
Abstract
In focal cerebral ischemia the plasminogen-plasmin system plays a role in the fibrinolysis of vessel-occluding clots and also in the proteolysis of extracellular matrix components, which potentially contributes to brain edema and bleeding complications. The authors investigated the plasminogen activation after middle cerebral artery occlusion with and without reperfusion (reperfusion intervals 9 and 24 hours) in rats by histologic zymography and compared areas of increased plasminogen activation to areas of structural injury, which were detected immunohistochemically. After 3 hours of ischemia, increased plasminogen activation was observed in the ischemic hemisphere. The affected area measured 5.2% ± 8.5% and 19.4% ± 30.1% of the total basal ganglia and cortex area, respectively. Reperfusion for 9 hours after 3 hours of ischemia led to a significant expansion of plasminogen activation in the basal ganglia (68.8% ± 42.2%, P < 0.05) but not in the cortex (43.0% ± 34.6%, P = 0.394). In the basal ganglia, areas of increased plasminogen activation were related to areas of structural injury (r = 0.873, P < 0.001). No such correlation was found in the cortex (r = 0.299, P = 0.228). In this study, increased plasminogen activation was demonstrated early in focal cerebral ischemia. This activation may promote early secondary edema formation and also secondary hemorrhage after ischemic stroke.
Keywords
Plasmin is a potent proteolytic enzyme that hydrolyzes fibrin and fibrinogen to their respective degradation products. The proenzyme of plasmin, plasminogen, is converted by plasminogen activators (PAs). Two 'distinct physiologic PAs are known: the tissue-type PA (tPA) and the urokinase-type PA (uPA). Both tPA and uPA can be inhibited by PA inhibitor 1 and 2. At the level of plasmin, inhibition also may be caused by alpha-2-antiplasmin (Lijnen and Collen, 1995).
Plasminogen activation also can be induced exogenously by intravenous application of recombinant tPA (rtPA). The functional outcome of patients with acute ischemic stroke is improved if rtPA is given up to 3 hours after the onset of symptoms (The National Institutes of Neurological Disorders and Stroke rtPA Stroke Study Group, 1995). This positive effect may result from early recanalization of occluded cerebral arteries and consecutive salvage of endangered tissue. A main risk of rtPA treatment is an increased rate of symptomatic cerebral hemorrhage. A large European trial of rtPA in acute stroke showed that the rate of symptomatic hemorrhage is increased by threefold in patients receiving rtPA (19.8% versus 6.5%) (Hacke et al., 1995).
The pathophysiologic mechanisms of hemorrhagic transformation in ischemic stroke are not fully understood (del Zoppo et al., 1998). Experimental work has shown that focal ischemia has detrimental effects on microvascular integrity. Depending on the intervals of reperfusion after experimental ischemia, the antigenicity of extracellular matrix (ECM) components (laminin, fibronectin, collagen type IV) is reduced (Hamann et al., 1995). Double-staining techniques reveal microvascular hemorrhage around injured microvessels after focal ischemia (Hamann et al., 1996). These asymptomatic microbleeds may expand to symptomatic hemorrhage.
Possible mechanisms of microvascular injury during focal ischemia and reperfusion include the proteolytic functions of the plasminogen-plasmin system (del Zoppo et al., 1998). Plasmin directly degrades ECM components such as laminin and fibronectin (Liotta et al., 1981). The ECM component collagen type IV is degraded mainly by the matrix metalloproteinases (MMP) 2 and 9 (gelatinase A and B). It is plasmin that converts the preform of MMP-9 (pro-MMP-9) to the active enzyme (Okada et al., 1992). Regarding MMP-2, there is indirect evidence of a plasmin-mediated activation: in cell line experiments, the conversion of pro-MMP-2 was inhibited by plasmin inhibitors and antibodies to uPA (Baramova et al., 1997). In experiments with gene-deficient mice (plasminogen −/−) activation of pro-MMP-9 (but not pro-MMP-2) was reduced in the absence of plasminogen (Lijnen et al., 1998). In rat experiments with hemorrhagic injury, increases of uPA and MMP-9 were seen after 16 to 24 hours (Rosenberg et al., 1994). This finding likewise suggests that interactions occur between the PA/plasmin system and the MMP-mediated ECM degradation. In another study, increased plasminogen activator activity was seen in spontaneously hypertensive stroke-prone rats, suggesting a role of PA in the development of stroke (Matsuo et al., 1992).
