
Abstract
To define the location and extent of microvascular damage of the basal lamina after cerebral ischemia and reperfusion in rats, the authors subjected animals (n = 16) to 3 hours of focal cerebral ischemia and 24 hours of reperfusion using the suture middle cerebral artery occlusion model; sham-operated animals served as controls (n = 6). The Western blot technique was used to define the collagen type IV protein content in various brain regions, whereas immunohistochemistry identified microvascular basal lamina loss (anticollagen type IV staining). The extent of damage was quantified by automatic morphometric video-imaging analysis. Statistical analysis was based on the Mann-Whitney test and the paired Student's t-test. The ischemic hemisphere showed a reduction of the collagen type IV protein content after ischemia and reperfusion in the Western blot (reduction compared with the nonischemic side: total hemisphere, 33% ± 6%; basal ganglia, 49% ± 4%; cortex, 25% ± 7%; P < 0.01). There was also a decrease in the number of cerebral microvessels between the ischemic and nonischemic hemispheres (16% ± 3%), cortical (14% ± 3%), and basal ganglia areas (21% ± 4%) (P < 0.01). Besides a reduction of the vessel number, there was also a loss in basal lamina antigen-positive stained area in ischemic areas (hemisphere, 20% ± 2%; cortex, 8% ± 3%; basal ganglia, 31% ± 3%; P < 0.001). Cortical areas had a less pronounced basal lamina loss than basal ganglia (P < 0.05). For the first time, microvascular basal lamina damage, indicated by collagen type IV loss, is proven in rats by biochemical and morphometric analysis. These changes are comparable with those found in nonhuman primates. The authors report novel data regarding microvascular ischemic changes in the cortex. These data provide a basis for future experiments to determine the mechanisms of ischemic microvascular damage and to devise new therapeutic strategies.
Keywords
Experimental studies have shown that ischemia and reperfusion cause a network of detrimental metabolic and morphologic alterations (Dietrich 1998; Dirnagl et al., 1999). Because most studies originally focused on neuronal tissue or glial cells, the changes in cerebral microvessels have only recently become an area of growing interest (del Zoppo and Hallenbeck, 2000). Using the awake stroke model in baboons (del Zoppo et al., 1986), Hamann et al. (1995, 1996) showed that cerebral microvessel walls become gradually bleached and lose basal membrane constituents, which are finally destroyed depending on the duration of ischemia and reperfusion. A quantitative analysis of immunohistochemically stained vessels within the ischemic basal ganglia and the corresponding regions of the nonischemic side showed a significant reduction of the content of diverse basal lamina antigens (laminin, fibronectin, and collagen-type IV). Microvascular basal lamina damage is also important for the blood–brain barrier disturbances that occur after ischemic stroke (Betz 1996; Rosenberg 1999, Neuwelt et al., 1999). Well-known experimental and clinical consequences of these microvessel wall alterations are edema as a result of endothelial integrity loss followed by extravasation of noncellular blood elements as hemorrhages (del Zoppo et al., 1998; Hamann et al., 1996, 1999a). Findings of proteolytic cascades involved in the basal lamina damage and their prevention may in the long-term permit the adoption of strategies useful in the human setting of acute stroke to prevent secondary bleeding complications in stroke (Hamann et al., 1999a,b).
Unfortunately, previous results of nonhuman primate studies do not allow us to test these strategies in experimental protocols using rodents. The benefit of nonhuman primate models in terms of their proximity to the human situation is counteracted by the problematic amounts of experimental drugs used in these large animals and of concerns regarding animal rights. However, a broad knowledge of proteolytic mechanisms of potential basal lamina-relevant systems has been accumulated in rats (Rosenberg et al., 1996, 2001). Therefore, data specifying the basal lamina damage in rodents are urgently needed. No experimental data regarding these events in cortical regions are available in nonhuman primates or rodents. Available data regarding the basal ganglia are from nonhuman primates (Hamann et al., 1995). Besides morphometric analysis, no protein analytic data are available.
Our study tested four main hypotheses: that (1) microvascular damage occurs during cerebral ischemia in the rat; (2) protein analysis confirms the morphometric histologic data regarding basal lamina protein loss; (3) the loss in collagen type IV content and expression are comparable with changes observed in nonhuman primates; and (4) cortical and basal ganglia microvascular changes are similar in extent to basal lamina damage.
MATERIALS AND METHODS
Experimental groups
All experiments used male Wistar rats (250 to 300 g), which were allowed free access to food and water. In ischemia experiments (n = 16), a constant 3-hour period of ischemia was followed by 24-hour reperfusion. The middle cerebral artery was occluded by advancing an intraluminal thread rostrally into the internal carotid artery (ICA) and up to the origin of the middle cerebral artery. After ischemia, withdrawing the thread into the lumen of the ICA induced the onset of reperfusion. A second group of animals (n = 6) that underwent sham operation served as controls.
