
Abstract
Microvascular integrity is lost during focal cerebral ischemia. The degradation of the basal lamina and extracellular matrix are, in part, responsible for the loss of vascular integrity. Matrix metalloproteinases (MMPs) may play a primary role in basal lamina degradation. By using a sensitive modification of gelatin zymography, the authors investigated the activity of MMP-2 and MMP-9 in frozen 10-µm sections of ischemic and nonischemic basal ganglia and plasma samples of 27 non-human primates after middle cerebral artery occlusion/reperfusion (MCAO/R) for various periods. The gelatinolytic activities were compared with parallel cell dUTP incorporation in the ischemic zones of adjacent sections. In the brain, the integrated density of MMP-2 increased significantly by 1 hour after MCAO and was persistently elevated thereafter. Matrix metalloproteinase-2 expression was highly correlated with the extent of neuron injury and the number of injured neurons (r = 0.9763, SE = 0.004, 2P < 0.0008). Matrix metalloproteinase-9 expression only was significantly increased in subjects with hemorrhagic transformation. In plasma, only MMP-9 increased transiently at 2 hours of MCAO. These findings highlight the early potential role of MMP-2 in the degradation of basal lamina leading to neuronal injury, and an association of MMP-9 with hemorrhagic transformation after focal cerebral ischemia.
Keywords
Focal cerebral ischemia is responsible for loss in microvascular integrity manifested by major alterations in vascular permeability and vessel wall basal lamina structure (Hamann et al., 1996; Okada et al., 1994). The basal lamina, a constituent of the extracellular matrix (ECM), contributes to microvascular integrity by providing an immobilized scaffold of ligands for cellular adhesion and an anatomic barrier to cellular transmigration (Aumailley, 995). The variety and distribution of ECM components conforms architectural and functional heterogeneity among different tissues (Yurchenco and Schittny, 1990). In the central nervous system, the microvascular basal lamina is the primary vascular contribution to the ECM. The cerebral capillary wall contains type IV collagen, laminins, and fibronectin as the major basal lamina components, in addition to entactin, thrombospondin, nidogen, and heparan sulfates (Webersinke et al., 1992). The major components decrease roughly in parallel during experimental middle cerebral artery occlusion and reperfusion (MCAO/R) (Hamann et al., 1995). Loss of basal lamina integrity has been postulated to be the primary cause of microvascular hemorrhage during focal cerebral ischemia (Hamann et al., 1996; Okada et al., 1994).
The mechanisms responsible for basal lamina degradation after MCAO/R are not yet defined. Noncellular proteolytic systems, including plasmin, certain serine proteases, matrix metalloproteinases (MMP), and proteases secreted by activated polymorphonuclear (PMN) leukocytes, such as collagenase, gelatinase, elastase, and cathepsin-G, hydrolyze components of the basal lamina. The MMPs, Zn2+ endopeptidases that are secreted in latent form, require activation for proteolytic activity and are inhibited by specific tissue inhibitors of metalloproteinase (TIMPs) (Nagase, 1997). Matrix metalloproteinase-2 (gelatinase A, 72-kDa type IV collagenase) and MMP-9 (gelatinase B, 92-kDa type IV collagenase), which degrade type IV collagen and the laminins, have been implicated in the physiologic conditions of differentiation, ECM and tissue remodeling, and wound healing. In the inflammatory and invasive conditions of tumor metastasis and osteoarthritis, MMPs are involved when matrix alterations are central features (Aumailley, 1995). In the central nervous system, changes in MMP-2 or MMP-9 expression have been associated with brain tumor growth (Rao et al., 1993; Nakagawa et al., 1994), multiple sclerosis (Cuzner et al., 1996; Maeda and Sobel, 1996; Anthony et al., 1997), Alzheimer's disease (Peress et al., 1995), and amyotrophic lateral sclerosis (Lim et al., 1996).
Considering the distribution of ECM in the central nervous system, and the known venues of MMP-2 and MMP-9 activity, their contribution to brain tissue injury during ischemic stroke is likely to involve the microvasculature. In one recent study, MMP-9 was increased by 7 days in association with leukocyte invasion in the injury zone of patients who died shortly after stroke (Anthony et al., 1997). Rosenberg et al. (1996) demonstrated increased MMP-2 and MMP-9 activity during MCAO in Wistar-Kyoto rats and spontaneously hypertensive rats (SHR). Furthermore, experiments using large concentrations of bacterial collagenase (type VII, Sigma, St. Louis, MO, U.S.A.) suggest that MMP-9 may contribute to brain edema and hemorrhage subsequent to the disruption of the basal lamina (Rosenberg et al., 1990). The late appearance of MMP-2 and MMP-9 and the need for substantial exogenous MMP in those experiments are, thus far, inconsistent with the development of much earlier neuron, microvascular, and glial injury. In contrast, the disruption of normal cell—matrix adhesion may induce cell death (Frisch and Francis, 1994), edema (Rosenberg and Navratil, 1997), and loss of cell viability (Re et al., 1994), as demonstrated when adhesion of human endothelial cells to ECM is inhibited. Those findings suggest the possibility that endothelial cell, astrocyte, and neuron injury could follow from degradation of the microvascular basal lamina, and that mechanisms that contribute to early disruption of cell-matrix integrity may accentuate neuron injury. It would first be necessary to show that MMP secretion is enhanced after MCAO in concert with neuron injury.
In the present study, using a sensitive modification of gelatin zymography, we demonstrate rapid and significant increases in MMP-2 and MMP-9 content in the ischemic basal ganglia very early after MCAO in the awake non-human primate, in parallel with evidence of neuron injury. The hypothesis tested in this study is that the upregulation of MMP-2 or MMP-9 is related to neuron injury and to hemorrhagic transformation.
MATERIALS AND METHODS
The procedures used in this study were approved by the institutional Animal Research Committee and were performed according to standards published by the National Research Council (The Guide for the Care and Use of Laboratory Animals) and the U.S. Department of Agriculture Animal Welfare Act. Every effort was made to ensure that the subjects were free of pain and discomfort. All procedures were attended by the veterinarians, the primate handling staff, and principal investigator.
Experimental stroke model in the non-human primate
Cerebral tissues from adolescent male baboons (Papio anubis/cynocephalus) were used for these studies. All animals were neurologically normal before MCAO and apparently free of infections or inflammation during the experiments. The experimental approaches to the awake MCAO/R stroke model have been detailed in previous studies of this group (del Zoppo et al., 1986; Hamann et al., 1995). All animals were allowed a 7-day procedure-free interval after surgical implantation of the balloon device before entry into the experiment. The experimental procedure required inflation of the MCA balloon for 1 hour or 2 hours, or for 3 hours with various periods of subsequent MCA reperfusion. Seventeen subjects underwent MCAO for 1 hour (n = 3) or 2 hours (n = 6), or 3 hours MCAO with subsequent reperfusion for 1 hour (n = 3), 4 hours (n = 2), or 24 hours (n = 3). In addition, a control group (n = 5) did not undergo any preparation procedure, and a group of subjects (n = 5) that suffered lenticulostriatal injury at surgical implantation were harvested 7 days later. An additional single sham-operated subject underwent the implantation procedure and the 7-day recovery period, but did not undergo MCAO.
Tissue processing
Tissues were removed from the animals during sodium thiopental anesthesia by left ventricular transcardiac perfusion of the cranial structures at 180 to 200 mm Hg with chilled (4°C) perfusate containing heparin (200 IU/L), nitroprusside (6.7 µmol/L), and bovine serum albumin (50 g/L; Sigma). Cranial tissues were removed en bloc within 15 minutes of complete perfusion and subdivided in 1-cm coronal slices. From each slice, symmetrically located blocks (1.0 × 1.0 × 0.2 to 0.5 cm) of the basal ganglia were cut and embedded in Tissue-Tek OCT compound (Miles, Inc., Elkhart, IN, U.S.A.), frozen in 2-methylbutane and dry ice, and stored at −80°C until sectioning.