The plasminogen-plasmin system also may have “nonvascular” effects on vulnerable cerebral tissue. Experimental work on knock-out mice (tPA−/−) demonstrates that tPA has neurotoxic effects after intracerebral injection of excitotoxins (Tsirka et al., 1995) and in focal ischemia, causing an increase in infarct size (Wang et al., 1998).
The positive effects of plasminogen activators (reperfusion) may compete with their negative effects (microvascular injury, neurotoxicity). To improve thrombolytic therapy for acute stroke, it is essential to increase our understanding of the role of the PA/plasmin system in focal ischemia. We investigated plasminogen activation in the basal ganglia and the cortex after 3 hours of focal cerebral ischemia followed by different intervals of reperfusion using an intraluminal thread rat model of middle cerebral artery occlusion.
EXPERIMENTAL PROCEDURES
Animal model
The experiments were performed on male Wistar rats weighing 250 to 300 g. A modified method of intraluminal vascular occlusion (Longa et al., 1989) was used, which was described in detail previously (Sporer et al., 1997). Three different groups (six animals each) were set up to investigate plasminogen activation after 3 hours of ischemia (I) with different reperfusion intervals (R) (Table 1). Briefly, animals of group 1 and 2 (I3R0 and I3R9; numbers indicate hours of ischemia and reperfusion, respectively) were anesthetized with thiopental (100 mg/kg intraperitoneally), tracheotomized, and artificially ventilated. Animals in group 3 (I3R24) were anesthetized during ischemia and at the end of the experiment with chloraldurate but were not tracheotomized. During reperfusion, rats in group 3 (I3R24) were allowed to wake up and were put back into their cages. To allow fluid substitution and to monitor blood pressure and blood gases, the femoral vein and artery were cannulated. Temperature was held constant by a feedback heating pad. To induce focal ischemia, the left common artery was ligated and cannuled with a polyethylene tube. A 3-0 surgical nylon suture was inserted into the tube and carefully pushed forward to 17 to 19 mm beyond the bifurcation of the carotid artery, where the ostium of the middle cerebral artery was occluded. Reperfusion was initiated by retracting the suture. To control focal ischemia and reperfusion in animals of groups 1 and 2 (I3R0, I3R9), a laser-Doppler probe was placed on the skull, where abrasions were created, over the ischemic hemisphere near the bregma. At the end of the experiments, the rats were killed and the brains were perfused with 1% albumin solution and frozen at −80°C. Animals showing no Doppler decrease were excluded from analysis. Signs of subarachnoid hemorrhage on inspection of the skull after removal of the brain also were a reason for exclusion.
Plasminogen activation and MAP-2 demarcation in focal ischemia/reperfusion

Mean values ± standard deviation (SD) of area of plasminogen activation and MAP-2 demarcation in percent of reference region; C, cortex, BG, basal ganglia. Animals of group 1 and 2 were anesthetized with thiopental, animals of group 3 with chloraldurate. Duration of ischemia (I) and reperfusion (R) are indicated in parentheses (numbers represent hours). Significant expansion of plasminogen activation in the basal ganglia is seen after 9 and 24 hours of reperfusion (Kruskal-Wallis, Mann-Whitney, P < 0.05, indicated as *). No significant expansion is seen in the cortex.
Preparation of cryostat sections
Cryostat sections of 10-μm thickness were taken from regions 0 to 1 mm behind the bregma (according to the atlas The Rat Brain by Paxinos and Watson [1998]), were prepared at −20°C, and stored at −80°C. To ensure comparability, neighboring sections were examined for microtubule associated protein 2 (MAP-2) immunohistochemical features and zymographic plasminogen activation.