Preparation protocol
Rats subjected to 24-hour reperfusion were intubated and anesthetized with isoflurane, nitrous oxide, and oxygen inhalation. Rats were kept in a state of narcosis until reperfusion began. Rectal temperature was maintained at 37.0°C ± 0.5°C with a feedback heating pad. All animals were subjected to 3 hours of ischemia. Transient ischemia and reperfusion were induced by a modified intravascular filament model based on the surgical procedure of Longa et al. (1989; Sporer et al., 1997). Briefly, a midline incision was made at the neck. The left common carotid artery, its bifurcation into the external carotid artery, and the ICA were exposed under an operating microscope. Special care was taken to avoid injuring the vagus nerve. After the left common carotid artery was ligated distally, blood flow within this artery was interrupted up to the bifurcation. A surgical nylon monofilament thread (Ethicon 4/0; Ethicon, Norderstedt, Germany) with a silicone-covered tip was used to occlude the middle cerebral artery. A small incision was made in the left common carotid artery to insert the monofilament thread; the tip of the sheath was placed just before the bifurcation into the external carotid artery and ICA. The filament was gently advanced 18 to 20 mm into the intracranial part of the ICA.
Advancement of the suture thread stopped when a notable increase in vascular resistance was detected. After the thread was withdrawn by approximately 4 or 5 mm and reperfusion began, rats were allowed to awake from anesthesia. They were returned to their cages and permitted food and water ad libitum. At the end of the reperfusion period, rats were killed by transcardial perfusion with cold physiologic saline containing bovine serum albumin (BSA; 10 g/L), heparin (10 IU/L), and 2-mL/L nitroprussin solution (1.8 g/L) diluted in 1 L isotonic sodium chloride solution (9 g/L) with deep anesthesia. Brains were removed and the skull base was inspected to exclude hemorrhage.
Control animals were sham-operated by advancing the thread for only 12 mm toward the intracranial part of the ICA so that it reached the lumen of the ICA but did not occlude the middle cerebral artery.
Consecutive coronary cryosections (10-μm thick) in the region of 0.0 to 1.0 mm distant from the bregma were prepared for immunohistochemistry. Stereotactic coordinates followed that of Paxinos and Watson (1998).
Protein isolation and Western blotting
All steps for brain tissue homogenization and centrifugation were performed at 4°C. Brain areas in region of the cortex, the basal ganglia of the same size in the ischemic and nonischemic hemispheres, and whole hemispheres were excised from 10-μm-thick cryosections.
Brain material was homogenized in a lysis solution of 20-mmol/L Tris (pH 7.3), 1-mmol/L edetic acid, and 2% sodium dodecyl sulfate. Protease inhibitors phenyl-methyl-sulfonyl fluoride (100 μg/mL), aprotinin (10 μg/mL), and leupeptin (10 μg/mL) were added to prevent protein degradation. Samples were then sonicated for 10 seconds and spun at 13,000 g for 10 minutes. Protein concentrations were routinely determined by a Bradford protein assay (TEBU GmbH, Frankfurt, Germany) using BSA as a standard.
Proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Equal amounts of protein were diluted in 2x sodium dodecyl sulfate–polyacrylamide gel electrophoresis, heated to 95°C for 5 minutes, loaded on a 7.5% polyacrylamide gel, and run for 2 hours at 100 V. The proteins were then transferred at 4°C to a polyvinyl difluoride membrane (Sequi-Blot; Biorad, Munich, Germany) for 1 hour at 400 mA in a transfer buffer containing 10-mmol/L 3-[cyclohexyl-amino]-1-propanesulfonic acid (pH 11.0) with 10% methanol. After immobilization, membranes were stained with Ponceau S to confirm equal loading and transfer of proteins in all samples.
The blots were blocked in 3% BSA in Tris-buffered saline with 0.2% Tween 20 at room temperature for 1 hour. Blots were incubated in goat anticollagen type IV antibodies at a dilution of 1: 1000 (Southern Biotechnology, Birmingham, AL, U.S.A.), made up in blocking solution (3% BSA in Tween 20) overnight at 4°C. The blots were then washed three times for 10 minutes with Tris-buffered saline and incubated at room temperature for 1 hour with biotinylated antigoat antibody (Vector Laboratories, Burlingame, CA, U.S.A.) in Tween 20. Vectastain ABC reagent was added for 30 minutes at room temperature after rinsing the sections with Tween 20. After additional washing (three times for 10 minutes with Tween 20) the Western blot procedure was completed with an ECL development kit (Amersham, Beckinghamshire, U.K.). Each blot is representative of at least three experiments.
Analysis of the Western blot results
An optical analysis program (Tina version 2.08, Raytest Iso-topenmessgeräte GmbH, Munich, Germany) was used to compare the different bands by optical densitometry. Results were displayed on an arbitrary optical density scale. The relation between optical density on the ischemic to that on the nonischemic side was determined.
Immunohistochemistry
Microvascular damage was localized by immunohistochemical methods using a pooled antiserum against bovine and human collagen type IV from goat (DUNN; Southern Biotechnology Associates, Birmingham, AL, U.S.A.), which also reacts with rat collagen type IV.
Staining protocol
Sections were fixed with cold acetone and chloroform (1:1) for 5 minutes at room temperature, immersed in 10-mmol/L glycine in phosphate-buffered saline (PBS: 8.3-mmol/L disodium hydrogen phospate, 3.2-mmol/L potassium dihydrogen phospate, and 123-mmol/L sodium chloride diluted in 1 L distilled water (pH 7.4) for 5 minutes, and rinsed three times for 5 minutes in PBS before a 20-minute incubation at 4°C with blotto (50 g nonfat dry milk, 1 mL horse serum, and 0.3-mmol/L sodium azide diluted in 1 L Tris-saline stock, 38.5-mmol/L Tris, and 150-mmol/L sodium chloride diluted in 1 L distilled water) to reduce unspecific binding.