Blood sampling and preparation
Blood samples were drawn from a peripheral vein into plain, EDTA, and heparinized tubes before surgical implantation, before MCAO, serially during MCAO, and during reperfusion. The total white cell count, white cell differential count, platelet count, and hematocrit were determined on commercial whole-blood analyzers. Plasma was isolated from blood in EDTA and heparinized tubes, by centrifugation at 3,000 rpm for 15 minutes, and stored at −80°C until analysis.
Preparation of samples for zymography
For zymography, heparinized plasmas from eight subjects at baseline (before MCAO) and 2 hours of MCAO were analyzed together with plasmas from other times (3 hours of MCAO with 1, 3, and 24 hours reperfusion). Separately, consecutive 10-µm cryostat sections from the ischemic and nonischemic basal ganglia were minced and dissolved in 120 µL of homogenizing buffer (50 mmol/L Tris-Hcl [pH 7.5], 75 mmol/L NaCl, 1 mmol/L phenylmethyl sulfonyl fluoride [PMSF; Sigma)). The homogenate was centrifuged at 4°C for 20 minutes at 9,000 rpm. Supernatants were divided into aliquots and stored at −80°C until analyzed. The protein concentration of each sample was measured with the Bio-Rad protein assay kit based on the method of Bradford (Bio-Rad Laboratories, Hercules, CA, U.S.A.) using a bovine gamma globulin standard.
Gelatin zymography
Gelatin zymography was performed based on methods modified from those previously published (Hibbs et al., 1985; Kleiner and Stetler-Stevenson, 1994; Nakagawa et al., 1994). For brain samples, the volume of supernatant containing 18 µg of protein was diluted in homogenizing buffer and mixed with an equal volume of sample buffer (80 mmol/L Tris-HCl [pH 6.8], 4% sodium dodecyl sulfate [SDS], 10% glycerol, 0.01 % bromphenol blue). For plasma samples, 2.5 µL heparinized plasma was diluted in 7.5 µL collagenase buffer (50 mmol/L Tris [pH 7.5], 20 mmol/L NaCl, 0.01% Brij-35) and mixed with an equal volume of sample buffer. The appropriate amount of plasma required was determined after a preliminary zymographic study with serial dilutions of plasma, such that the amounts provided gelatinolytic activities in the linear range and yielded approximately 2.2 µg protein. For standardization, 50 pg recombinant human MMP-2 and MMP-9, which were diluted in collagenase buffer and mixed with equal volumes of sample buffer, were loaded on each gel. For electrophoresis, 8% SDS-polyacrylamide resolving gels containing 1 mg/mL gelatin were overlaid with 5% stacking gels, and samples were loaded and run at 4°C (25 mA per gel). After electrophoresis, gels were rinsed with distilled water briefly and washed three times in 150 mL 2.5% Triton X-100 solution (15 minutes each) on a rotary shaker. The gels were then incubated in 250 mL of 50 mmol/L Tris-HCl (pH 7.5) containing 10 mmol/L CaCl2 and 0.02% NaN3 at 37°C for 42 hours. In separate experiments, 10 mmol/L EDTA, 5 mmol/L 1,10-phenanthroline (Sigma), 2 mmol/L PMSF, or 1 µmol/L trans-epoxysuccinyl-
Scanning of gels and quantitation of gelatinolytic bands
Gels were scanned using a Personal Densitometer SI (Molecular Dynamics Inc., Sunnyvale, CA, U.S.A.) and quantified by the NIH Image 1.61 Program staged on a Macintosh platform. Each gel was scanned with a step tablet (Kodak Scanner Step Tablet, ST-34; Eastern Kodak Co., Rochester, NY, U.S.A.) under the mode of 12 bits per pixel digital resolution and 100-µm pixel size, after calibration. This mode provides 4,096 levels of digital resolution and 1,000 data points/cm2. The band intensities were determined as described in the manual of the NIH Image Program. Briefly, the software was calibrated each time for each gel using the simultaneously scanned step tablet and linearized. After calibration, each band was curved using the gel-plotting macros. The measured value of the height of the curve at any point is the mean of the optical densities of a given row of pixels in the marked line and is expressed as the integrated density.
Intragel and intergel reproducibility and normalization of the measured integrated density
Preliminary experiments examining serial concentrations of recombinant human MMP-2 and MMP-9 (from 1 to 1,000 pg) demonstrated that this technique detected as little as 5 pg of recombinant human MMP. The integrated densities were linear with regard to MMP concentration (Fig. 1), and brain section extracts or plasma MMP activities were all within that range.
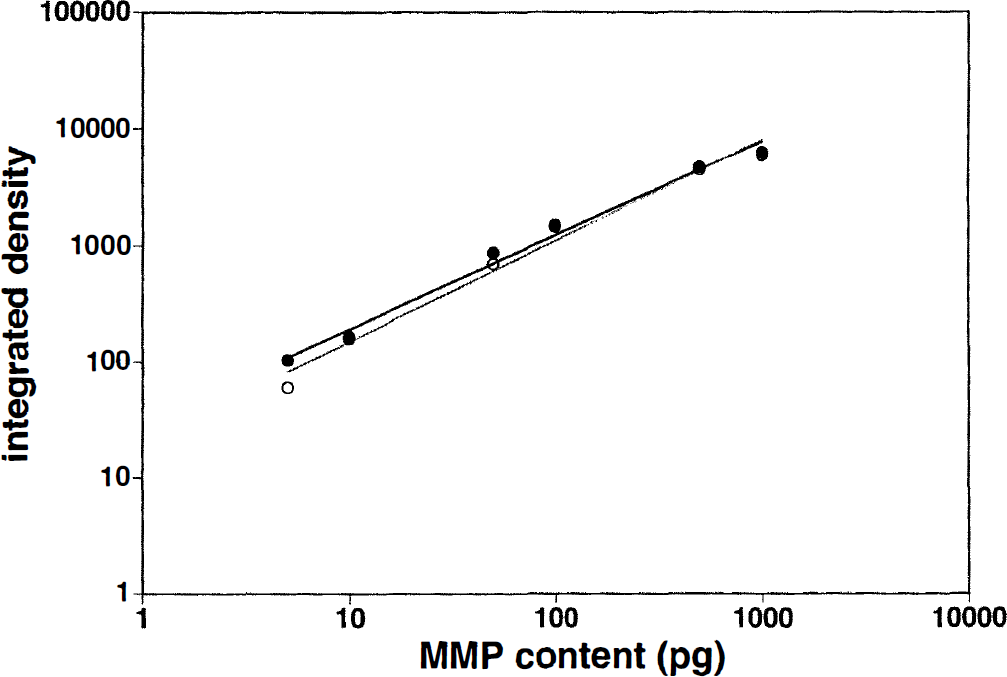
Examples of the relationship between the content of recombinant MMP-2 (closed circles) and MMP-9 (open circles) and the measurements of gelatinolytic activities (integrated density; see Materials and Methods).
Intragel reproducibility for protein extracts and plasmas was acceptable, and coefficients of variance of 4.8% ± 3.3% (n = 4; range, 0.48% to approximately 10.7%) were obtained. Standardization of electrophoresis running times and staining and destaining procedures was made to insure intergel reproducibility. The intergel reproducibility of 50 pg recombinant human MMP-2 and MMP-9 standards was satisfactory, providing coefficients of variance of 13.9% and 12.6%, respectively (n = 10).
