
Abstract
Increasing evidence supports a role for oxidative stress, proinflammatory cytokines, and apoptosis in the pathophysiology of focal ischemic stroke. Previous studies have found that the multi-action drug, carvedilol, is a mixed adrenergic antagonist, and that it behaves as an antioxidant and inhibits apoptosis. In the current study, the authors investigated whether carvedilol provides protection in focal cerebral ischemia and whether this protection is associated with reduced apoptosis and the downregulation of the inflammatory cytokines, tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β). Male Sprague-Dawley rats were subjected to transient middle cerebral artery occlusion (MCAO) by an intraluminal filament technique. Carvedilol (1, 3, and 10 mg/kg) was injected daily subcutaneously 2 or 4 days before the induction of ischemia. Neurologic scores, infarct volumes, TUNEL staining, and mRNA levels of TNF-α and IL-Iβ were assessed at 24 hours reperfusion. The effect of carvedilol on microvascular cortical perfusion was studied with continuous laser—Doppler flowmetry. Twenty-four hours after MCAO, carvedilol at all three doses reduced infarct volumes by at least 40% and reduced neurologic deficits on average by 40% compared with vehicle-treated controls when given 2 or 4 days before the induction of ischemia. This protection was not mediated by changes in temperature or blood flow. Treatment with all three dose regimens resulted in fewer TUNEL positive cells compared with controls. At 24 hours reperfusion, carvedilol decreased TNF-α and IL-1β expression by 40% to 50% in the ipsilateral ischemic cortex compared with the contralateral controls. The results of the current study indicate that carvedilol is neuroprotective in focal cerebral ischemia and may protect the ischemic brain by inhibiting apoptosis and attenuating the expression of TNF-α and IL-1β.
Keywords
The pathophysiology of ischemic stroke involves complex, interconnected processes including glutamate excitotoxicity, calcium overload, and free radical formation. In cerebral ischemia, increasing evidence supports a role for apoptosis (Savitz and Rosenbaum, 1998), which may represent a common pathway triggered by these events. In contrast to necrosis, apoptosis is an active process requiring the expression of death promoting genes to initiate a program of cellular autodestruction. In focal ischemia, necrosis occurs within the core of the infarct, whereas cell degeneration surrounding the core in the penumbra has some features of apoptosis (Linnik et al., 1994; Li et al., 1995; Du et al., 1996). In global ischemia, the delayed cell death that occurs several days afterward also has many features suggestive of apoptosis (Nitatori et al., 1995; Rosenbaum et al., 1998).
The inflammatory response after ischemia also contributes to neuronal damage, mediated in part by free radical injury. In addition, free radical generation can activate cell stress pathways and increase the expression of proinflammatory cytokines and apoptosis (Barone and Feuerstein, 1999). The inflammatory cytokines, tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), are up-regulated in models of focal ischemia (Barone and Feuerstein, 1999). Pretreatment with TNF-α or IL-1β exacerbates focal ischemic injury and neurologic deficits (Yamasaki et al., 1995; Barone et al., 1997; Stroemer and Rothwell, 1998), whereas blocking endogenous TNF-α or IL-1β after focal ischemia reduces neuronal damage (Rothwell et al., 1993; Barone et al., 1997). One potentially effective therapeutic intervention in stroke could interfere with apoptosis and proinflammatory cytokines, thus impacting on brain injury and inflammation.
Carvedilol [1-[carbazolyl-(4)-oxy]-3-[(2-methoxyphenoxyethyl)amino]-2-propanol], a multi-action drug indicated for hypertension and congestive heart failure, is an α and β adrenoceptor antagonist. It also behaves as a neuroprotective agent by exerting antioxidant properties in an in vitro model of free radical mediated neuronal injury (Lysko et al., 1992a; Yue et al., 1994a). Neuroprotection has also been shown in an in vivo global ischemic model in which pretreatment with carvedilol limited injury in the CA1 hippocampal zone (Lysko et al., 1992a; Yue et al., 1994b). In addition, carvedilol exerts antiapoptotic effects in the myocardium. Carvedilol prevents apoptosis in a model of myocardial ischemia—reperfusion injury (Yue et al., 1998), and the therapeutic effects of carvedilol for congestive heart failure have been attributed in part to an antiapoptotic mechanism (Ruffolo and Feuerstein 1998; Yue et al., 1999).
Given carvedilol's multiple actions, the authors investigated whether it would afford protection in transient focal ischemia, and whether this protection might be associated with reduced apoptosis and reduced expression of the inflammatory cytokines, TNF-α and IL-1β.
MATERIALS AND METHODS
Transient middle cereberal artery occlusion
Transient focal ischemia was induced by the intraluminal suture model (MCAO, middle cerebral artery occlusion) for 3 hours. Fasting adult male Sprague-Dawley rats (Taconic Farms, Germantown, NY, U.S.A.) weighing 250 to 275 g received anesthesia by inhalation of 3% halothane through a face mask in oxygen-enriched air. Body core temperature was maintained rectally at 37.0 ± 0.5°C by a heating blanket and heat lamp and followed until the animals became mobile.