MAP-2 immunohistochemistry
For MAP-2 detection, a monoclonal mouse antibody (Boehringer Mannheim, Mannheim, Germany; dilution 1:800, incubation for 2 hours at 37°C, followed by 24 hours at 4°C) was used. Before incubation, sections were fixated in a 1:1 mixture of acetone and chloroform, washed in phosphate-buffer solution (PBS), and incubated with blotto (50 g nonfat dry milk, 10 mL horse serum, 0.3 mmol/L sodium azide diluted in TRIS saline stock [38.5 mmol/L TRIS, 150 mmol/L NaCl]) to reduce unspecific binding. After repeated washing in PBS, incubation with the secondary biotinylated antibody (horse, anti-mouse, Vector Laboratories, Burlingame, CA, U.S.A.; 1:200, incubation 2 hours at 37°C) was followed by incubation with the ABC-Complex (Vectastain-Elite-Kit, Vector Laboratories; incubation 30 minutes at 37°C) after blocking endogenous peroxidase with H2O2. Peroxidase signal was developed with AEC (AEC-Kit, Biomeda Corporation, Foster City, CA, U.S.A.). Nuclei were counterstained with Mayer's hematoxylin (Sigma, Deisenhofen, Germany), and tissue was blued in saturated sodium bicarbonate.
Zymographic detection of plasminogen activation
We used the method described by Sappino et al. (1993): briefly, after thawing and drying at room temperature, sections were incubated over 22 hours at 37°C with an overlay consisting of commercial nonfat milk powder (containing the plasmin substrate casein), agarose, and human plasminogen. The overlay was prepared in several steps. Three different solutions were prepared. Solutions 1 (100 mL PBS + 24.6 mg MgSO4 + 13.2 mg CaCl2) and 2 (10 mL dH2O + 1.6 g commercial milk powder) were separately heated to 50°C, solution 3 (10 mL dH2O + 250 mg agarose) to 70°C. The solutions were mixed (0.75 mL of solution 1, 0.70 mL of solution 2, 0.50 mL of solution 3), and the mixture was cooled to 37°C. Then 100 μL of plasminogen (1.5 mg/mL, Sigma) were added. This mixture was evenly spread over the heated sections (37°C) and immediately covered by a coverslip. After incubation at 37°C for 22 hours, the sections were immediately inspected for zones of overlay lysis and digitally scanned for later computer analysis. The incubation period of 22 hours was chosen, since plasmin-mediated lysis then had reached a maximum. Longer incubation periods up to 36 hours showed no further expansion of lytic regions. To exclude unspecific overlay lysis, control sections of all groups were incubated with an overlay lacking plasminogen.
Measurements
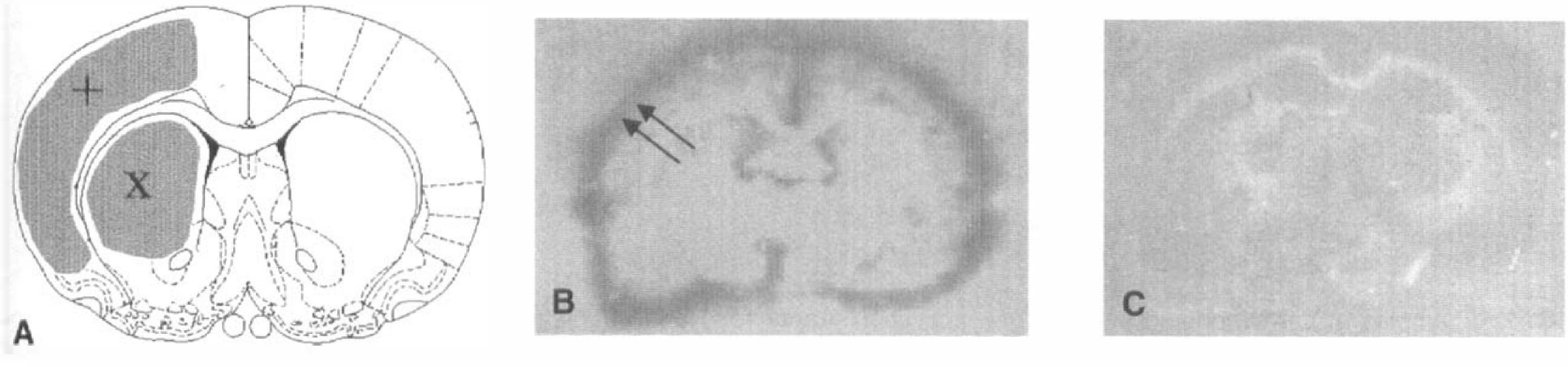
To evaluate areas of visible plasminogen activation and decreased MAP-2 antigenicity, a morphometric program (Soft Imaging System, Stuttgart, Germany) was used. Areas of the total basal ganglia and cortex on both the ischemic and control hemispheres were measured, as well as zones of plasmin-mediated lysis and MAP-2 demarcation in these areas. Measurements were performed in a blinded fashion. The person taking the measurements had no information regarding the groups to which individual sections belonged. Affected regions were expressed as a percentage of total hemispheric basal ganglia or cortex area (Fig. 1A).