The consecutive sections were incubated with the primary antibodies against bovine or human collagen type IV from goat. Antibodies were diluted 1:800 in reagent (400 mg bovine serum albumin and 0.06-mmol/L thimerosal diluted in 1 L Tris-saline stock), first for 2 hours at 37°C and then additionally for 12 hours at 4°C. Sections were washed several times in PBS between the different incubation steps. The biotinylated secondary antibodies (antigoat, Jackson ImmunoResearch Laboratories, West Grove, PA, U.S.A.) were further incubated for 30 minutes at 37°C. They were diluted 1:200 in a solution of PBS, horse serum, and 10% Tween (1000:15:1). For subsequent development with the peroxidase technique, it was necessary to block the activity of endogenous peroxidases by incubating them with hydrogen peroxide (1-ml/L PBS 30% hydrogen peroxide) for 20 minutes at room temperature. The Vectastain-Elite-Kit (Vector Laboratories) was used to incubate sections with the avidin–biotin complex (concentrations according to the manufacturer's instructions) for 30 minutes at 37°C. Subsequently the peroxidase activity was detected with AEC (3-amino,9 ethyl-carbazole; concentrations according to the manufacturer's instructions; AEC-Kit, Biomeda Corporation, Foster City, CA, U.S.A.).
The same staining protocol using an antibody against microtubili-associated protein 2 (MAP2) (Chemicon, Harrow, U.K.) was used to define the region of interest (ROI) for the morphometric analysis.
Quantification of microvascular damage by video-imaging microscopy
To quantify the microvessels, their number per area unit (vascular density) and percentage of the area (vascular fraction) were calculated in the area of infarction and the contralateral control side as described below. First, an MAP2-stained, adjacent section was digitalized in a conventional flatbed scanner and the respective ROI delineated (e.g., the MAP2-demarcated area to define the infarct) using the OPTIMAS 6.5 image analysis system (Media Cybernetics, Silver Spring, MD, U.S.A.). In addition, a contralateral area that mirrored the ROI was delineated as a control. Next the complete collagen-stained section was digitalized in a Zeiss (Gottingen, Germany) Axiophot microscope with a fivefold objective using a Sony Power HAD 3CCD color video camera (Sony, Tokyo, Japan) and then was imported into OPTIMAS. Subsequently, the ROI masks were transferred to the collagen-stained section and the vessel density and fraction in each ROI were calculated by OPTIMAS and EXCEL (Microsoft, Seattle, WA, U.S.A.).
Statistics
Data are expressed as mean ± SD. For each group of animals, the ipsilateral was compared with the contralateral side by a paired Student's t-test with a level of significance of 1%. Comparisons between the experimental groups were made with the Mann-Whitney test using a level of significance of 5%.
RESULTS
A total of 12 rats survived the 3-hour ischemia and 24-hour reperfusion period, 9 of which had confirmed cerebral infarction in the MAP2 stain. These 9 animals were subjected to further comparisons with the sham-operated animal group.
The Western blot protein quantification revealed a significant loss of collagen type IV content on the ischemic brain side. The reduction in collagen type IV was 33% ± 6% for the ischemic hemisphere (P < 0.006), 49% ± 4% for the cortex (P < 0.008), and 25 ± 7% for the basal ganglia region (P < 0.004). Figure 1 shows the differences in the Western blot results. Figure 2 shows a typical example of the Western blot lines comparing ischemic and nonischemic tissue. The differences between the sham-operated and the ischemia group were significant for the total hemisphere (0.98 ± 0.04 vs. 0.67 ± 0.06, P < 0.01) and the basal ganglia (0.98 ± 0.01 vs. 0.75 ± 0.07, P < 0.01) but less so for the cortical regions examined (0.98 ± 0.04 vs. 0.51 ± 0.04, P < 0.05).