For MCAO, the left common carotid artery and left external carotid artery were exposed through a midline neck incision. The left external carotid artery was coagulated. Then, the common carotid artery and internal carotid artery were separated from the adjacent vagus nerve. A 4–0 monofilament suture (Ethicon, Summerville, NJ, U.S.A.), whose tip had been rounded by heating near a flame, was inserted into the external carotid artery and advanced gently into the internal carotid artery past the MCA origin until the tip reached the proximal anterior cerebral artery, thus occluding the origin of the MCA (Zea Longa et al., 1989). After three hours of MCAO, the filament was removed and blood flow was restored as observed by laser doppler flow signal.
The right femoral artery was cannulated to monitor mean arterial blood pressure. A thermocouple was placed into the temporalis muscle to monitor possible changes in brain temperature as a consequence of drug or ischemia. All procedures followed the guidelines of the National Institutes of Health (Guide for the Care and Use of Lab Animals; DHEW DHHS Publication No. (NIH) 85–23, revised 1985, Office of Science and Health Rep, DRR/NIH, Bethesda, MD 20205).
Treatment with carvedilol
Carvedilol was dissolved in 100% DMSO, then diluted in a 0.9% saline solution (final concentration of DMSO 55%). DMSO/saline vehicle was administered as a control. Both control and drug were administered daily by subcutaneous injection 2 or 4 days before ischemia. Three sets of groups (total, n = 72) were studied. In each set, one group received vehicle (n = 8), the second group received carvedilol 2 days before ischemia (n = 8), and the third group received carvedilol 4 days before ischemia (n = 8). Animals received 1 mg/kg carvedilol in the first set of groups, 3 mg/kg in the second set, and 10 mg/kg in the third set.
Cortical microvascular perfusion
Cortical microvascular perfusion was measured by laser—Doppler flowmeter (Periflex PF3; Perimed, OH, U.S.A.). The laser—Doppler probe was positioned in the MCA territory at 1.5 mm posterior and 5 mm lateral to the bregma at the dural surface in the left hemisphere. Microvascular perfusion changes were continuously recorded on a multichannel chart recorder and normalized to basal (that is, pre-MCAO or 100%) perfusion.
Evaluation of infarct volume with TTC staining
Twenty-four hours after occlusion, the rats were anesthetized by inhalation of 3.0% halothane. The rats were then killed by decapitation, and brains were immediately removed, placed in a brain matrix, and cut in 2-mm coronal sections beginning 2 mm posterior to the anterior pole. A total of 5 slices were immersed in a saline solution containing 2.0% triphenyltetrazolium-chloride (TTC) at 37°C for 30 minutes and then fixed with 10.0% phosphate-buffered formalin solution. Each brain slice was scanned, and the unstained area in each image was delineated and quantified from a video image analyzing system (NIH Image, version 1.55). Infarct size was calculated as percentage of infarct area to total hemispheric area for each slice and by calculating infarct volume.
Evaluation of neurologic behavior
A graded neurologic exam as described by Bederson et al. (1986) was performed on all rats 24 hours after ischemia and before death by an observer who had no knowledge of which drug the animal received. A grading scale of 0 to 3 was used to assess the effects of occlusion. Scoring was graded as follows: 0 = no observable effect; 1 = forelimb flexion; 2 = decreased resistance to lateral push (and forelimb flexion) without circling; 3 = the same behavior as grade 2 with circling.
TUNEL staining
To visualize DNA fragmentation in a separate group of animals (n = 4) the TUNEL method, as described by Gavrieli et al. (1992), was used with these modifications: biotinylated-11-dUTP was obtained from Sigma, and Extra-Avidin-Peroxidase (Sigma, St. Louis, MO, U.S.A.) was diluted 1:200. Paraffin-embedded tissue was sectioned into 10-μm slices. Sections were incubated with 20 μg/mL proteinase K at room temperature. As a positive control, sections of normal brain were preincubated with DNase 1 (Sigma; 1 μg/mL in TM buffer; 50 mmol/L Tris-Hcl [pH 7.4], 10 mmol MgSO4, 0.1 mm dithiothrentol) for 10 minutes at 37°C. After three rinses in distilled water, the slides were processed according to the standard procedure. As a negative control, sections of the ischemic brain were used and processed after the standard procedure but biotinylated dUTP was omitted. Adjacent slices were stained with hematoxylin and eosin. The sections were examined by light microscopy in three random MCA areas in the inner border of the infarct, and the number of stained cells were counted per high-powered field by a blinded observer. TUNEL positive cells were considered apoptotic if undergoing cellular shrinkage and chromatin condensation based upon observations under light microscopy.