Statistical analysis
For statistical description, data were presented as mean ± SD. Group-to-group comparison was performed using nonparametric tests (Kruskal-Wallis, Mann-Whitney). To test the correlation between areas of MAP-2 demarcation and plasminogen activation, a nonparametric bivariate correlation analysis (Spearman) was performed.
RESULTS
Plasminogen activation in controls
Limited areas of overlay lysis were seen in sham-operated animals and in the control (nonischemic) hemispheres of all sections. This lysis uniformly occurred in two different patterns: (1) in small, pinpoint-shaped regions with no preference for distinct anatomic structures; and (2) in a circular rim of lysis following pial structures around the sections (Fig. 1B).
Plasminogen activation after 3 hours of ischemia (I3R0)
As in the controls, background plasminogen activation (pinpoint regions, circular rim) was seen after 3 hours of ischemia. However, additional plasminogen activation was observed in the basal ganglia and the cortex of the ischemic hemisphere (5.2% ± 8.5% and 19.4% ± 30.1% of total basal ganglia and cortex area of the ischemic side, respectively). There was no significant difference in the extent of plasminogen activation between the basal ganglia and the cortex (Wilcoxon test, p = 0.18). In contrast to the background activation, the areas of additional plasminogen activation presented as coherent zones of overlay lysis (Fig. 2). The observed lysis of the casein containing overlay clearly depended on plasminogen. When an overlay lacking plasminogen was used, the overlay remained unaffected (Fig. 1C). The direct overlay lysis by added plasminogen was excluded, since lysis on the slides depended on the presence of brain tissue. Therefore, overlay lysis unquestionably represented plasminogen activation by enzymes located in the examined brain sections.