Normalization of integrated densities was performed by dividing values obtained from each sample by the fraction of recombinant human MMP-2 and MMP-9 standards run in parallel in the same gel.
dUTP incorporation and determination of the region of dUTP-incorporation
Evidence of nuclear DNA scission and repair was taken as an indication of significant cellular injury as previously described (Tagaya et al., 1997). Incorporation of digoxigenin-dUTP on 10-µm cryosections was detected by the DNA polymerase I-based procedure (Tagaya et al., 1997). Cryosections adjacent to those used for zymography were fixed with 10% neutral-buffered formalin. After washing, sections were treated with 2% H2O2 for 5 minutes and incubated with 0.1 U/µL DNA polymerase I (Promega, Madison, WI, U.S.A.) and digoxigenin DNA labeling mixture (Boehringer Mannheim Corp., Indianapolis, IN, U.S.A.) in translation buffer (50 mmol/L Tris-HCl [pH 7.5], 10 mmol/L MgSO4 50 µg/mL bovine serum albumin) at 37°C for 60 minutes. After washing with phosphate-buffered saline, the sections were incubated with horseradish peroxidase-conjugated antidigoxigenin antibody (Boehringer Mannheim Corp.) for 30 minutes. After further washing with phosphate-buffered saline, color was developed with 3-amino-9-ethyl carbazole (AEC kit, Biomeda Corp., Foster City, CA, U.S.A.). The extent of regions of dUTP incorporation (Ic) was marked on the slides by an investigator not involved in these studies, and the slides were scanned under the mode of 12 bits per pixel digital resolution and a 50-µm pixel size. This mode provided satisfactory images of sections with 4,096 levels of digital resolution and 4,000 data points/cm2. The total section and Ic region surface areas were measured using the NIH Image Program.
Statistical analysis
All data are presented as the mean ± SD. The Mann-Whitney U Test was used for comparison between hemorrhagic and nonhemorrhagic subjects. Associations between MMP (integrated density) and ischemic injury or leukocyte counts were performed by means of linear regression. Jackknifed standard errors (SE) of reported correlation coefficients are provided for assessment of the precision of the estimated correlations. As in the past (Tagaya et al., 1997; Wagner et al., 1997), for temporal studies data were subject to square root transformation (to in duce approximate normality) to which analysis of variance with Bonferroni corrections for multiple comparisons was applied. Significance was set at 2P < 0.05.
RESULTS
MMP-2 and MMP-9 during focal cerebral ischemia
Gelatin zymographic studies of frozen 10-µm sections from ischemic and nonischemic basal ganglia displayed clear evidence of MMP-2 and MMP-9 activity (Fig. 2). Four bands were identified, each of which was inhibited by EDTA or the Zn2+ chelator 1,10-phenanthroline, but not by the serine protease inhibitor PMSF, or the cysteine protease inhibitor E-3132. These findings indicate that the activities in these bands were exclusively because of MMP. Furthermore, MMP-9 activity was inhibited by a function-blocking antibody against human MMP-9 (no equivalent antibody was available for MMP-2).
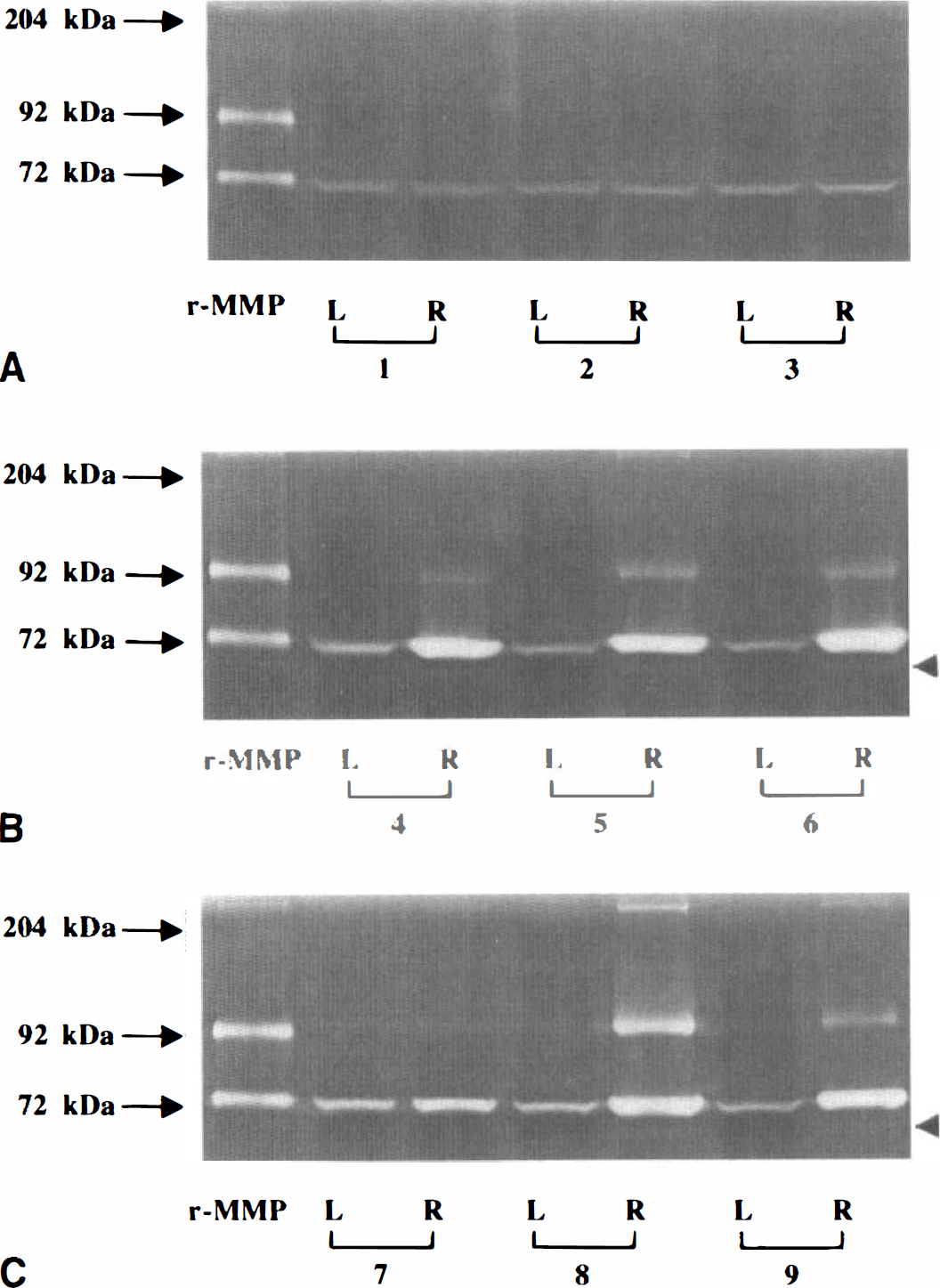
Gelatinolytic activities in protein extracts from frozen 10-µm sections of basal ganglia. Representative zymograms of nonischemic (L) and ischemic (R) basal ganglia from three subjects each undergoing 2 hours of MCAO
An approximately 72-kDa (70-kDa) band representing the inactive form of MMP-2 (Kenagy et al., 1996) was observed in all nonischemic tissues from control subjects and those undergoing MCAO. The other three bands were observed only in subjects undergoing MCAO. These included lysis zones above the 204-kDa myosin marker, just above the 92-kDa band of recombinant human MMP-9 (100 kDa), and below the 72-kDa band of recombinant human MMP-2 (60 kDa) (Kenagy et al., 1996) (Fig. 2; Table 1). The slowest migrating band, seen in 22.7% (5 of 22) of nonischemic and 72.7% (16 of 22) of ischemic basal ganglia samples most likely represents a dimer of MMP-9. The approximately 100-kDa band corresponds to the latent form of MMP-9 and was recognized in 68.2% (15 of 22) of nonischemic and 95.5% (21 of 22) of ischemic basal ganglia samples. The 60-kDa band was often very faint and corresponds to the active form of MMP-2. It was identified in 59.1% (13 of 22) of ischemic basal ganglia samples, but not in nonischemic basal ganglia samples. The integrated density of the active form of MMP-2 comprised 2.1% ± 1.6% (range, 1.03% to 6.88%) of all forms of MMP-2 and was correlated directly with that of latent MMP-2 (r = 0.612, 2P = 0.0007). The active form of MMP-9 was not identified in this study.
