cheng et al., 1994; Schwartz et al., 1991). The dual capacity of TNF-α to convey injurious or beneficial effects have been emphasized previously (Tracey and Cerami, 1994). The capacity of TNF-α to provide beneficial effects in brain injury is inferred from studies with TNF-receptor knock-out mice (p55 and p75 knockout) which display increased sensitivity to brain ischemia (Bruce et al., 1996; Rothwell and Luheshi, 1996) and the capacity of TNF and IL-1 to elicit states of ischemic tolerance on repeated administration (Ohtsuki et al., 1996; Nawashiro et al., 1997b). A later expression of TNF-α in macrophages involved in resolution of ischemic brain injury has been described (Clark et al., 1993, 1994; Liu et al., 1994). However, the data to date show that augmenting the acute effects of TNF-α and IL-1B is not protective (and in fact increase ischemic injury) and that blocking the acute surges in TNF-α and IL-1B that occur after focal stroke is neuroprotective (see below).
Figure 6 illustrates the relative timing of the dynamic changes that occur after stroke. This scheme suggests a progression of events through repair and remodeling and into neuronal plasticity and recovery of function (Jenkins and Merzenich, 1987; Li et al., 1998a). Note that brain inflammation spans across all these processes and can be expected to influence early injury and later postinjury repair processes. Postinjury plasticity and functional recovery has been shown to be activity-dependent and is influenced by training (Johansson and Grabowski, 1994) and facilitated by growth factors (Kawamata et al., 1997). Although initial brain inflammation can contribute to the degree of brain damage after injury, anti-inflammatory interventions to limit the degree of damage have been shown to interfere with nervous regeneration and recovery (Hirschberg et al., 1994). In addition, recent data (Moalem et al., 1999) indicate that activated T cells can protect neurons from secondary degeneration after central axonomy. Also, recruitment of activated macrophages in lesioned spinal cord was linked to regeneration failure after injury in the CNS (Lazarov-Spiegler et al., 1998). Interleukin-1 has been associated with dopaminergic sprouting in denervated striatum (Ho and Blum, 1998) and increased neuronal TNF-α observed during spinal cord recovery in experimental allergic encephalomyelitis also suggests additional reparative recovery roles for TNF-α in the CNS (Villarroya et al., 1997). Better regeneration occurs in the CNS associated with more marked inflammation (Guest et al., 1997) and activated macrophage and microglia facilitation of neuronal plasticity/recovery after injury is apparently associated with secretion of neurotrophic factors from the macrophages (Batchelor et al., 1999). Therefore, the inflammation that does occur in response to injury in the CNS appears to serve multiple purposes. While certain strategies may be required to intervene early in brain inflammation to reduce injury and neurodegeneration, other intervention may be necessary to facilitate repair and recovery of regeneration processes after CNS injury (Dopp and deVillis, 1998; Lazarov-Spiegler et al., 1998). Clearly, more must be learned about these complex interactions. In addition, timing of specific interventions may be critical to development of significant neuroprotective anti-inflammatory therapy. A recent, novel anti-inflammatory cytokine experimental approach has shown the neuroprotective effects of IL-10 administration in both stroke (Spera et al., 1998) and neurotrauma (Knoblach and Faden, 1998). These latter reports may suggest that certain anti-inflammatory modulatory cytokines may be of therapeutic potential in ischemic or traumatic brain injury.