TNF-α and IL-1 expression
RNA isolation. In a separate group of animals (n = 3), 24 hours after transient focal ischemia, unfixed rat brain was removed. Tissue was sampled from areas that showed nonviable tissue as revealed by TTC staining in vehicle-treated control animals. The contralateral (control) and ipsilateral (reperfused) cortex was dissected from vehicle- and carvedilol 4 day treated (10 mg/kg) animals, fresh frozen, and stored at −80°C. To extract total RNA, samples were homogenized in approximately 1 mL of Trizol Reagent (Life Technologies, Grand Island, NY, U.S.A.) per 100 mg of tissue, according to manufacturer's instructions. Typical RNA yield was 0.5 to 0.75 μg/mg tissue.
Analysis of cytokine expression
Levels of cytokine mRNA expression in cortical samples were measured by Real Time Quantitative PCR (TaqMan PCR) as described previously (Wang et al., 2000a,b). Primers and probes for detection of mRNA were designed in Primer Express 1.0 (Perkin-Elmer Applied Biosystems, Norwalk, CT, U.S.A.) from available rat sequences [TNF-α (S40199), IL-1β (M98820), rpL32 (X06483)]: TNF-α forward (bp 495–515) 5′-CCA GGA GAA AGT CAG CCT CCT-3′; TNF-α reverse (bp 561–581) 5′-TCA TAC CAG GGC TTG A GC TCA-3′; TNF-α TaqMan probe (bp 527–552) 5′-(FAM)AGA GCC CTT GCC CTT GCC CTA AGG ACA CCC CT(TAMRA)-3′; IL-1β forward (bp 793–814) 5′-CAC CTC TCA AGC AGA GCA CAG A-3′; IL-1β reverse (871–849) 5′-GGG TTC CAT GGT GAA GTC AAC-3′; IL-1β TaqMan probe (823–848) 5′-(FAM)TGT CCC GAC CAT TGC TGT TTC CTA GG(TAMRA)-3′; rpL32 forward (bp 314–336) 5′-TGT CCT CTA AGA ACC GAA AAG CC-3′; rpL32 reverse (bp 365–385) 5′-CGT TGG GAT TGG TGA CTC TGA-3′, rpL32 TaqMan probe (bp 338–363) 5′-(FAM)TCG TAG AAA GAG CAG CAC AGC TGG CC-(TAMRA)-3′.
Reverse transcription reactions were conducted according to manufacturer's instructions with TaqMan Reverse Transcription Reagents and TaqMan Gold RT-PCR (reverse transcription polymerase chain reaction) kit (Perkin-Elmer Applied Biosystems) with pooled total RNA samples (3 samples from carvedilol-treated and 5 samples from vehicle-treated). Five milligrams pooled RNA was added to the final reaction mixture containing 1× TaqMan ET buffer, 5.5 mmol/L MgCl2, 500 mmol/L each dNTP, 2.5 mmol/L oligo d(T)16 primer, 0.4 U/mL RNase Inhibitor, and Multiscribe Reverse Transcriptase. Reverse transcription was carried out at 25°C for 10 minutes, 48°C for 30 minutes, and 95°C for 5 minutes. After reverse transcription, real time PCR was performed using the TaqMan Gold RT-PCR kit according to manufacturer's instructions. One-tenth of RT products were added to 1× TaqMan buffer, 5.5 mmol/L MgCl2, 200 mmol/L dATP/dCTP/dGTP, 400 mmol/L dUTP, 200 nmol/L primers, 100 nmol/L TaqMan probe, 0.01 U/mL AmpErase, and 0.025 U/mL AmpliTaq Gold DNA Polymerase. After initial incubations at 50°C for 2 minutes and 95°C for 5 minutes, thermal cycling proceeded at 95°C for 15 seconds and 60°C for 1 minute for 40 cycles in an ABI PRISM 7700 Detection System (Perkin Elmer Applied Biosystems). mRNA quantitation was determined by comparison with standard curves generated with known amounts of each target, and cytokine mRNA levels were normalized to the expression rpL32 (Wang et al., 2000a,b). Results are expressed as the mean ± SD from three PCR reactions.
Statistical analysis
Within each dosage group, the authors conducted pair-wise comparisons among the 2 day treated carvedilol group, 4 day treated carvedilol group, and the control group. Groups were compared using Student's t-test. A Mann—Whitney U test was used for the neurologic scores to make inferences regarding pair-wise comparisons. Differences were considered significant at P < 0.05. All data are expressed as mean ± SD.