Pattern of matrix metalloproteinase expression
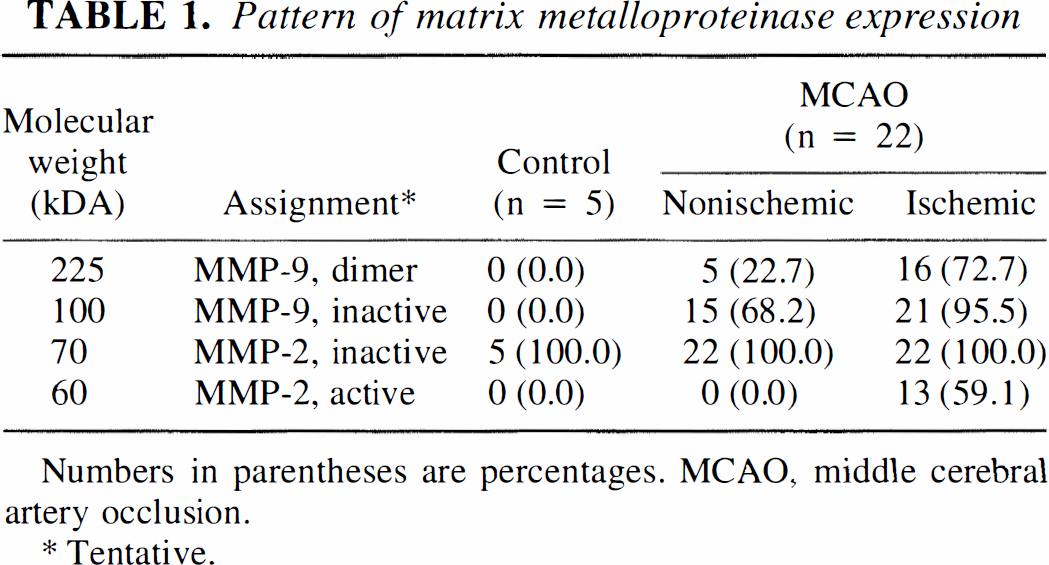
Numbers in parentheses are percentages. MCAO, middle cerebral artery occlusion.
Tentative.
Relationship of MMP to ischemic injury
Latent MMP-2 was significantly increased in the ischemic basal ganglia 1 hour after MCAO compared with baseline (controls), and remained persistently elevated thereafter (Table 2; Figs. 2 and 3). No significant change in MMP-9 activity was apparent in the ischemic basal ganglia during the same period (2P = 0.11).
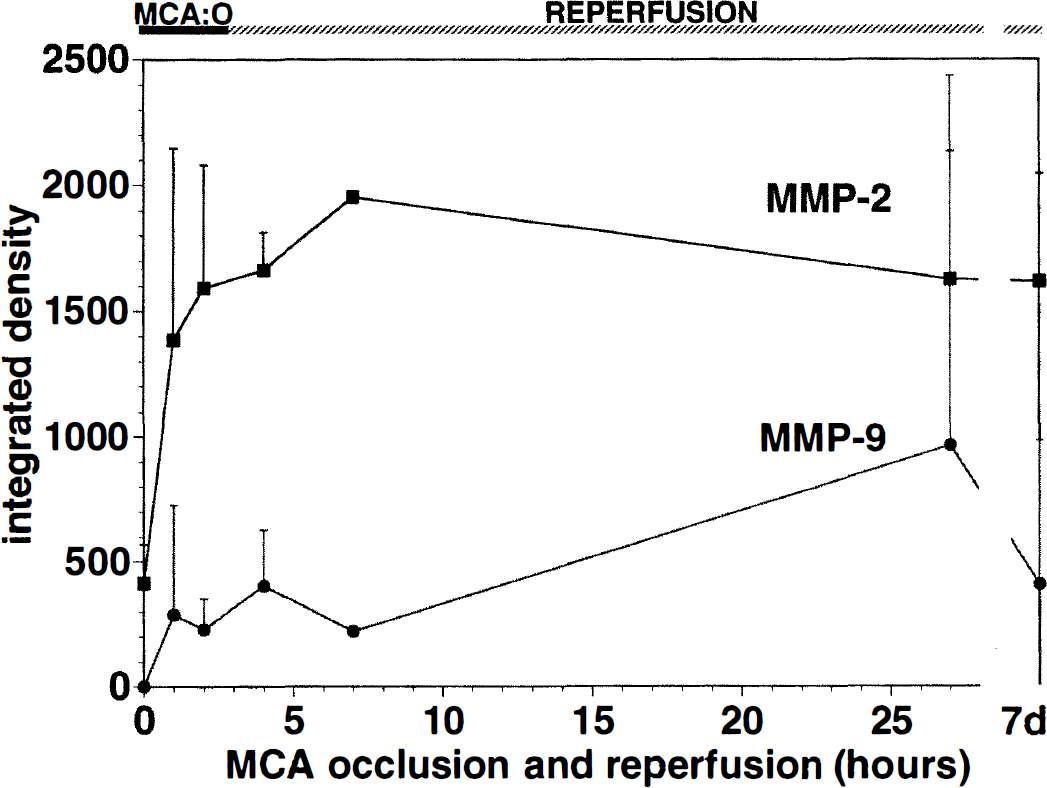
Effect of MCAO/R on the integrated densities of latent MMP-2 and MMP-9 in the brain. A very early significant increase in MMP-2 is shown (see Table 2).
Activity of matrix metalloproteinases in the brain
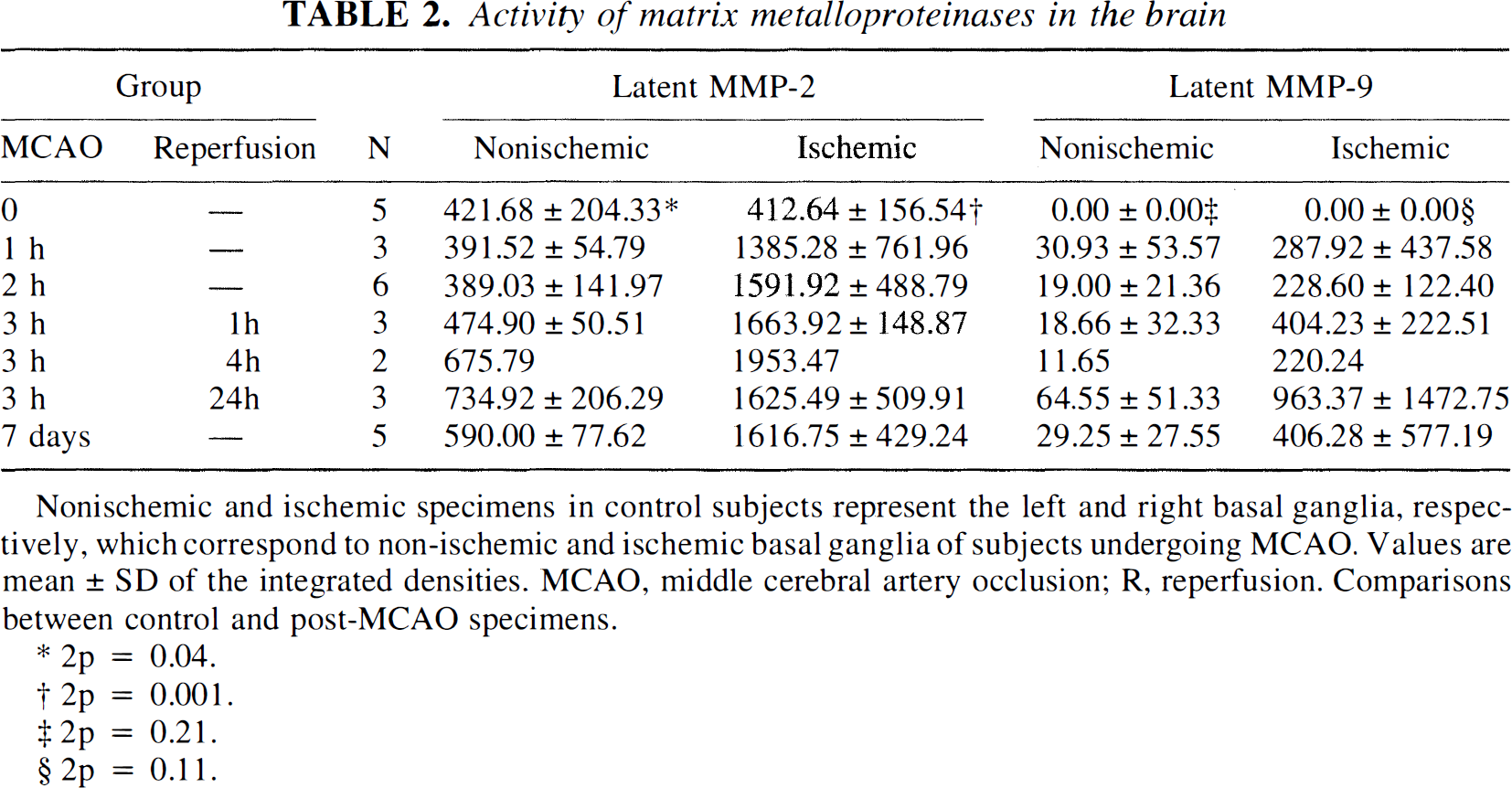
Nonischemic and ischemic specimens in control subjects represent the left and right basal ganglia, respectively, which correspond to non-ischemic and ischemic basal ganglia of subjects undergoing MCAO. Values are mean ± SD of the integrate'd densities. MCAO, middle cerebral artery occlusion; R, reperfusion. Comparisons between control and post-MCAO specimens.