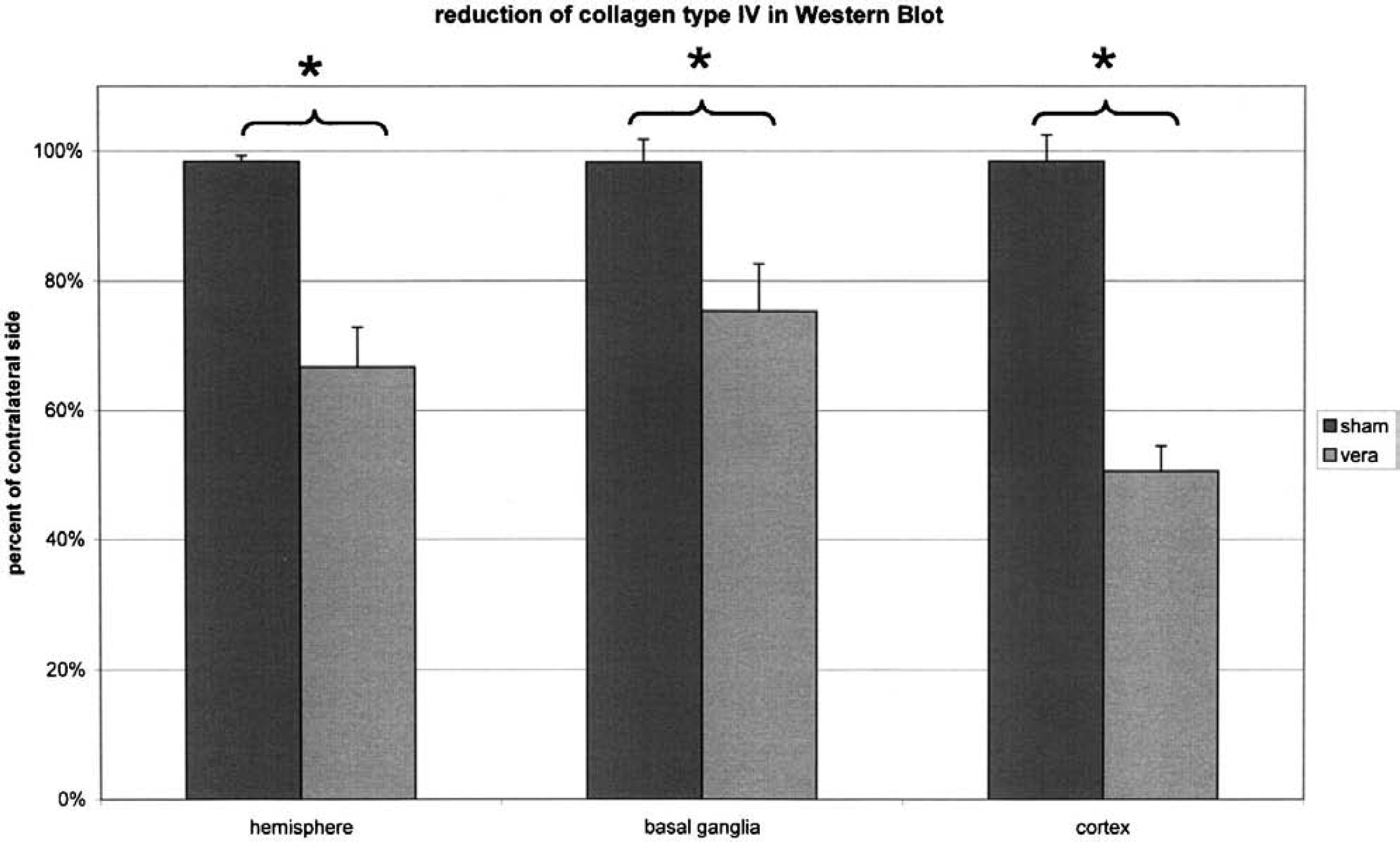
The reduction of Western blot collagen type IV quantification. The difference between cortical and basal ganglia was not significant. *P < 0.01.
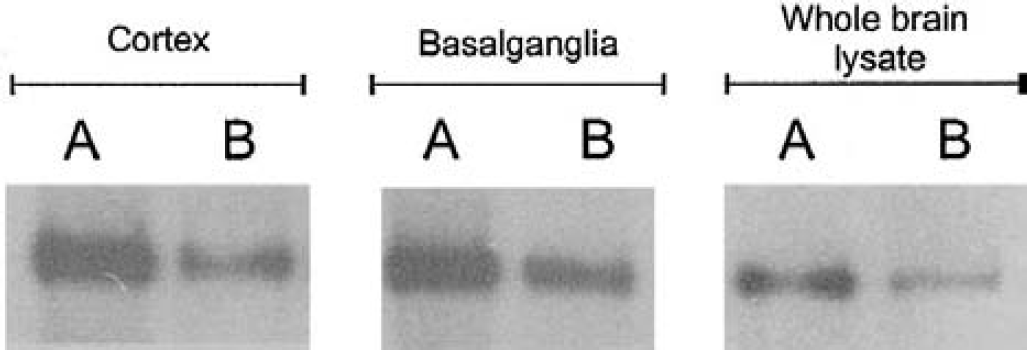
Western blot of rat brain areas after 3-hour ischemia and 24-hour reperfusion. The membrane was probed with a monoclonal antibody to collagen type IV at a dilution of 1:1000. Protein expression of collagen type IV in nonischemic
The number of detectable microvessels and the total stained area for collagen type IV were significantly reduced within the ischemic hemisphere, the cortex, and the basal ganglia of rats subjected to 3-hour ischemia and 24-hour reperfusion. The reduction in area of stained collagen type IV (Fig. 3) is 16% ± 3% for the total hemisphere, 14% ± 3% for the cortex, and 21% ± 4% for the basal ganglia (P < 0.01). The differences between the ischemia and the sham-operated groups were also significant for the total hemisphere and basal ganglia region (0.84 ± 0.03 vs. 1.01 ± 0.02, P < 0.001; 0.79 ± 0.04 vs. 1.04 ± 0.02, P < 0.001) and the cortical area (0.86 ± 0.03 vs. 1.02 ± 0.04, P < 0.05).