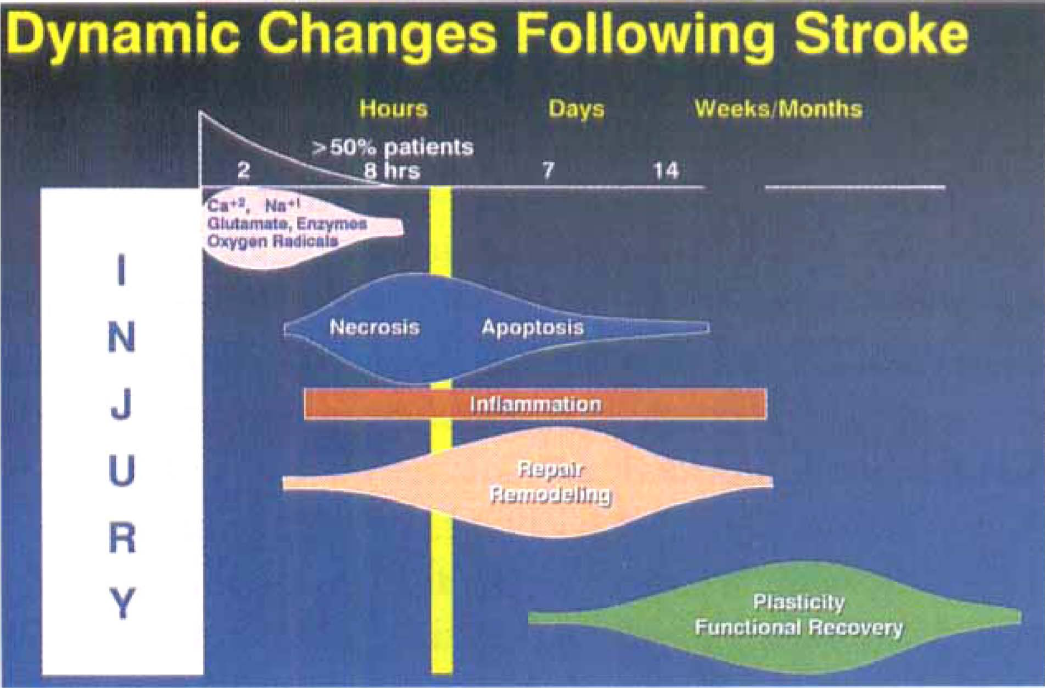
Schematic diagram depicting relative time sequence of the dynamic changes that occur after ischemic stroke. Of particular interest is the brain inflammation that occurs throughout all other phases of brain changes in response to initial ischemia/injury. Text describes the “yin/yang” involvement of brain inflammation in exacerbation and evolution of initial ischemic injury, and in the repair and recovery of function of neuron tissue following injury.
ANTI-LEUKOCYTE STRATEGIES FOR NEUROPROTECTION
Matsuo et al. (1994) have used the RP-3 monoclonal antibody that selectively depletes leukocytes in the rat (about 90% to 95%) and reported a dramatic reduction in both neutrophil accumulation in focal ischemic brain tissue and infarct size (decreased by 45% to 50%). Although some controversy exists (Hayward et al., 1996), other studies have verified these effects in various experimental models (Chen et al., 1992; Jaing et al., 1998; Dutka et al., 1989; Shiga et al., 1991; Heinel et al., 1994; Bowes et al., 1995; Bednar et al., 1991; Vasthare et al., 1990; Hartl et al., 1996). However, although these “depletion” experiments support the hypothesis that early inflammatory cell infiltration in stroke exacerbates ischemic injury, this strategy is unlikely to translate into clinical use.
ANTI-ADHESION MOLECULE ANTAGONISTS FOR NEUROPROTECTION
Another attractive approach is the inhibition of endothelial interactions with the leukocyte. Chen et al. (1994) treated MCAO rats intravenously with an antibody against MAC-1, the leukocyte counterpart of ICAM-1 binding and demonstrated reduction in infarct size by 45% to 50% in a rat transient MCAO model. Zhang et al. (1994b) used the intravenous administration of an anti-ICAM-1 antibody to demonstrate a 40% reduction of infarct size in a similar model. Blocking adhesion molecules can also reduce apoptosis induced by focal ischemia (Chopp et al., 1996). Other studies verified these effects but also illustrated that these antibodies could not reduce infarct size when the ischemia was permanent (Chopp et al., 1994; Jiang et al., 1994; Bowes et al., 1993; Clark et al., 1991; Zhang et al., 1995). However, the strategy may work if both leukocyte and endothelial adhesion proteins are blocked in permanent focal stroke. The combination of t-PA and anti-CD 18 provides significantly improved outcome, and may increase the therapeutic time window in stroke (Zhang et al., 1999). In a rabbit embolic model of stroke, anti-ICAM-1 antibody was shown to increase the amount of clot necessary to produce permanent damage (Bowes et al., 1993). In addition, in a baboon model of transient focal ischemia anti-CD 18 monoclonal antibody administered 25 minutes before reperfusion led to an increase in reflow in microvessels of various sizes (Mori et al., 1992). However, in contrast to the demonstrated anti-ischemic effect of anti-adhesion molecules in animal models, the recent failure of the murine anti-ICAM monoclonal antibody (enlimomab) in human stroke (DeGraba, 1998), and its ability to activate human neutrophils (Vuorte et al., 1999) show the difficulties in extrapolating encouraging data derived from animal models to clinical reality.