2p = 0.04.
2p = 0.001.
2p = 0.21.
2p = 0.11.
The activities of the latent forms of MMP-2 and MMP-9 displayed distinctly different relationships to ischemic injury. The integrated densities of latent MMP-2 at any time showed a very strong linear correlation with the extent of the Ic region defined by the region containing dUTP-labeled cells (r = 0.8535, SE = 0.078, 2P < 0.0001) (Fig. 4). The relationship of latent MMP-2 to the total number of dUTP-positive cells at 2 hours of MCAO, when 80.0% ± 6.6% were microtubule-associated protein-2-positive neurons in this territory, was very strong (r = 0.9763, SE = 0.004, 2P = 0.0008) (Tagaya et al., 1997). In contrast, latent MMP-9 showed a much weaker correlation (r = 0.455, SE = 0.289, 2P = 0.033) with respect to the Ic region at all times, but a very strong correlation with the number of dUTP-positive cells at 2 hours of MCAO (two subjects had evidence of hemorrhage). In the sham-operated control, there was no difference in the levels of latent MMP-2, the only recognized band, between the operated and nonischemic basal ganglia.
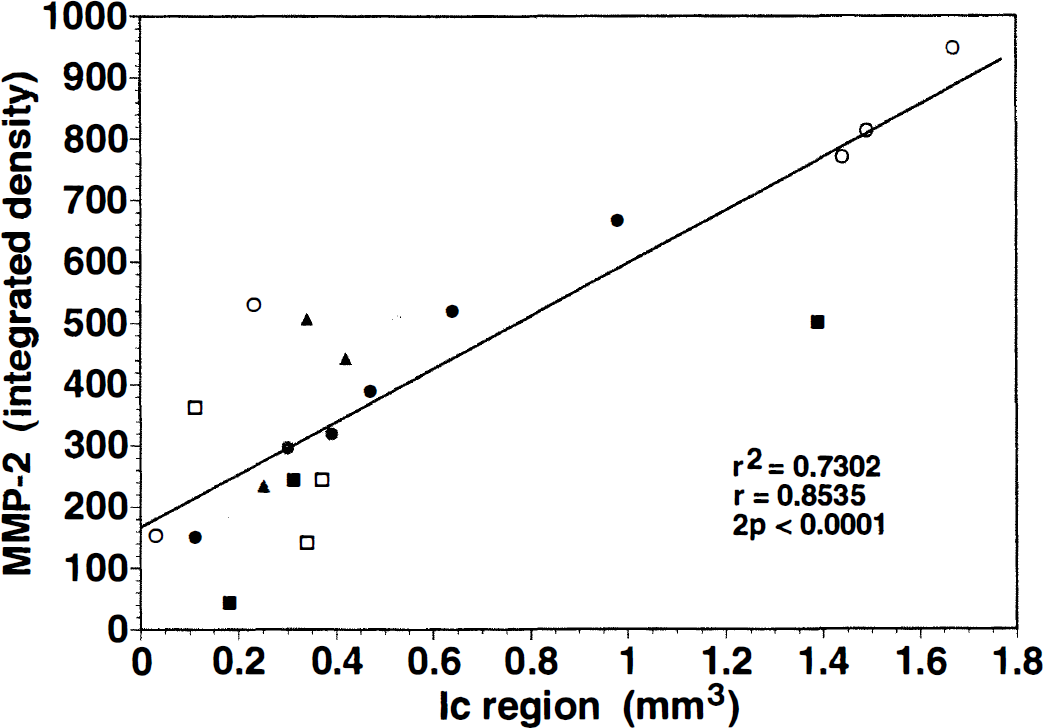
Correlation between MMP-2 activity and the extent of ischemic injury, defined as the volume of an adjacent frozen 10-µm section displaying cellular dUTP incorporation (Ic region) (r = 0.8535, SE = 0.078, 2P < 0.0001).
Association of MMP with hemorrhagic transformation
At least one subject at each time, eight total (36.4%), demonstrated visible hemorrhagic transformation of the Ic region after MCAO (Fig. 5). The integrated density of the latent form of MMP-9 was significantly increased in the ischemic basal ganglia of those subjects displaying hemorrhagic transformation (773.10 ± 873.57) compared with those without hemorrhagic transformation (187.52 ± 174.27; 2P = 0.013) (Fig. 6). MMP-9 activity in the contralateral nonischemic basal ganglia was not significantly increased in the subjects with hemorrhagic transformation (37.97 ± 35.67 versus 23.02 ± 32.78, 2P = 0.33). No association of MMP-2 with hemorrhagic transformation was noted (1,685.43 ± 793.27 versus 1,504.79 ± 177.50, 2P = 0.57).
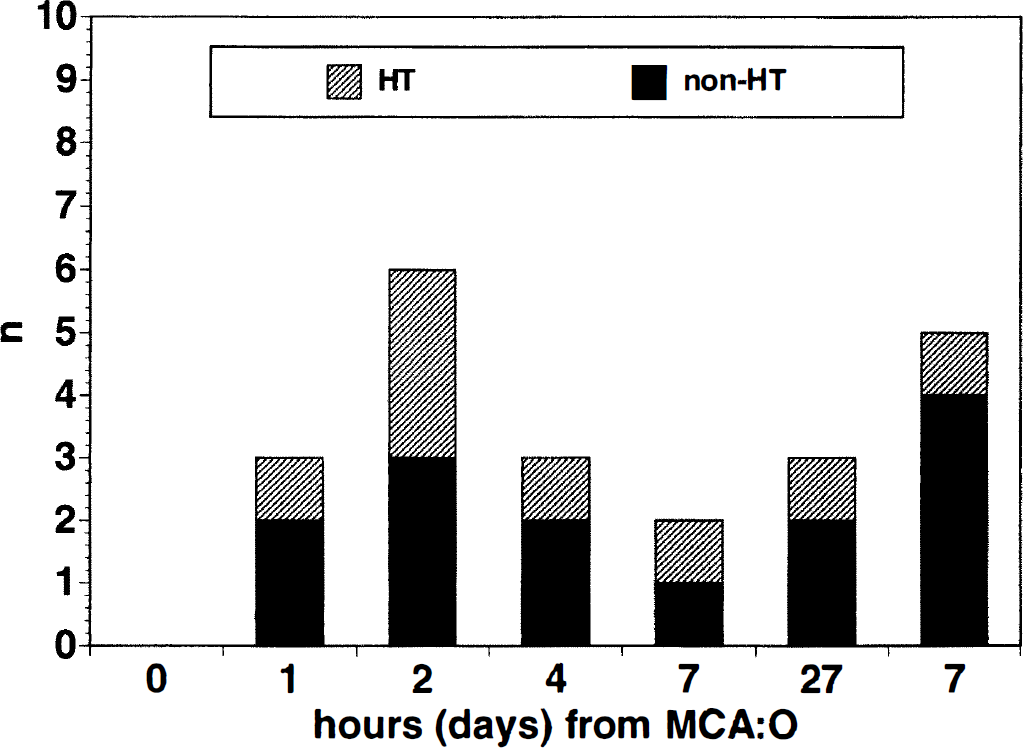
Distribution of subjects with hemorrhagic transformation. At least one MCAO subject at each time displayed evidence of hemorrhagic transformation.