RESULTS
Infarct volumes
Middle cerebral artery occlusion for 3 hours ischemia followed by 24 hours reperfusion resulted in extensive and reproducible hemispheric infarction throughout cortical and subcortical structures quantified 24 hours after MCAO. Treatment with carvedilol daily at 1 mg/kg, 3 mg/kg, or 10 mg/kg either 2 or 4 days before the onset of ischemia significantly reduced the infarct area and volumes compared with vehicle-treated controls at 24 hours. In the 1 mg/kg set, the mean volume of infarction was 285 ± 25 mm3 in the placebo group, 163 ± 19 mm3 in the 2 day carvedilol-treated group, and 164 ± 34 mm3 in the 4 day carvedilol treated group (Fig. 1A). For both time points, there was a 40% reduction in volume compared with controls (P < 0.05). In the 3 mg/kg set, the mean volume of infarction was 258 ± 15 mm3 in the placebo group, 145 ± 22 mm3 in the 2 day carvedilol-treated group, and 170 ± 26 mm3 in the 4 day carvedilol-treated group (Fig. 1B). For both time points, there was at least a 40% reduction in volume (P < 0.05). In the 10 mg/kg set, the mean infarct volume was 268 ± 20 mm3 in the placebo group, 112 ± 28 mm3 in the 2 day carvedilol-treated group, and 131 ± 40 mm3 in the 4 day carvedilol-treated group (Fig. 1C). For both time points, there was at least a 50% reduction in volume (P < 0.05).
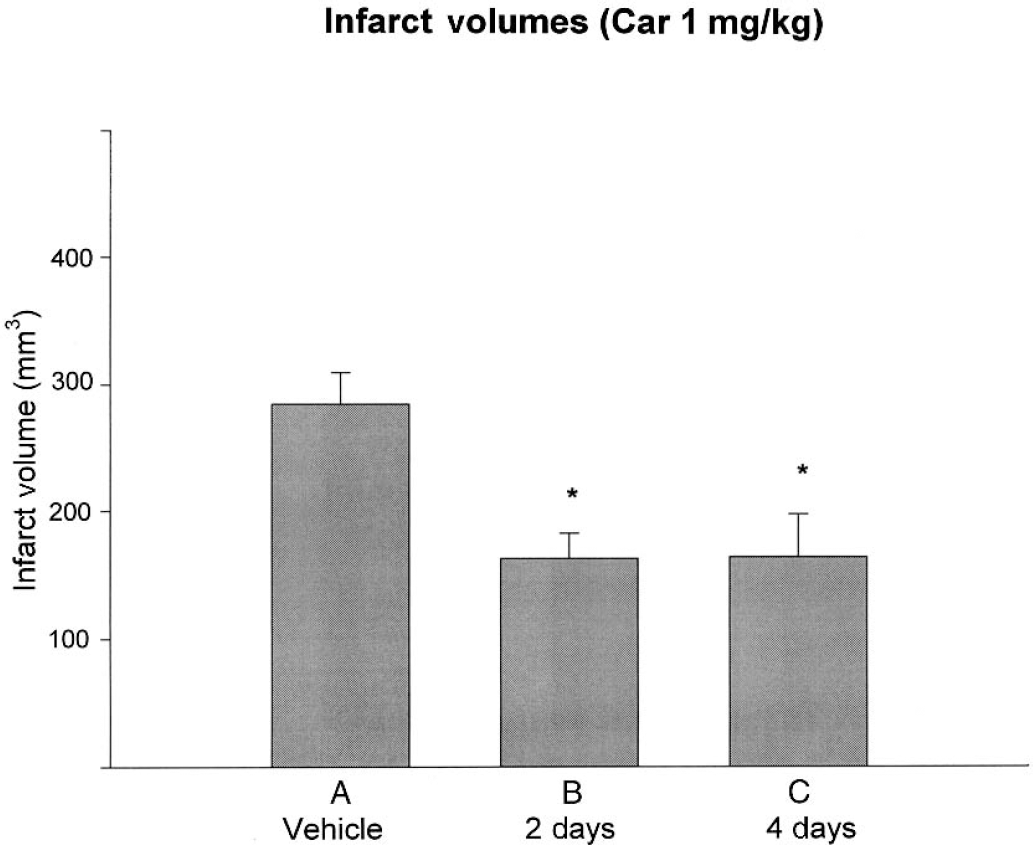
Effect of carvedilol at three different doses on infarct volume at 24 hours after 180 minutes of middle cerebral artery occlusion.
Neurologic scores
All vehicle-treated animals that underwent MCAO displayed behavioral defects before reperfusion of the ischemic territory. Clinical evaluation of the rats at 24 hours showed significantly reduced neurologic deficits in the animals treated with carvedilol compared with controls. In the 1 mg/kg set, the 2 day carvedilol group displayed a 33% improvement in neurologic function, and in the 4 day group there was a 46% improvement compared with controls (P < 0.05; Fig. 2A). In the 3 mg/kg set, the 2 day carvedilol group displayed a 42% improvement and the 4 day group displayed a 38% improvement compared with controls (P < 0.05; Fig. 4B). In the 10 mg/kg set, the 2 and 4 day carvedilol groups displayed a 45% improvement compared with controls (P < 0.05; Fig. 2C).