NEUROPROTECTION BY CYTOKINE INHIBITION SUPPORTS ANTI-INFLAMMATORY APPROACH
An alternate possibility to modulate inflammation is to aim directly for cytokine and chemokine suppressive agents. While proof for a role of TNF-α and IL-1β in ischemia brain damage has not been definitively established, the availability of selective and potent antagonists of cytokine production may aid in reaching this goal. Many studies have shown the protective effects of IL-1ra in brain injury. Thus, intracerebroventricular administration of recombinant IL-1ra produced a marked reduction in brain damage induced by focal stroke (Relton et al., 1996; Rothwell and Relton, 1993; Loddick and Rothwell, 1996), brain hypoxia-ischemia (Martin et al., 1995), or fluid percussion injury in the rat (Toulmond and Rothwell, 1995). This neuronal protective effect of IL-1ra in focal stroke was further supported by a recent study using an adenoviral vector that overexpressed IL-1ra in the brain (Betz et al., 1995). The excess of IL-1ra significantly reduced infarct size after focal stroke. While such modes of IL-1ra delivery are impractical in clinical terms, the studies point out a potential therapeutic remedy if delivery of IL-1ra can be achieved in a timely manner. In addition, IL-1ra expression increases after ischemic preconditioning in a manner that parallels the development of brain ischemic tolerance (Barone et al., 1998). Of interest is data showing that peripheral administration of IL-1ra reduces brain injury (Relton et al., 1996), suggesting a potential use of IL-1ra as a neuroprotective agent in human stroke and/or neurotrauma. Likewise, several studies have shown that blocking TNF-α results in improved outcome in brain trauma and stroke. Pentoxifyline (a methylxanthine that reduces TNF-α production at the transcriptional level) or soluble TNF receptor I (which acts by competing with TNF-α at the receptor) improves neurologic outcome, reduces the disruption of the blood-brain barrier, and protects hippocampal cells from delayed cell death after closed head injury in the rat (Shohami et al., 1996). In rat focal ischemia, an anti-TNF-α monoclonal antibody and the soluble TNF receptor I were neuroprotective (Barone et al., 1997a). In the latter studies, TNF-α was blocked by repeated intracerebroventricular administrations before and during focal stroke which significantly reduced infarct size. In murine focal stroke, topical application of soluble TNF receptor I on the brain surface significantly reduced ischemic brain injury (Nawashiro et al., 1997a). In addition, in another study evaluating TNF blockade on focal stroke in hypertensive rats, soluble TNF receptor I administered intravenously, before or after MCAO, significantly reduced the impairment in ischemic cortex microvascular perfusion and the degree of cortical infarction, strongly suggesting an inflammatory/vascular mechanism for TNF-α in focal stroke (Dawson et al., 1996).
The detrimental effects of TNF-α and its role as a mediator of focal ischemia may involve several mechanisms. For example, TNF-α increases blood-brain barrier permeability and produces pial artery constriction that can contribute to focal ischemic brain injury. Furthermore, a direct toxic effect of TNF-α on the capillaries was also noted (Goldblum and Sun, 1990; Beutler and Cerami, 1987). Also, by stimulating the production of matrix-degradating metalloproteinase (gelatinase B) (Kim et al., 1995; Rosenberg et al., 1996; Romanic et al., 1998; Rosenberg et al., 1995), TNF-α may further exacerbate capillary integrity. Tumor necrosis factor-α also causes damage to myelin and oligodendrocytes (Robbins et al., 1987; Selmaj and Raine, 1988) and increases astrocytic proliferation thus potentially contributing to de-myelination and reactive gliosis. In addition, TNF-α activates the endothelium for leukocyte adherence and procoagulation activity (i.e., increased tissue factor, von Willebrand factor, and platelet activating factor) that can exacerbate ischemic damage (Pober and Cotran, 1990). Indeed, increased TNF-α in the brain and blood in response to lipopolysaccharide seems to contribute to increased stroke sensitivity/risk in hypertensive rats (Feuerstein et al., 1998; Siren et al., 1992; Hallenbeck et al., 1988; Barone et al., 1992). Tumor necrosis factor-α activates neutrophils (Shalaby et al., 1985) increases leukocyte-endothelial cell adhesion molecule expression (Balasingam et al., 1994), leukocyte adherence to blood vessels, and subsequent infiltration into the brain (for review see Feuerstein et al., 1998).