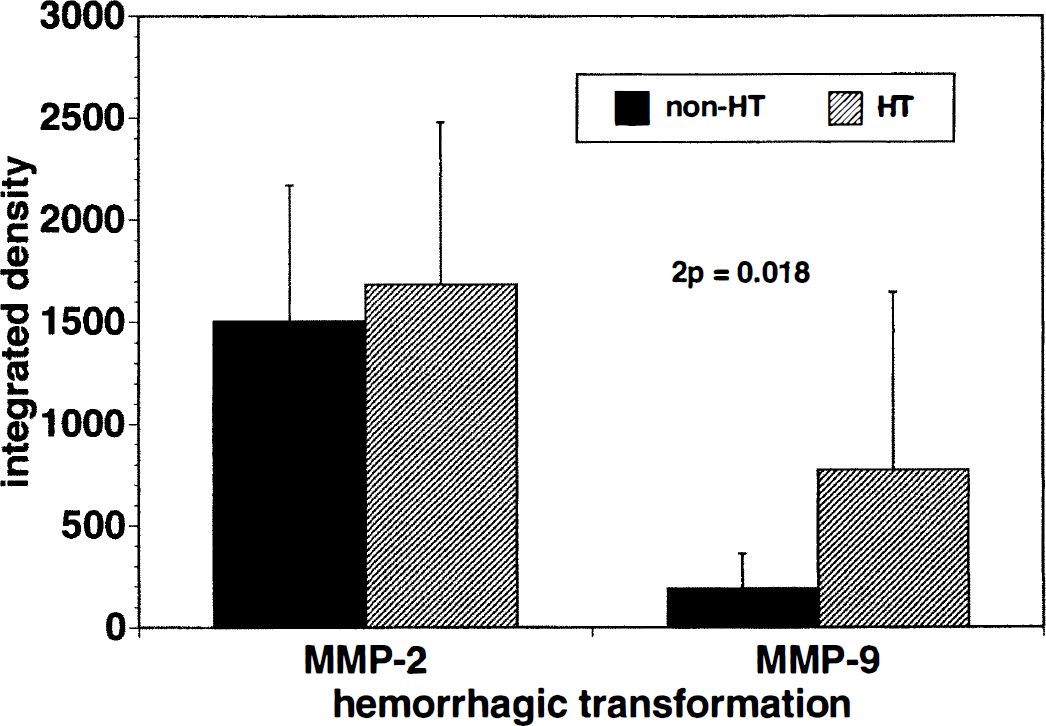
Matrix metalloproteinase activities in subjects with and without hemorrhagic transformation. Activity of MMP-9 was significantly increased in subjects with hemorrhagic transformation (2P =0.018).
MMP-2 and MMP-9 in plasma
The four MMP bands were identified in all plasma samples. Besides these, a band above 121 kDa (approximately 130 kDa), whose identity is uncertain, was seen in 60.0% (15 of 25) of the plasma samples (Fig. 7). A complex of 100-kDa MMP-9 and neutrophil gelatinase-associated lipocalin is conceivable (Kjeldsen et al., 1993). Alternatively, the dimer of active MMP-2 (Vartio and Baumann, 1989) or a complex of MMP-9 and TIMP-1 (Moll et al., 1990) may contribute to this band. The latent forms of MMP-2 and MMP-9 were found in the plasmas of all nonischemic control subjects, and did not differ substantially.
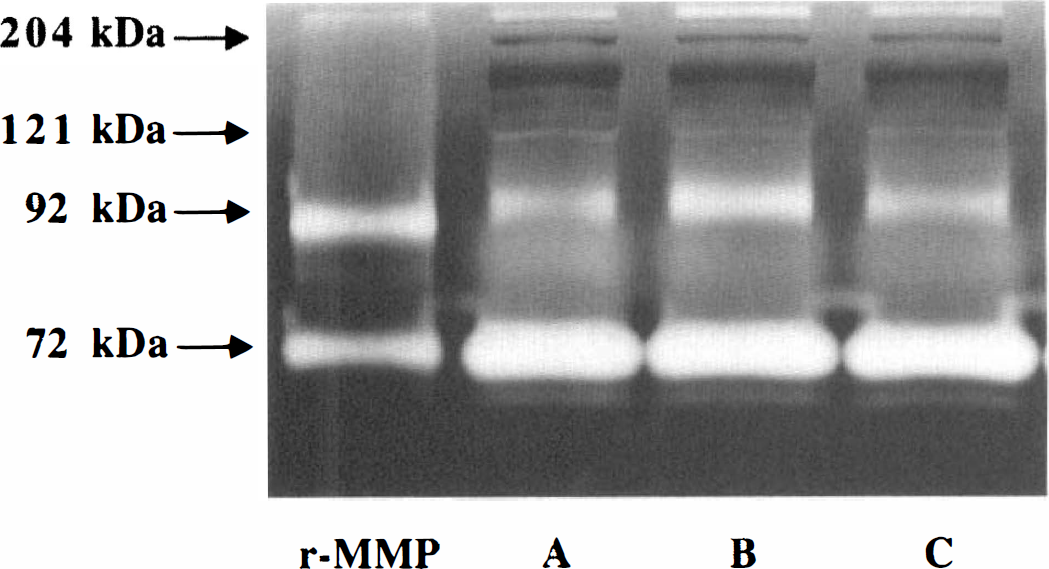
Representative zymogen of plasma samples from one subject undergoing MCAO. Gelatinolytic activity of MMP-9 was transiently increased at 2 hours of MCAO. Besides the four bands that were identified in the brain samples (see Fig. 2), a band just above the 121-kDa marker (approximately 130 kDa) is seen. Each point represents the time when the blood was drawn: A, baseline; B, 2 hours of MCAO; C, 1 hour of reperfusion after 3 hours of MCAO.
The surgical implantation procedure did not affect the presence of MMP-2 or MMP-9 in plasma. The integrated densities of latent MMP-2 before and after the surgical implantation (n = 6) were 1,704.99 ± 492.67 and 1,936.81 ± 469.53, and those of latent MMP-9 were 552.49 ± 295.59 and 583.62 ± 280.59, respectively. After MCAO, no change in the constitutive levels of latent MMP-2 in plasma was seen. However, the latent form of MMP-9 in the plasma increased transiently at 2 hours of MCAO (2P = 0.002), and decreased thereafter (Fig. 8).