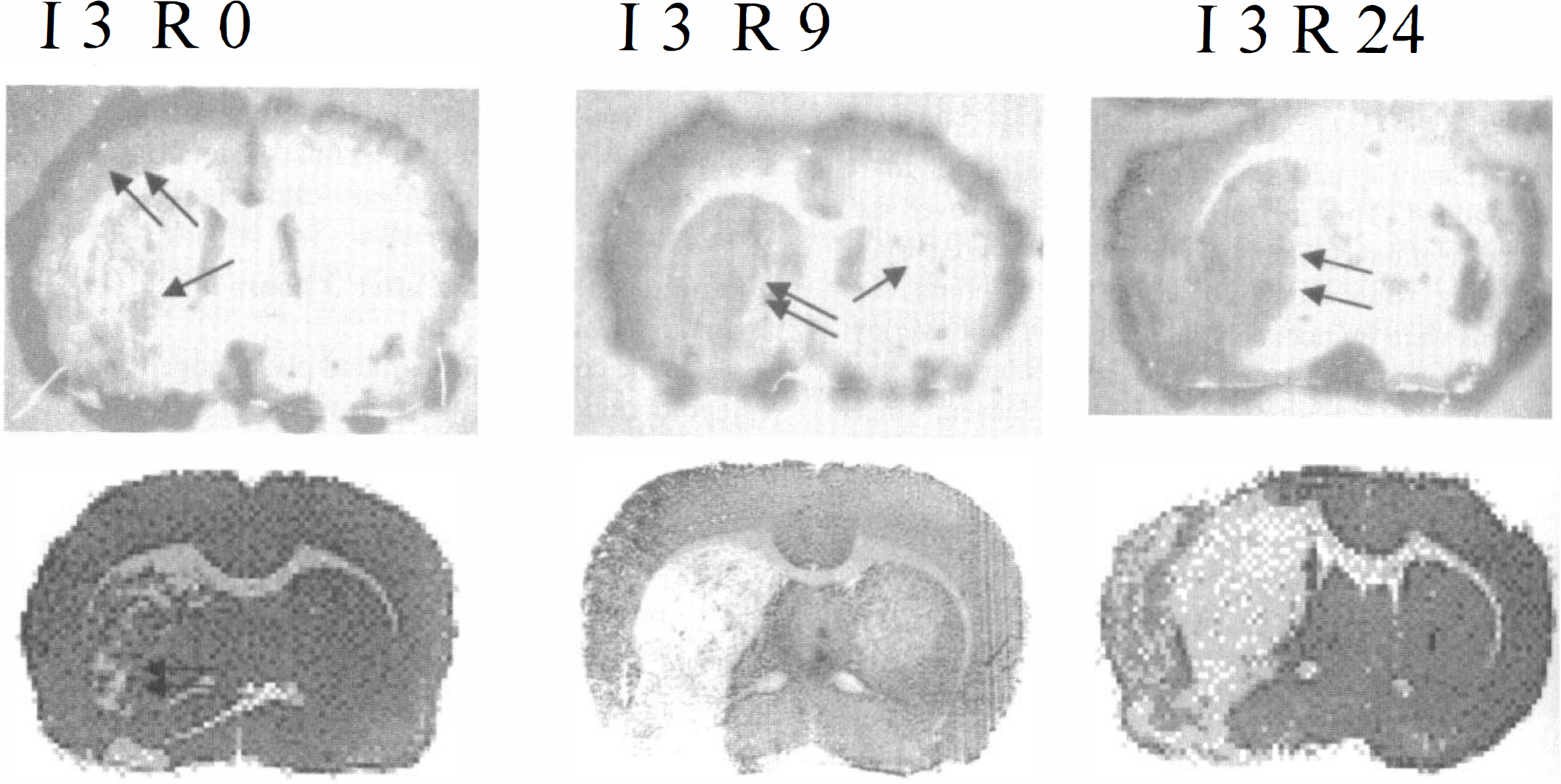
Examples of corresponding plasminogen activation (upper row) and MAP-2 immunohistochemical study (lower row). Zymographic plasminogen activation and MAP-2 immunohistochemical study were performed in neighboring sections of the same animal for each example. I3R0: Notice areas of overlay lysis mainly in cortical regions (double arrow) of the ischemic hemisphere. There also is some lysis in the basal ganglia (arrow) corresponding to loss of MAP-2 antigenicity (double arrow). Area of cortical overlay lysis exceeds area of MAP-2 demarcation in this example. I3R9: Expansion of overlay lysis, now affecting most of the basal ganglia (double arrow) and the cortex. Also, notice small pinpoint-shaped zones of background overlay lysis in the nonischemic hemisphere (arrow). I3R24: Extensive overlay lysis (double arrow) that corresponds well with loss of MAP-2 antigenicity.
Plasminogen activation after ischemia and reperfusion (I3R9, I3R24)
Compared with 3 hours of ischemia alone, the area of plasminogen activation in the basal ganglia of the ischemic hemisphere had significantly increased after 9 and 24 hours of reperfusion (68.8% ± 42.2% and 36.6% ± 29.3%, respectively, versus 5.2% ± 8.5%, p < 0.05; Table 1). However, the duration of reperfusion (9 versus 24 hours) had no significant effect. Regarding cortical regions, reperfusion intervals had no significant influence on the area of plasminogen activation (I3R0, 19.4% ± 30.1%; I3R9, 43.0% ± 34.6%; I3R24, 35.0% ± 23.9%; Table 1).