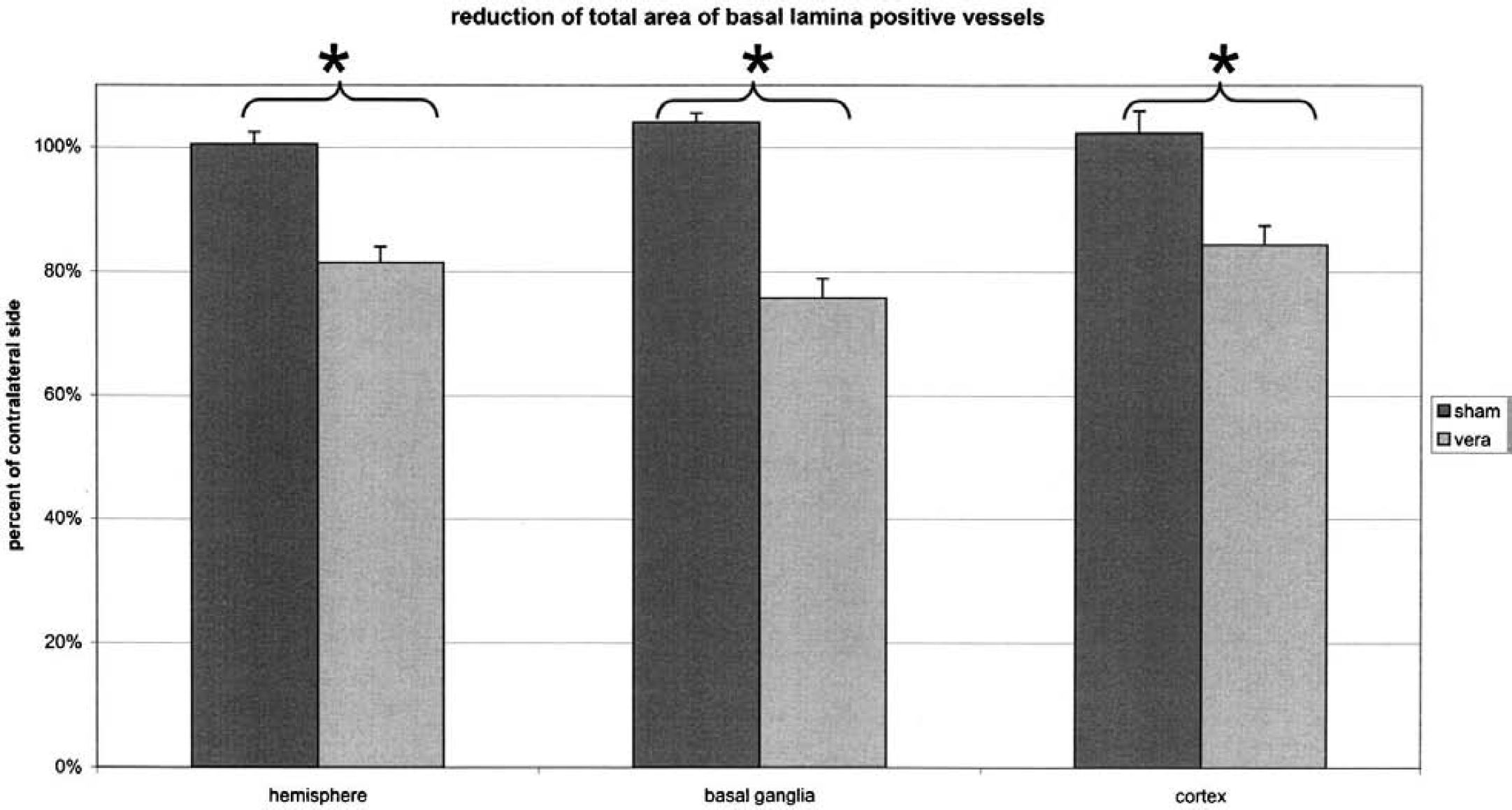
The reduction of the area of collagen type IV–positive stain. *P < 0.01.
The total number of microvessels identified by a collagen type IV stain was reduced in the ischemic hemisphere by 20% ± 2% (P < 0.001), the cortex by 8 ± 3% (P < 0.05), and the basal ganglia by 31 ± 3% (P < 0.001) (Fig. 4). Again, the differences between the ischemia and the sham-operated groups were also significant for the total hemisphere and basal ganglia region (0.80 ± 0.02 vs. 0.96 ± 0.01, P < 0.01; 0.69 ± 0.03 vs. 0.99 ± 0.02, P < 0.00) and the cortical area (0.92 ± 0.02 vs. 1.00 ± 0.03, P < 0.05). The difference in reduction between the cortical and basal ganglia regions was significant for the vessel density; there was a much slighter reduction in the number of vessels detected by collagen type IV stain in the cortex (P < 0.05).