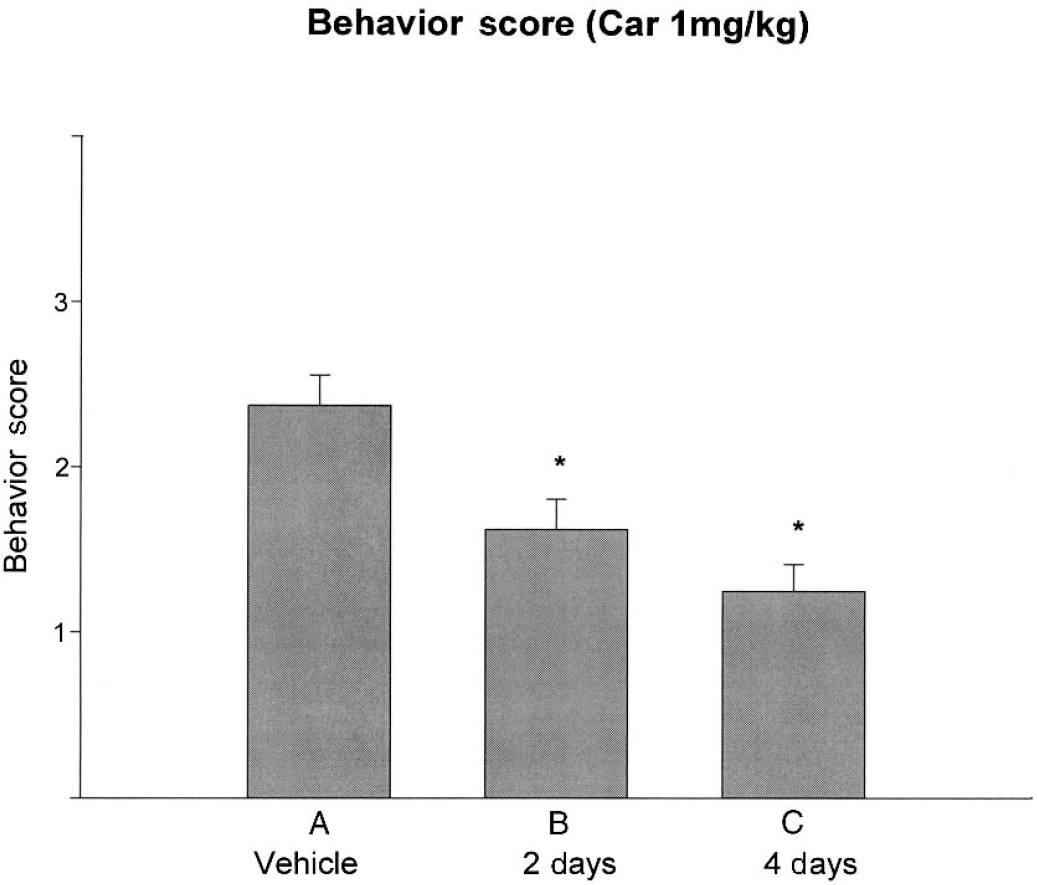
Deficits were quantified according to a neurologic scale widely used in the rat.
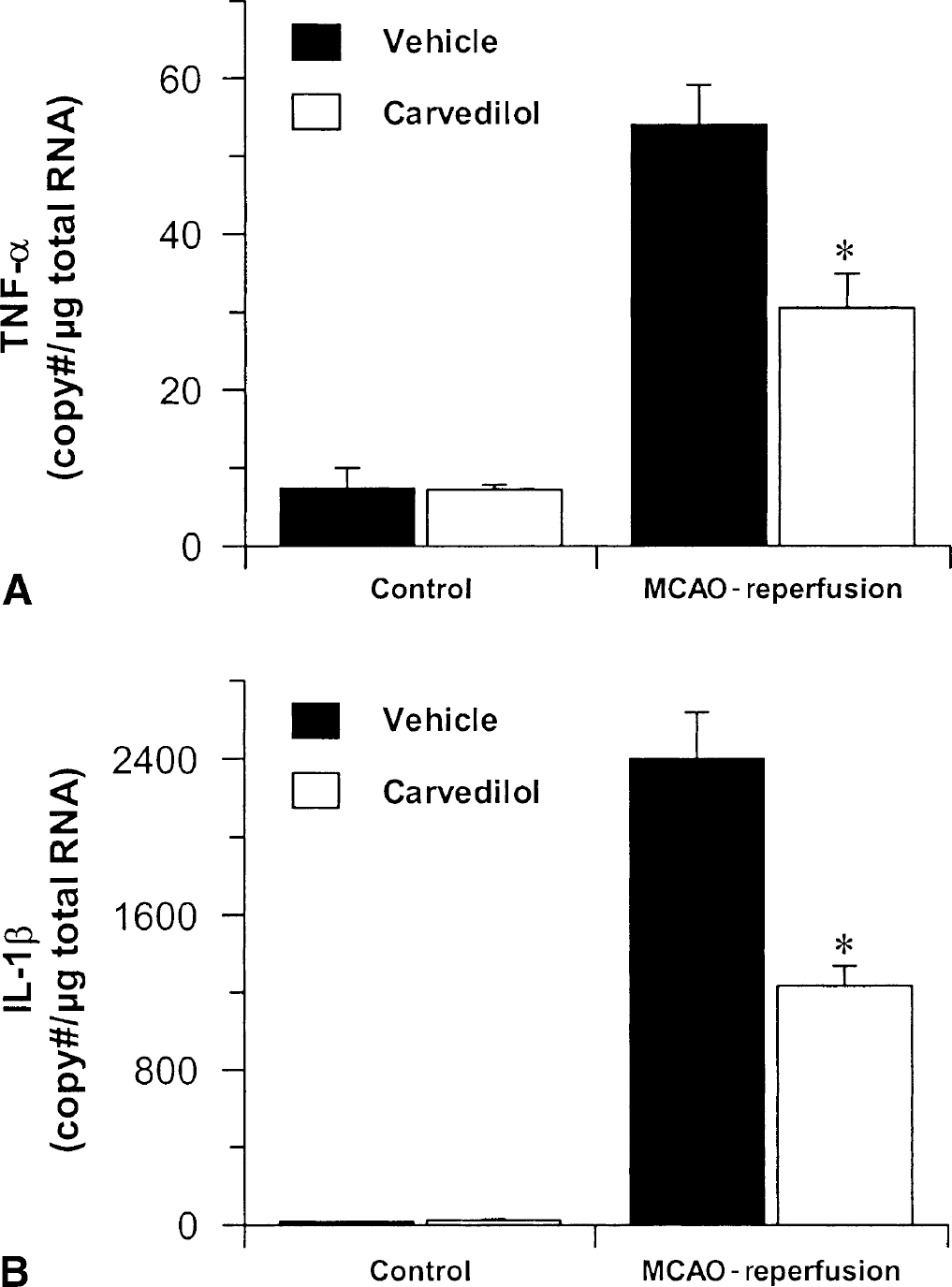
Effect of carvedilol on increased
Physiologic parameters
Up to 10 mg/kg carvedilol given daily 4 days before ischemia had no significant effect on arterial blood pressure (Table 1) or core temperature compared with vehicle-treated controls during MCAO.
Effect of carvedilol (10 mg/kg) on arterial blood pressure (mm Hg)

Cortical microvascular perfusion
Middle cerebral artery occlusion resulted in an 85% decrease in cortical perfusion as compared with baseline values. Treatment with carvedilol 10 mg/kg demonstrated no significant changes in percent decrease in cortical perfusion compared with vehicle-treated controls (Table 2).
Effect of Carvedilol (10 mg/kg) on microvascular cerebral perfusion (% baseline) as determined by laser Doppler

TUNEL staining
After 24 hours reperfusion, there was evidence of apoptotic cells as determined morphologically by light microscopy and by the TUNEL method. TUNEL positive cells were seen throughout the area of infarction (cortical and subcortical). Treatment with carvedilol at 1, 3, or 10 mg/kg resulted in fewer TUNEL positive cells compared with corresponding sections of control (Fig. 3). The vehicle-treated group had a mean number of TUNEL positive cells per high-powered field of 26.0 ± 17.3, which differed significantly from each of the carvedilol groups: 1 mg/kg (5.6 ± 4.7; P < 0.05), 3 mg/kg (8.0 ± 8.3; P < 0.05), and 10 mg/kg (4.6 ± 4.1, P < 0.05).