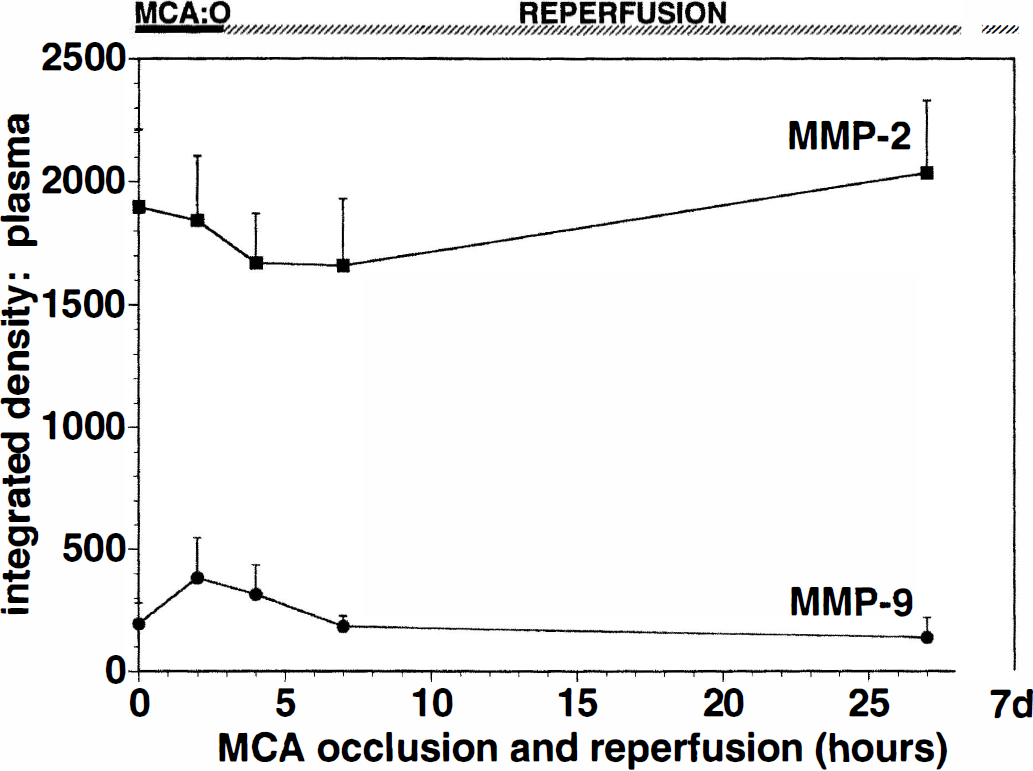
Effect of MCAO on plasma MMP integrated densities. Matrix metalloproteinase-9 activity was transiently increased at 2 hours of MCAO, whereas MMP-2 did not change.
No relationship between the integrated density of plasma MMP-9 and the total peripheral blood leukocyte, PMN leukocyte, or monocyte counts was demonstrated. For instance, the correlation between latent MMP-9 and PMN leukocyte count did not reach statistical significance (r = 0.425, SE = 0.238, 2P = 0.079) (Fig. 9). Separately, plasma MMP-2 and MMP-9 levels did not show any association with hemorrhagic transformation.
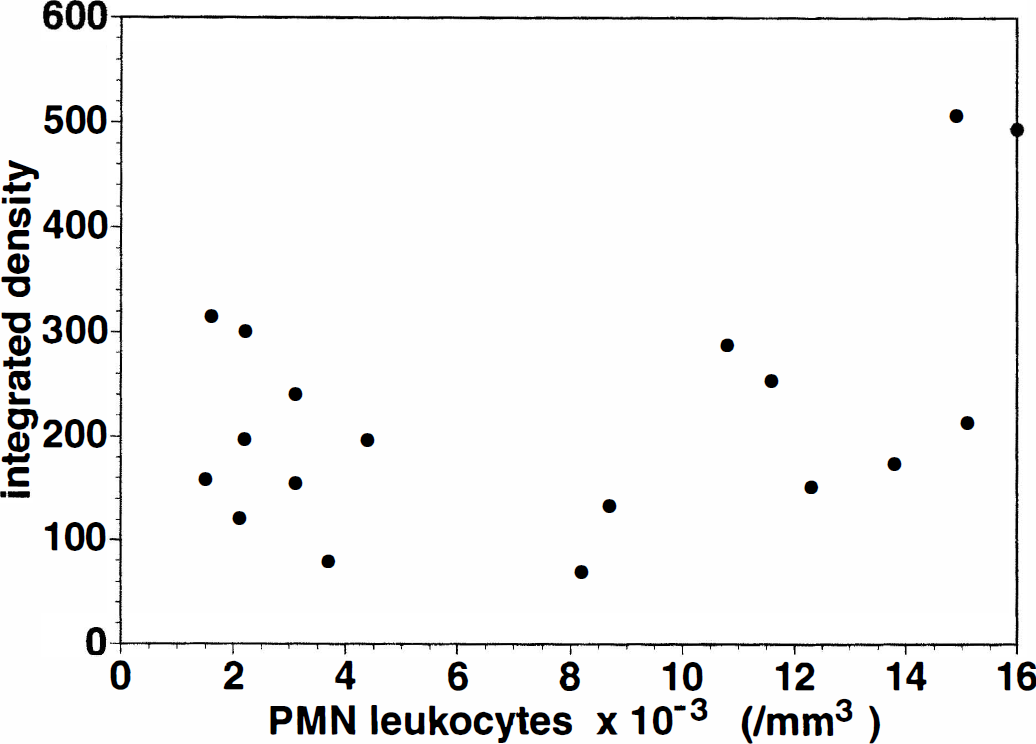
Comparison of plasma MMP-9 and PMN leukocyte counts. No significant correlation could be demonstrated (r = 0.425, SE = 0.238, 2P = 0.079).
DISCUSSION
Focal cerebral ischemia is associated with (1) a very early significant increase in latent MMP-2 within the ischemic basal ganglia after MCAO, (2) a significant correlation between the increase in MMP-2 and the extent of the Ic region and of neuron injury, (3) a significant increase of latent MMP-9, but not MMP-2, in the ischemic basal ganglia of subjects displaying hemorrhagic transformation, and (4) a transient increase of latent MMP-9 during MCAO in plasma.
Gelatinase activities can be identified by zymographic techniques, which have the advantages of detecting several proteases and variable forms of the same enzyme simultaneously within the same samples and estimating their activity. Although the reproducibility and quantitation of zymography have been questioned (Kjeldsen et al., 1992), several modifications have allowed an acceptably reproducible display technique of MMP-2 and MMP-9 activity to be developed (Kleiner and Stetler-Stevenson, 1994). Extension of the gel incubation period allows excellent sensitivity to detect small quantities of specific MMP (Kleiner and Stetler-Stevenson, 1994). As little as 5 pg of MMP-2 and MMP-9 derived from less than 20 µg protein was detectable in the present experiment. This provided detection of MMP-2 and MMP-9 activity in unfixed frozen sections suitable for immunohistochemical, dUTP incorporation, or in situ hybridization studies.
The inactive form of MMP-2 was observed in perfused nonischemic basal ganglia of control and MCAO subjects, indicating the constitutive expression of MMP-2 in non-human primate brain. This observation is consistent with previous reports of constitutive MMP-2 expression by most cell types (Yu et al., 1996). In contrast, MMP-9 was not increased in the nonischemic tissues of control subjects. Matrix metalloproteinase-9 was detected only in the ischemic basal ganglia of subjects undergoing MCAO. Anthony et al. (1997) reported the appearance of MMP-9 antigen on PMN leukocytes in fixed human brain within 1 week of stroke, and MMP-2 variably on monocyte-macrophages. Those findings are in accord with the presence of MMP-9 in granulocytes (Hibbs et al., 1985) and both MMP-2 and MMP-9 in cells of monocyte-macrophage lineage (Xie et al., 1994).
In this study, the significant increase in latent MMP-2 expression and the appearance of MMP-91 hour after MCAO in the ischemic basal ganglia indicate that both MMP did not arise from blood cellular sources because (1) the perfusion techniques remove more than 99% blood elements and plasma (a source of MMP-9), (2) the approximately 130-kDa band found in most plasma samples was not seen in any brain samples, and (3) substantial infiltration of leukocytes occurs 12 to 24 hours to several days after focal cerebral ischemia (Garcia et al., 1994). Although this study does not locate the cellular source of MMP-2 and MMP-9 activities in primate brain, both MMP have been identified with endothelial cells (Nakagawa et al., 1994; Cuzner et al., 1996; Maeda and Sobel, 1996), astrocytes (Cuzner et al., 1996), and microglia (Cuzner et al., 1996; Maeda and Sobel, 1996). The endogenous expression of MMP is regulated at several levels. At the transcriptional level, the synthesis of MMP-9 is upregulated by cytokines including interleukin-β and tumor necrosis factor-α (Unemori et al., 1991; Hanemaaijer et al., 1993), whose mRNA are detected as early as 1 hour after MCAO in rats (Liu et al., 1993, 1994). An AP-1 promoter site, to which the products of immediate early genes bind, appears to cooperate with SP1, PEA3, and nuclear factor-κB sites to induce MMP-9 transcription (Gum et al., 1996). Although the promoter sites of MMP-2 are not fully characterized, a recent report describes an AP-1 site on the MMP-2 gene (Bian and Sun, 1997). However, the exact transcriptional mechanisms remain to be illuminated.