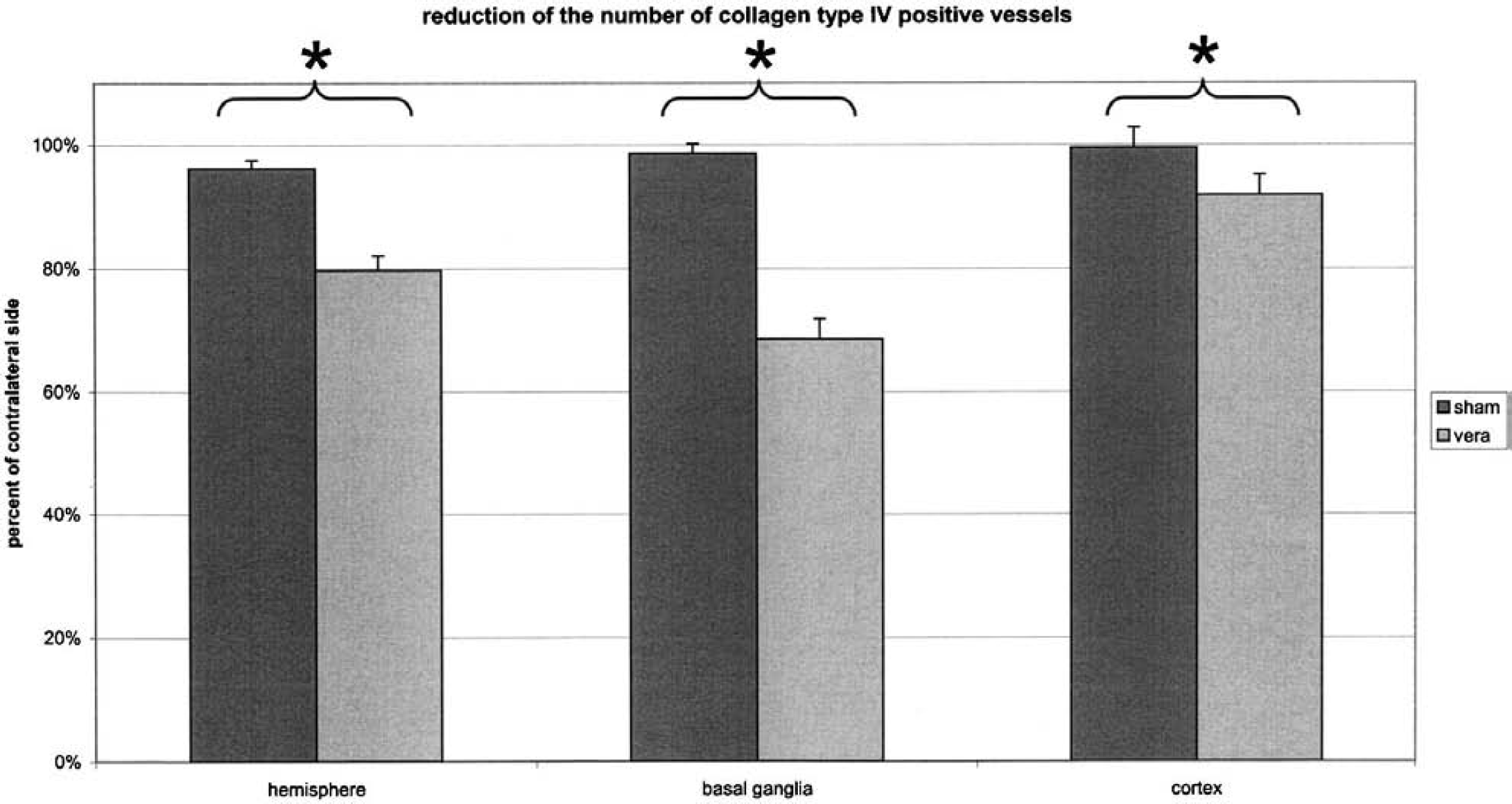
The number of identifiable microvessels is significantly different (*) in ischemic cortical, basal ganglia, and total hemisphere regions of interest.
Figure 5 shows a typical example of microvascular density reduction in the ischemic compared with the nonischemic basal ganglia. Microvascular abnormalities detected by immunohistochemistry were never seen in any other structures ipsilateral to the occluded vessels or in the contralateral hemisphere.