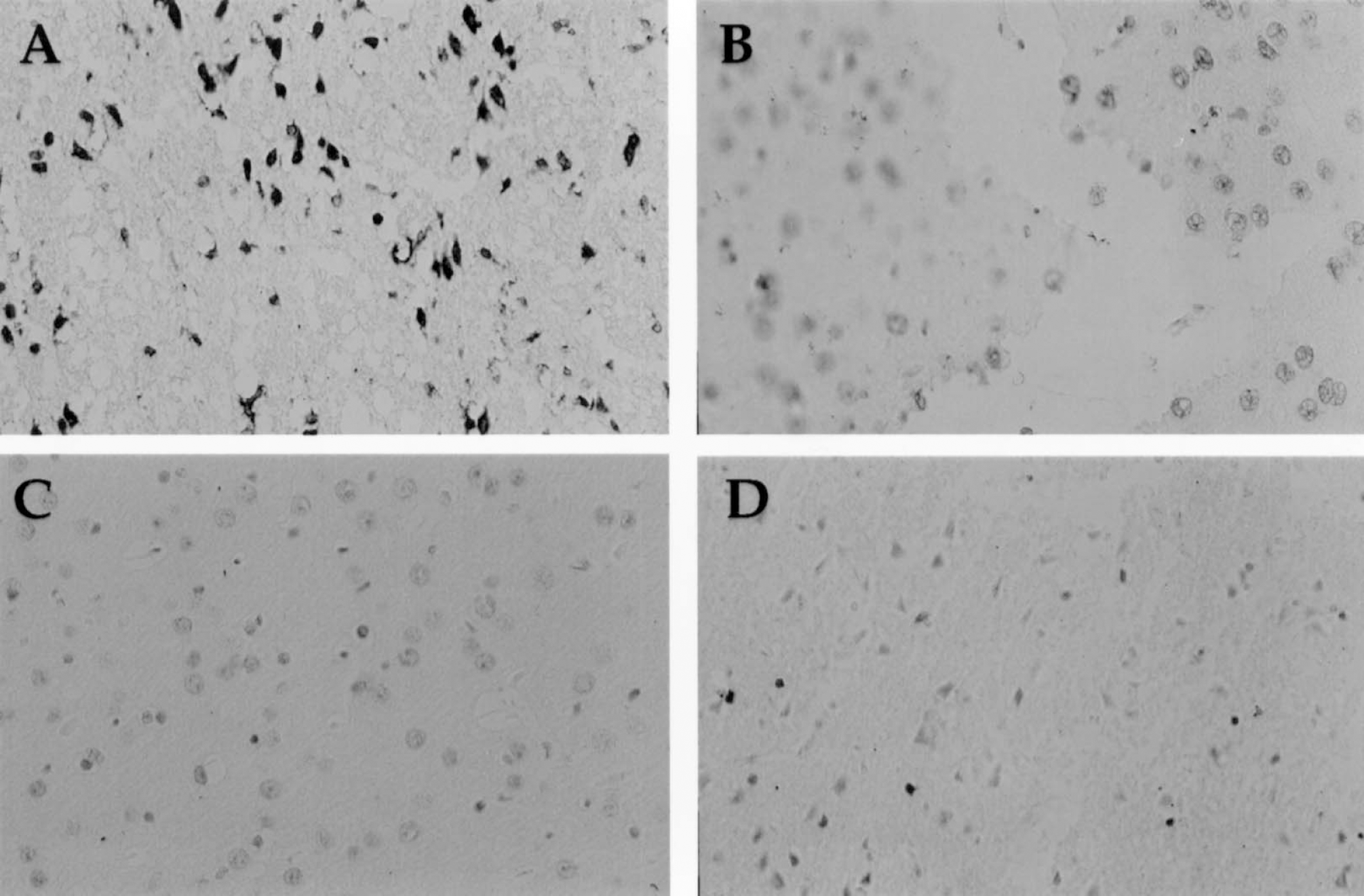
Representative TUNEL staining at 24 hours after 180 minutes of transient middle cerebral artery occlusion
TNF-α and IL-1 expression
To determine proinflammatory cytokine expression after transient focal ischemia, quantitative RT-PCR was performed on total RNA samples. A significant increase in TNF-α and IL-1β message was detected in the reperfused cortex in vehicle-treated animals compared with the control cortex. Tumor necrosis factor-α was expressed at a level of 54.0 (± 5.1) copies/μg RNA in reperfused cortex, compared with 7.3 (± 2.7) copies/μg RNA in the control cortex (Fig. 4A). Similarly, IL-1β expression in reperfused cortex was increased to 2396.0 (± 237.2) copies/μg RNA compared with 15.4 (± 1.2) copies/μg RNA in the normal cortex (Fig. 4B). Treatment with carvedilol (10 mg/kg) significantly attenuated the increase in cytokine expression after ischemia—reperfusion. Levels of TNF-α and IL-1β were approximately 40% to 50% less in carvedilol-treated reperfused cortex (30.5 (± 4.4) copies/μg RNA and 1232.0 (± 107.0) copies/μg RNA, respectively) compared with vehicle-treated controls (Fig. 4A and B).
DISCUSSION
The current study demonstrated that carvedilol over a relatively large range of doses markedly reduced infarct size and improved functional recovery after transient focal cerebral ischemia. There was no difference in the effects of treatment among the experimental groups and all dose regimens-pretreatment durations studied were