Interference with either IL-1β or TNF-α has now been shown repeatedly to result in reduced deficits in focal stroke and head trauma models. The evaluation of additional potent and specific anti-cytokine therapeutics in proper models of brain injury is clearly warranted. Much evidence has accumulated that indicates TNF-α production is regulated at both transcriptional and translational levels (Tracey and Cerami, 1993). Thus, TNF-α mRNA synthesis inhibitors such as rolipram (Semmler et al., 1993), a phosphodiesterase IV inhibitor, could be of benefit in the treatment of brain inflammation. In addition, selective inhibition of this phosphodiesterase a predominant isoform in cerebral vessels, mediates significant vasorelaxation that can also improve outcome in stroke (Willette et al., 1997). Other novel classes of drugs include highly specific protein kinase C inhibitors such as calphostin C (Kobayashi et al., 1990), which has been shown to potently inhibit lipopolysaccharide endotoxin-stimulated TNF-α production from human monocytes in vitro (Prabhakar et al., 1993) as well as lipopolysaccharide endotoxin and viral stimulated TNF-α production in astrocytic cell lines (Lieberman et al., 1989, 1992). Interestingly, protein kinase C also serves as a “focal point” in the regulation of production of IL-1β (Gorospe et al., 1993) and endothelial cell-derived adhesion molecules (Pober and Cotran, 1990). However, because of the ubiquitous nature of protein kinase C, nonselective inhibitors may result in toxic consequences. However, the finding that liposome-entrapped staurosporine greatly enhanced survival in a rat model of endotoxemia possibly related to the suppression of serum TNF-α levels (Tschaikowsky and Brain, 1994), supports the feasibility of such a pharmacologic paradigm to prevent brain inflammation. Further advances in this approach will be derived from the development of isozyme-specific protein kinase C inhibitors (Wilkinson and Hallam, 1994). The utility of blocking inflammatory cytokines in conjunction with thrombolysis using tissue plasminogen activator has been discussed recently (Garcia et al., 1997).
MITOGEN-ACTIVATED PROTEIN KINASES: A NOVEL APPROACH TO CYTOKINE SUPPRESSION
Figure 7 illustrates the general features of the mitogen-activated protein kinase (MAPK) family. Many neurotrophins/growth factors bind to Trk receptors and signal through Ras MAPK pathway via ERK/MAPK (for review see Skaper and Walsh, 1998). Stress-activated protein kinases (e.g., p38 and JNK, Fig. 7) play an important role in transducing stress-related signals by a cascade of phosphorylation of intracellular kinases and transcription factors (Robinson and Cobb, 1997) that regulate cell survival, apoptosis, and inflammatory cytokine production (Kummer et al., 1997; Lee et al., 1994; Lee and Young, 1996; Xia et al., 1995). Sustained activation of JNK and p38 MAPK inhibitors promote (in vitro) survival of sensory, sympathetic, ciliary, dorsal root ganglia, and motor neurons (Horstmann et al., 1998; Skaper and Walsh, 1998). In PC 12 cells, withdrawal of nerve growth factor causes apoptosis that is preceded by decreased ERK and increased JNK/p38 activity (Xia et al., 1995), suggesting a balance between ERK and stress-activated MAPKs under some conditions to mediate cell survival. Apoptosis of PC12 cells is associated with p38 activation and is reduced by inhibiting p38/MAPK activity (Kummer et al., 1997; Xia et al., 1995). In addition, insulin promotes survival of cultured forebrain neurons concomitant with inhibition of p38 (Heidenreich and Kummer, 1996), and p38/MAPK activation has been shown to be involved in glutamate toxicity-induced neuronal apoptosis (Kawasaki et al., 1997). In global forebrain ischemia, p38/MAPK activation has been identified in microglial cells adjacent to dying, vulnerable neurons (Barone et al., 1999; Walton et al., 1998).