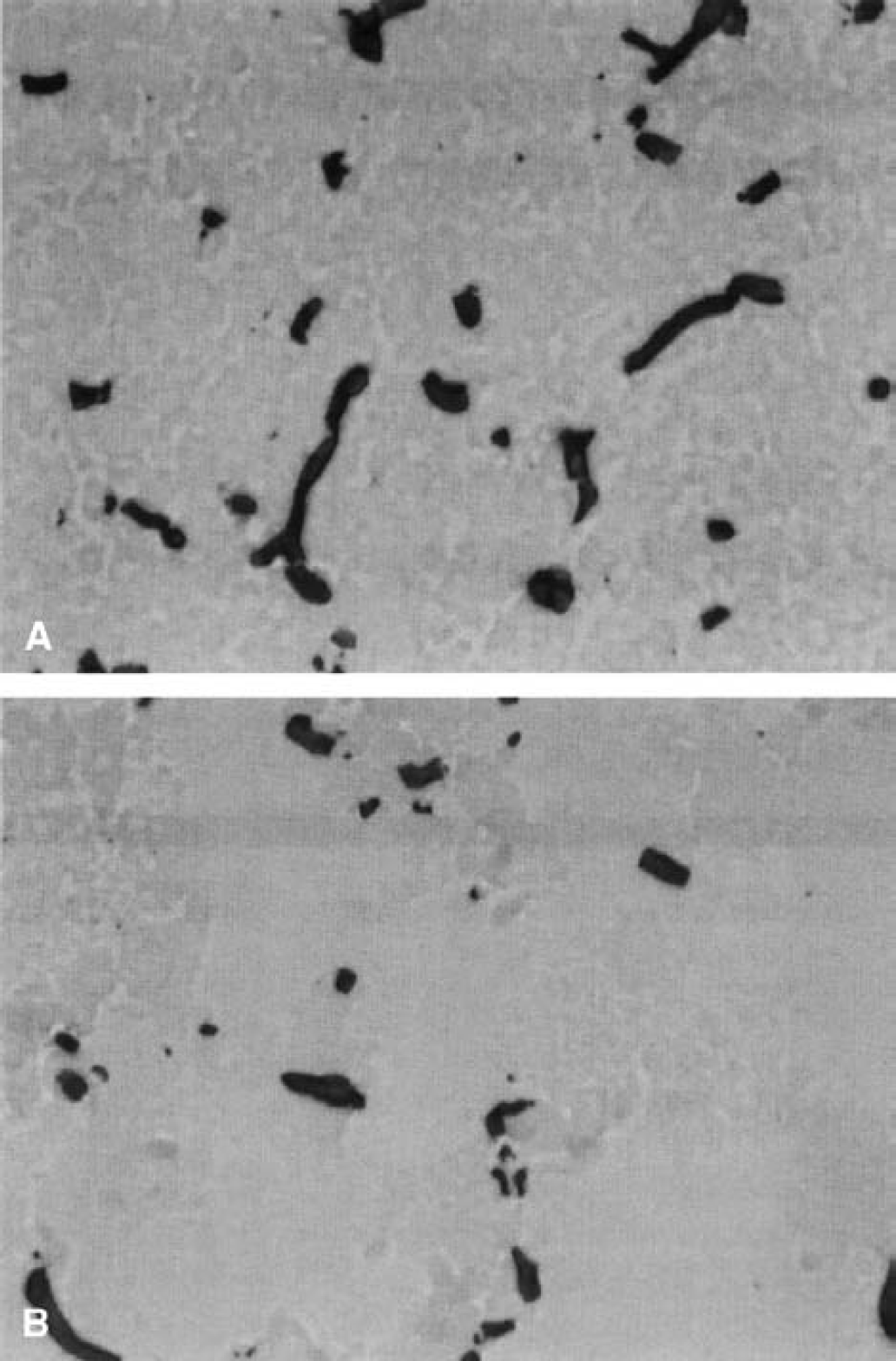
Typical example of a basal ganglia area with nonischemic microvessels
DISCUSSION
This study shows that substantial microvascular basal lamina damage occurs in rats after transient cerebral ischemia and reperfusion (hypothesis 1). This damage is proven by the significant reduction in vessel number and collagen type IV-stained areas observed in the immunohistochemical and morphometric analyses, and by the reduced collagen type IV content in the Western blot analysis. The implications of these findings are relevant for an explanation of proteolytic postischemic changes (Rosenberg et al., 1996) and for the development of strategies to prevent edema formation or hemorrhage (Hamann et al., 1999a,b). Protein analysis confirmed the results of immunohistochemistry and morphometric analysis.
This combination of independent methods shows the significance of microvascular basal lamina protein loss (hypothesis 2). The biologic importance of the basal lamina of cerebral microvessels is based on the formation of a second integrity barrier, and on the well-known interendothelial cell tight junctions. The endothelial (blood–brain) barrier is responsible for regulating the transport of fluids and soluble substances (del Zoppo and Hallenbeck, 2000). During cerebral ischemia, the functional and structural integrity of the endothelial barrier is rapidly destroyed (Hamann et al., 1999a,b; del Zoppo and Hallenbeck, 2000). Endothelial tight junctions open and transendothelial transport rapidly increases (Betz et al., 1989). The basal lamina is the only barrier that prevents large amounts of protein-rich fluids and cellular blood elements from entering into the brain during ischemia (Hamann et al., 1999a). This double layer is mainly formed by a sheet of collagen type IV polymer and a net of laminin, both of which are interconnected by entactin (Yurchenco and Schittny 1990; Risau et al., 1998). Additional important proteins in the extracellular matrix and the basal lamina are fibronectin, various proteoglycans, and glucosaminoglycans (Jaffe 1987; Yurchenco and Schittny, 1990). The ischemic, stimulated proteolytic enzymes from both the blood and the brain tissue start digesting the basal lamina. Once this integrity barrier is dissolved, hemorrhage can occur (Hamann et al., 1996, 1999a).
These digestive mechanisms involve at least three major proteolytic pathways (Hamann et al., 1999a,b). The first pathway, the plasminogen–plasmin system, is especially interesting because the first licensed drug for acute stroke treatment was recombinant tissue plasminogen activator (The NINDS and Stroke rt-PA Stroke Study Group, 1995). The plasminogen–plasmin system is important for endothelial cells, which produce tissue plasminogen activator to prevent wall thrombosis and plasminogen activator inhibitor to counteract the fibrinolytic effects (Lijnen and Collen, 1997; Lijnen et al., 1998). A local balance in this system seems to be relevant for the microvasculature (del Zoppo, 1994). Unfortunately, plasmin is a potent protease than can cleave various components of the extracellular matrix (e.g., laminin) (Liotta et al., 1981; Lijnen et al., 1998). Although the plasminogen–plasmin system cannot directly cleave collagen type IV, it activates the matrix metalloproteinases (Lijnen et al., 1999; Pfefferkorn et al., 2000).