neuroprotective in this model. These treatment doses result in plasma levels relevant to the clinically effective range (McPhillips et al., 1988). No significant side effects were noted in the treatment groups; in particular, the arterial blood pressure remained the same among the groups. Other studies have reported the neuroprotective effects of carvedilol in a global ischemia model, and it has also been shown to rescue the myocardium from ischemic injury (Lysko et al., 1992; Yue et al., 1994,1999). The current study is the first to demonstrate carvedilol's protective effects in focal ischemic stroke. This protection was not mediated by changes in temperature or blood flow (Table 2), which might have been expected because carvedilol is an alpha-1 adrenoceptor antagonist.
Oxygen free radicals play an important role in ischemic brain injury (Chan, 1996), and carvedilol is a known antioxidant in addition to being a vasodilator. Carvedilol prevents Fe-induced lipid peroxidation (Tadolini and Franconi, 1998), suppresses superoxide release from neutrophils (Yue et al., 1992), and scavenges hydroxy radicals (Yue et al., 1992). Carvedilol also protects cultured neurons from excitotoxicity and oxidative stress (Yue et al., 1994). The metabolites of carvedilol, SB 211475 and SB 209995, are more potent than the parent compound for inhibiting lipid peroxidation in rat brain homogenates (Yue et al., 1994; Feuerstein et al., 1994). Therefore, these metabolites may also be responsible for carvedilol's neuroprotective effects.