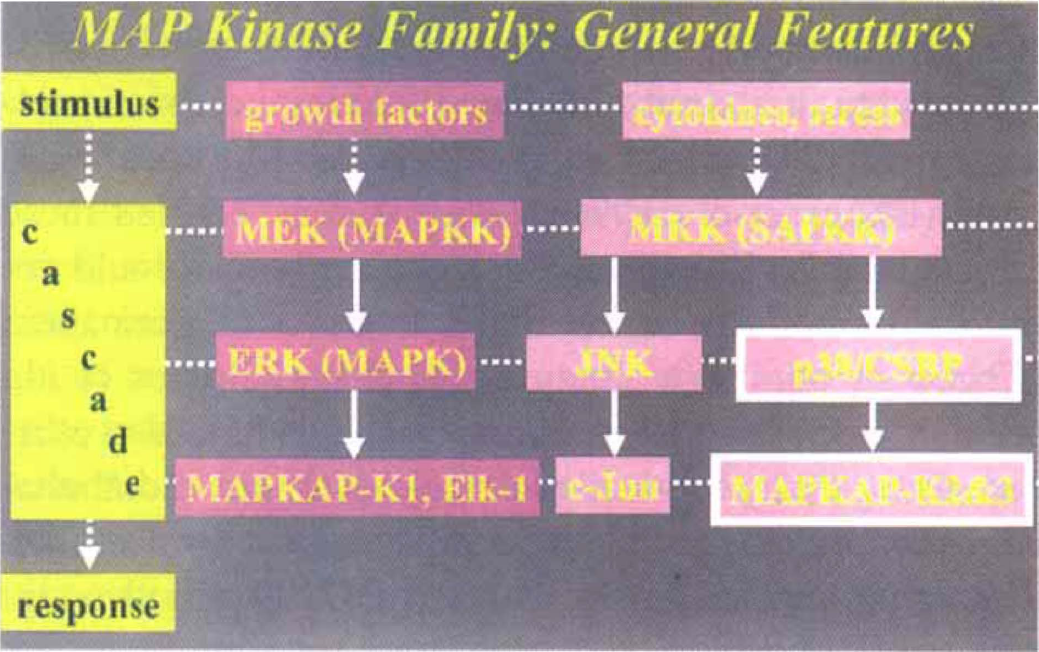
General features of the mitogen-activated protein kinase (MAPK) family. Stimuli, typically acting at the cell surface, provide second messenger signaling via a cascade of protein phosphorylations to result in a response, typically at the level of protein transcription or translation. Of particular interest is the stress activated MAP kinase pathway involving p38 MAPK which is selectively inhibited by cytokine suppressive anti-inflammatory drugs (CSAIDs). p38 MAPK was originally referred to as the CSAID Binding Protein; CSBP). CSAIDs inhibit the production of the inflammatory cytokines tumor necrosis factor (TNF) and interleukin (IL)-1. CSAIDs/p38 MAPK inhibitors act after p38 MAPK activation (via its phosphorylation from upstream kinases) to prevent p38 activation/phosphorylation of downstream kinases (e.g., MAPKAPK2). Thus, the cellular inflammatory cytokine production and apoptosis in response to various cellular stressors (e.g., ischemia or trauma) can be blocked. See text for details.
Recently, a new class of compounds, cytokine-suppressive anti-inflammatory drugs, have been developed which are potent inhibitors of TNF-α and IL-1β production (Young et al., 1993, 1997; Lee et al., 1993, 1994, 1996; Kassis and Prabhakar 1993; Kumar et al., 1997). Interestingly, these pyridinyl imidazole drugs have been shown to block translation of both TNF-α and IL-1β by mechanisms independent of cAMP. Cytokine suppressive anti-inflammatory drugs block the ability of activated (phosphorylated) p38 to phosphorylate downstream kinases in the signaling cascade (e.g., cytokine suppressive anti-inflammatory drugs block the activation of MAPKAP-K2&3; see Fig. 7) (Kumar et al., 1997). Not only do cytokine suppressive anti-inflammatory drug/p38 MAPK inhibitors block the production of IL-1β and TNF-α, but they also block the expression of several inflammatory proteins, including inducible nitric oxide synthase (Badger et al., 1998; Bhat et al., 1998) and the chemokine IL-8 (Lee et al., 1993).
Taken together, several aspects of the inflammatory cascade might be targeted by the inhibition of p38/MAPK. Certainly, apoptosis contributes significantly to ischemic stroke injury and its outcome (Li et al., 1998b; MacManus and Linnik, 1997; Morrison et al., 1998). In addition, the convergence of inflammatory cytokines and apoptotic pathways may result in reduced delayed neuronal death. (Hara et al., 1997; Sidotidefraisse et al., 1998). Thus, targeting p38/MAPK might be expected to provide an opportunity for intervention not only in early cytokine production but also to block delayed neuronal death by apoptosis. Very recently, we have identified significant neuroprotection by selectively inhibiting significant activation of p38 (Irving et al., 1999) and in focal brain ischemia, In Vivo (Barone et al., 1999).
SUMMARY
Throughout the last decade, a well-defined inflammatory reaction to diverse insults to the brain has been defined. Many aspects of this in situ and de novo inflammatory response has been enabled by application of molecular biologic techniques including PCR, cDNA library strategies and genetics in conjunction with highly specific probes (antibodies, DNA/RNA) that illustrate the comprehensive inflammatory capacity of the brain. Tumor necrosis factor-α and IL-1β emerge as reasonable candidates for pharmacologic manipulation in view of modulating the inflammatory response induced by stroke. The emerging understanding of the discrete molecular pathways in cytokines synthesis and their signaling mechanisms (e.g., p38/MAPK) already resulted in promising new opportunities for selective intervention in cytokine-mediated actions that are believed to contribute to histologic and functional outcomes of stroke and neurotrauma. It is hoped that rapid development of these compounds for clinical trials in stroke will result in new therapeutics for this devastating disease.