Another pathway, the matrix metalloproteinase system, can lead to postischemic brain edema and hemorrhage (Mun-Bryce and Rosenberg, 1998; Rosenberg, 1999; Rosenberg et al., 1996, 1998, 2001). The expression of matrix metalloproteinases is related to reperfusion, and the main peak of matrix metalloproteinase activity is seen after 12 to 24 hours of reperfusion (Rosenberg et al., 1996). The therapeutic efficacy of preventing secondary edema by inhibitors of this system has also been established (Rosenberg et al., 1992; Rosenberg and Navratil, 1997).
A third pathway involves polymorphonuclear granulocytes, invasive cells than can secrete digestive enzymes (e.g., elastases; del Zoppo et al., 1998) that digest the basal lamina (Armao et al., 1997). The invasion of leukocytes starts at 12 hours and peaks 24 hours after the onset of ischemia (Garcia et al., 1994).
The comparison of our results with those of nonhuman primate studies (Hamann et al., 1995) showed that the extent of basal lamina damage is similar (hypothesis 3). The absolute loss in collagen type IV staining-positive microvessels in the ischemic basal ganglia of the nonhuman primate amounted to a difference of approximately 45% ± 7% compared with the nonischemic basal ganglia (Hamann et al., 1995). We observed a 31% ± 3% loss of basal lamina-positive microvascular structures in the rat basal ganglia. This difference may be due to the different animal model used, which would suggest that the non-human primate has a slightly more vulnerable microvascular wall in the basal ganglia. Alternative interpretations include the consequences of CBF changes caused by middle cerebral artery occlusion in different animal models. Baboons may have a more profound CBF reduction (Young et al., 1996, 1997; Marchal et al., 1999). In view of the striking differences between stroke models in rodents and in nonhuman primates (del Zoppo et al., 1998; del Zoppo and Hallenbeck, 2000), it is rather surprising how similar the extent of microvascular integrity loss is. The loss of vessel wall proteins has now been established to be a typical ischemia-related consequence in different animal models.
Until data were available for cortical brain regions, this phenomenon was thought to be restricted to the basal ganglia area. Because the microvasculature is organized differently in the basal ganglia and cortex (del Zoppo, 1994), vessel wall damage was assumed to occur in the cortex (hypothesis 4), but cortical disturbance had not yet been proven (Hamann et al., 1995). Cortical microvascular networks are organized like parallel, main-supply arterioles that are crossed by smaller branches to form a gridlike structure. In contrast, the basal ganglia have the geometry of a treelike vascular bed (del Zoppo, 1994, 1997). Cortical areas may also have a different collateral blood supply and, therefore, a different level of blood flow reduction (Touzani et al., 1995, 1997; Marchal et al., 1999). We show for the first time that although extensive basal lamina damage occurs in cortical areas, this damage is significantly less severe than that observed in the basal ganglia area. Therefore, hypothesis 4 can be rejected: there is a significant difference in the vascular reaction between basal ganglia and cortex microvessels in rats. Leptomeningeal cortical anastomosis may help prevent severe ischemic CBF reduction.
CONCLUSIONS
To gain more insight into complex microvessel wall changes, it will be necessary to compare microvascular alterations and neuronal changes. There is evidence that secondary neuronal cell loss can be considered as a result of microvascular disturbances (Chen and Strickland, 1997; Grammas, 2000). These results broaden our knowledge of the relation between the loss of microvessel integrity and neuronal damage in degenerative diseases such as Alzheimer disease (Grammas, 2000).
Further work is needed to evaluate the relation between microvascular vessel wall disorders and the occurrence of hemorrhage. These consequences have been illuminated in nonhuman primate studies (Hamann et al., 1996), but not yet in rodent studies. It may also be interesting to compare microvascular and neuronal or glial changes that result from different (shorter) periods of ischemia and various reperfusion times. Artificially administered plasminogen activators may increase the rate of hemorrhages in this model. This effect has been seen in patients receiving thrombolysis.
Protective strategies that help prevent a secondary vessel wall breakdown with subsequent hemorrhage or the development of brain edema may prove (del Zoppo et al., 1998; Hamann et al., 1999a). Such strategies may include special antioxidants that have a prominent endothelial cell protective effect (Franko et al., 1999) or locally inhibit the plasmin or matrix metalloproteinase system at the damaged vessel wall (Hamann et al., 1999a). Antigranulocyte strategies may also help reduce secondary integrity changes in the cerebral circulation (Hartl et al., 1996). Promising new strategies may involve the restoration of intact endothelial cell–basal lamina and astrocytic crosstalk through the integrins (Wagner et al., 1997). These transmembrane-signaling proteins are early targets for ischemia, and preventing damage to them may save microvascular and tissue structures.
Our study identifies for the first time that the microvascular basal lamina antigen fading of collagen type IV is a common consequence of transient cerebral ischemia in a rat model. Cortical areas are less severely affected by these digestive processes than are basal ganglia areas.
Footnotes
Acknowledgments:
The authors thank Mrs. Judy Benson, for language editing of this manuscript.