Rosenberg et al. (1996) first reported increases in MMP-9 activity by 12 to 24 hours and MMP-2 activity by 5 days after permanent MCAO in anesthetized Wistar-Kyoto rats and SHR. The significantly earlier increase in MMP-2 in the non-human primate basal ganglia may have been related in part to the more sensitive zymographic assay used here, species differences in the models used, and ancillary effects including the impact of anesthetics in the rodent studies. Tagaya et al. (1997) demonstrated marked temporal and topographic differences in cellular injury after MCAO, displayed by nuclear incorporation of dUTP, between non-human primates and Wistar rats. In addition to species differences, anesthetics, including barbiturates, have been demonstrated to reduce cerebral injury after focal and global ischemia, whereas halogenated anesthetics significantly impair leukocyte chemotaxis in humans and primates, potentially delaying the inflammatory cell contribution to the infarct (Ember et al., 1994).
A very significant linear relationship between the increase in latent MMP-2 activity and the volume of dUTP incorporation (Ic region) throughout MCAO/R implies a potential direct relationship between MMP-2 activity and cell injury after MCAO in the primate. This relationship was strongest at 2 hours of MCAO, when 80% ± 6.6% of cells that incorporate dUTP are neurons in this model (Tagaya et al., 1997). Here, early MMP-2 secretion may result in or be related to early neuron injury. An intact basal lamina matrix and integrin-mediated matrix adhesion are required for cell survival, so that disruption of cell and matrix contacts may lead to changes in cell viability (Frisch and Francis, 1994; Re et al., 1994). Although yet unproven for the cerebral microvasculature, alterations in endothelial cell and astrocyte endfoot processes that occur in the time of increased MMP-2 expression and evident neuron injury may result from loss of those matrix contacts. Astrocytes suffer cytoplasmic alterations, and swelling of the endfeet occur very early after MCAO (Garcia et al., 1993). We have shown that integrins α1β1 on endothelium (Tagaya et al., unpublished data) and α6β4 on astrocytes (Wagner et al., 1997) are lost at the same time as their ligands within the basal lamina (Hamann et al., 1995). Consequently, disruption of the integrin—matrix contacts by degradation of basal lamina is likely to separate endothelium from astrocytes and therefore their interactions with neurons. This suggests the hypothesis that degradation of microvascular basal lamina by MMP-2 (and other MMP and proteases) initiated by cerebral ischemia may play a significant role in neuron injury, by disrupting cell—matrix integrity. However, our findings do not directly prove the causal link between MMP-2 activity and neuron injury. Further studies are required to validate their causal relationship.
One of the notable findings in the present study is the significant increase of latent MMP-9 in subjects with hemorrhagic transformation. Hemorrhagic transformation develops as a consequence of evolving ischemic injury and is associated with the use of plasminogen activators in cerebral ischemia (del Zoppo et al., 1990, 1998). Okada et al. (1994) and Hamann et al. (1996) demonstrated that degradation of microvascular basal lamina during cerebral ischemia allows emigration of plasma components and hemoglobin in the non-human primate. Although the predisposing or triggering factors responsible for hemorrhagic transformation are not well understood, major alterations in vascular permeability and vessel wall integrity occur (Okada et al., 1994; Hamann et al., 1996; del Zoppo et al., 1995). On the basis of experiments with bacterial collagenase-induced intracerebral hemorrhage in rats, Rosenberg et al. (1990) have suggested that MMP may cause brain edema and hemorrhage by breaking down vascular matrix and opening the blood—brain barrier. Our findings specifically implicate early MMP-9 expression in hemorrhagic transformation. It is not possible to determine whether an involvement of MMP-9 is causal or resultant to hemorrhage in the present experiments, however. Generation of thrombin during the hemorrhage may also stimulate MMP secretion from myointimal smooth muscle cells (Fabunmi et al., 1996) and provide a stimulus for the increases in MMP-9 levels. The absence of a parallel increase in MMP-9 levels from nonischemic tissue rules out an increased risk of hemorrhage on the basis of a genetic predisposition. A causative involvement of MMP-9 in these events remains to be tested.
Focal cerebral ischemia may influence plasma MMP activities and vice versa. No detectable change in plasma MMP-2 or MMP-9 activities occurred during MCAO/R, except for a transient elevation of latent MMP-9 at 2 hours of MCAO. Although leukocytes, including granulocytes, are major sources of MMP-9 in the plasma (Hibbs et al., 1985; Kjeldsen et al., 1992; Vartio and Baumann, 1989) and increase early during MCAO in this model (Ember et al., 1994), no clear correlation between MMP-9 activity and leukocyte count could be seen here. This lack of correlation also has been noted in human plasma (Kjeldsen et al., 1992). Significant interindividual differences in leukocyte MMP-9 content or differences in leukocyte activation are possible explanations (Kjeldsen et al., 1992). Removal of activated leukocytes from the circulation or decreases in cell responsiveness after MCAO in this model may be alternative explanations (Ember et al., 1994). The possibility that a modest contribution of MMP-9 from activated PMN leukocytes to MMP-9 activity in the ischemic Ic region cannot be excluded.
In this study, the inactive forms of MMP-2 and MMP-9 and the active form of MMP-2 were increased in ischemic brain. The inactive forms of MMP-2 and MMP-9 are secreted from cells into the extracellular space immediately after synthesis, and then activated by cleavage of their propeptides by specific proteases. Plasmin plays a central role in the activation of MMP-9 (Murphy et al., 1994; Nagase, 1997), and membrane-type MMPs activate pro-MMP-2 at cell surfaces (Sato et al., 1996). The presence of membrane-type MMP, known to activate pro-MMP-2 in the region of increased MMP-2 expression, may be taken as further evidence that the increased latent MMP-2 is locally activated. The activation of latent MMP-2 and MMP-9 and their final enzymatic reactions are regulated by TIMP. Tissue inhibitor of metalloproteinase-2 is an inhibitor of MMP-2 (Stetler-Stevenson et al., 1989), whereas TIMP-1 inhibits most MMP including MMP-9 (Woessner, 1991). In one report, TIMP-1 mRNA was induced at 12 hours and reached a peak level at 2 days after prolonged and transient MCAO in the SHR (Wang et al., 1998). However, the simultaneous quantitation of MMP and TIMP levels in tissue is problematic. In this study, the active forms of MMP-2 and MMP-9 in the ischemic tissues were relatively absent. This pattern of latent MMP-related activity also has been described in cerebral ischemia in rats (Rosenberg et al., 1996) and human stroke (Clark et al., 1997). However, the relative absence of active forms with each detection system does not rule out the presence of active forms locally. It is conceivable that MMPs are rapidly degraded once the latent forms are locally activated. Information on clearance mechanisms of the MMPs in ischemic brain after activation also would be essential to account for the relative absence of active forms of MMP-2 and MMP-9 in focal cerebral ischemia.
The observations presented here highlight the early potential roles of MMP-2 in the degradation of microvascular basal lamina to effect neuronal injury during experimental MCAO/R. The temporal and topographic codistribution of MMP-2 activity, neuron injury, and (in separate studies) loss of microvascular integrin receptors supports this hypothesis. Subsequent studies will be required to define the distribution of the inhibitor TIMP-2 and MMP-2 antigen. Furthermore, an association of MMP-9 with hemorrhagic transformation and MMP-2 with neuron injury after focal cerebral ischemia will require detailed immunohistochemical studies to assign cellular sources to both MMP and membrane-type MMP. The very early appearance of MMP-2 in particular raises the novel proposal that disturbances of the matrix contacts between endothelial cells and astrocytes may contribute to neuron injury.
Footnotes
Acknowledgments
The authors thank Dr. William G. Stetler-Stevenson for his very generous gift of recombinant MMP-2 and MMP-9. The authors also thank British Biotech Inc. for their generous gifts of the anti-MMP-2 and MMP-9 monoclonal antibodies used in these experiements to identify MMP-9 activity.