Oxidative stress is one of many signaling pathways that can activate neuronal apoptosis (Love, 1999). Increasing evidence supports a role for apoptosis in cerebral ischemia (Savitz and Rosenbaum, 1998). The current study supports an antiapoptotic role for carvedilol in focal ischemia as evidenced by fewer TUNEL positive cells in the treated groups compared with controls (Fig 3). There is also evidence that carvedilol prevents apoptosis in congestive heart failure (Yue et al., 1998). It should be noted that a TUNEL-positive reaction can occur in cells undergoing necrosis (Rink et al., 1995). However, in the current study we correlated the TUNEL staining with apoptotic morphology. It is possible, however, that the decrease in TUNEL positive cells emanates from reduced neuronal damage. Carvedilol's antiapoptotic effects may be related to its antioxidant properties. However, (Yue et al. 1998, 1999) have shown that carvedilol may inhibit ischemia-induced myocardial apoptosis by downregulating the stress-activated protein kinase (SAPK) pathway and the expression of the Fas receptor.
Another related mechanism to explain carvedilol's neuroprotective effects is the downregulation of inflammatory cytokine gene expression. The brain inflammatory response to injury is mediated in part by the proinflammatory cytokines, TNF-α and IL-1β. Both cytokines have been shown to be up-regulated and contribute to focal ischemic brain injury (Barone and Feuerstein, 1999). In the current study, we demonstrate that carvedilol significantly attenuates the focal ischemia-induced up-regulation of mRNA expression of TNF-α and IL-1β in the ischemic brain (Fig 4). However, we cannot discount the possibility that changes in cytokine expression produced by carvedilol may be secondary to smaller infarcts, and may not contributory to neuroprotection in the present studies.
Our results suggest a possible link between the downregulation of TNF-α and IL-1β and the prevention of apoptosis in focal cerebral ischemia. Tumor necrosis factor-α has been shown to promote apoptosis of cultured, differentiated neuroblastoma cells (SK-N-MC) (Talley et al., 1995) and the cytokine has been implicated as a mediator of neuronal death induced by HIV in AIDS complex dementia. New et al. (1998) have shown that the regulatory HIV protein Tat causes neuronal apoptosis of SK-N-MC in part through the release or synthesis of TNF-α. Other studies have shown that overexpressing the intracellular domain of the TNF receptor, p55, leads to apoptosis of PC12 cells (Haviv and Stein, 1998).
Tumor necrosis factor-α and IL-1β drive the brain inflammatory response and can produce toxic-free radicals. Tumor necrsosis factor-α activates microglia, circulating macrophages, and neutrophils, all of which can accelerate damage through the release of toxic oxygen free radicals and excitotoxins (Shalaby et al., 1985; McGeer et al., 1993). In addition to leukocyte activation, TNF-α and IL-1β participate in leukocyte recruitment to the ischemic site by up-regulating endothelial adhesion molecules. Interleukin-1β also increases the astrocytic release of arachidonic acid, which itself causes neuronal death (Okuda et al., 1994), potentiates glutamate-mediated toxicity (Miller et al., 1992), and leads to the release of free radicals. Evidence indicates that reactive oxygen species may play a physiologic role as second messengers by regulating the expression of important immunoregulatory transcription factors, such as NFkB and AP-1 (Schulze-Osthoff et al., 1995). Both IL-1 and TNF-α are among the genes responsive to NFkB (Savitz and Rosenbaum, 1999). Therefore, it is possible that Carvedilol decreases inflammatory cytokines through an antioxidant mechanism.
Carvedilol may also modify the release of inflammatory cytokines as an antiadrenergic agent. Immune cells express various adrenergic receptors sensitive to transmitters of the sympathetic nervous system (Hasko and Szabo, 1998). Beta-adrenergic stimulation prevents the release of IL-1 and TNF-α from activated macrophages and microglia (Hetier et al., 1991; Levi et al., 1993). Based on these observations, we might have expected Carvedilol to enhance neuroinflammatory damage; however, prolonged exposure to beta blockers supersensitizes the beta-adrenoceptor, enabling lower concentrations of neuroepinephrine and epinephrine to suppress cytokine release (Ignatowski and Spengler, 1995). It is also important to note that carvedilol is a calcium channel blocker (Lysko et al., 1992), an N-methyl-
In summary, the current study suggests that carvedilol administered for as short as 2 days in doses as small as 1 mg/kg reduces infarct volume and neurologic deficits after temporary focal cerebral ischemia. Carvedilol may not only provide effective antihypertensive therapy, but it may also provide additional benefits by protecting against free radicals, apoptosis, and/or the inflammatory cytokines and inflammatory response that contributes to tissue injury after stroke. Therefore, it may be particularly useful as a prophylactic agent for patients at risk for stroke.
Footnotes
Acknowledgment
The authors thank Dr. Giora Feuerstein for his advice and support in these studies.