Plasminogen activation and MAP-2 demarcation
A comparison of plasminogen activation with structural lesions detected by demarcation in MAP-2 immunohistochemical study (see Table 1 for mean values) revealed a highly significant positive correlation for the basal ganglia (r = 0.873, p > 0.001; Fig. 3) but not for the cortex (r = 0.299, p = 0.228; Fig. 4). Overlapping of MAP-2 demarcation and plasminogen activation (less than 20% difference in the affected area of MAP-2 demarcation and plasminogen activation) was seen in 18 of 36 individual pairs (50%). In the basal ganglia, the area of MAP-2 demarcation exceeded the area of plasminogen activation in 8/18 pairs (44%). The opposite (plasminogen activation exceeding MAP-2 demarcation) was seen in 0/18 pairs (0%). In the cortex, overlapping was seen in 8/18 pairs (44%). In 5/18 pairs (28%), MAP-2 demarcation exceeded plasminogen activation. Cortical plasminogen activation exceeded MAP-2 demarcation in 5/18 pairs (28%). Remarkably, extensive cortical plasmin activation (ranging from 58% to 100% of total cortex area) was seen in 3/18 pairs (17%) despite normal MAP-2 staining.
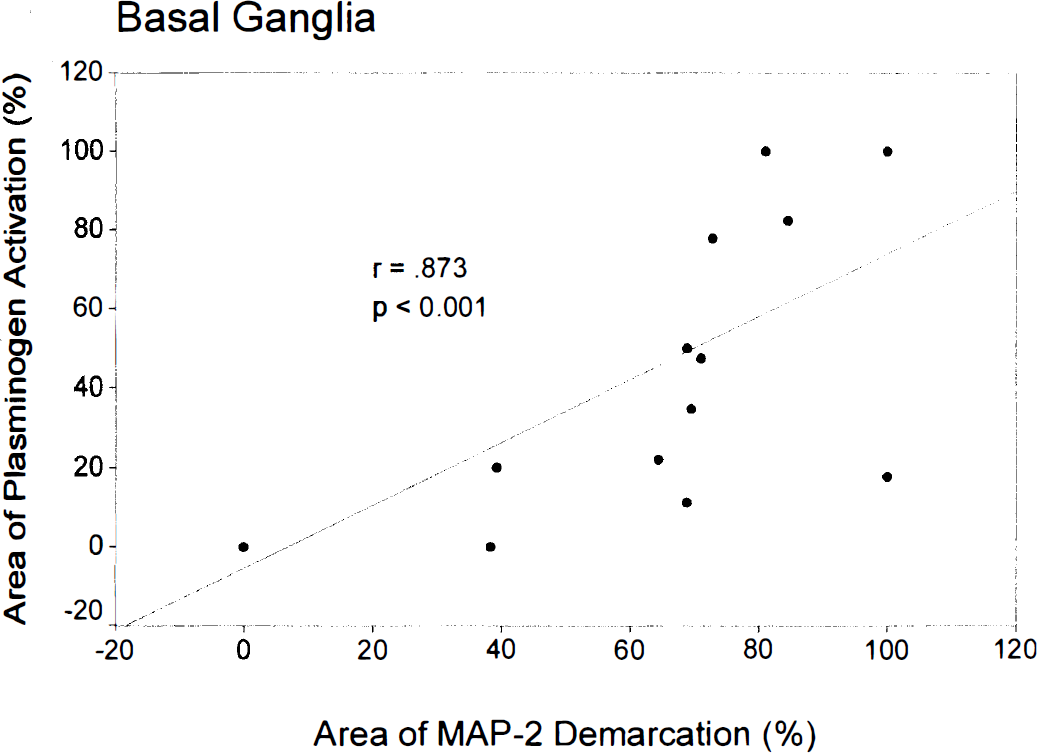
Correlation of plasminogen activation and MAP-2 demarcation in the basal ganglia (Spearman correlation). Only 13 markers are seen because of overlapping (several pairs of 0/0% and 100/100%).
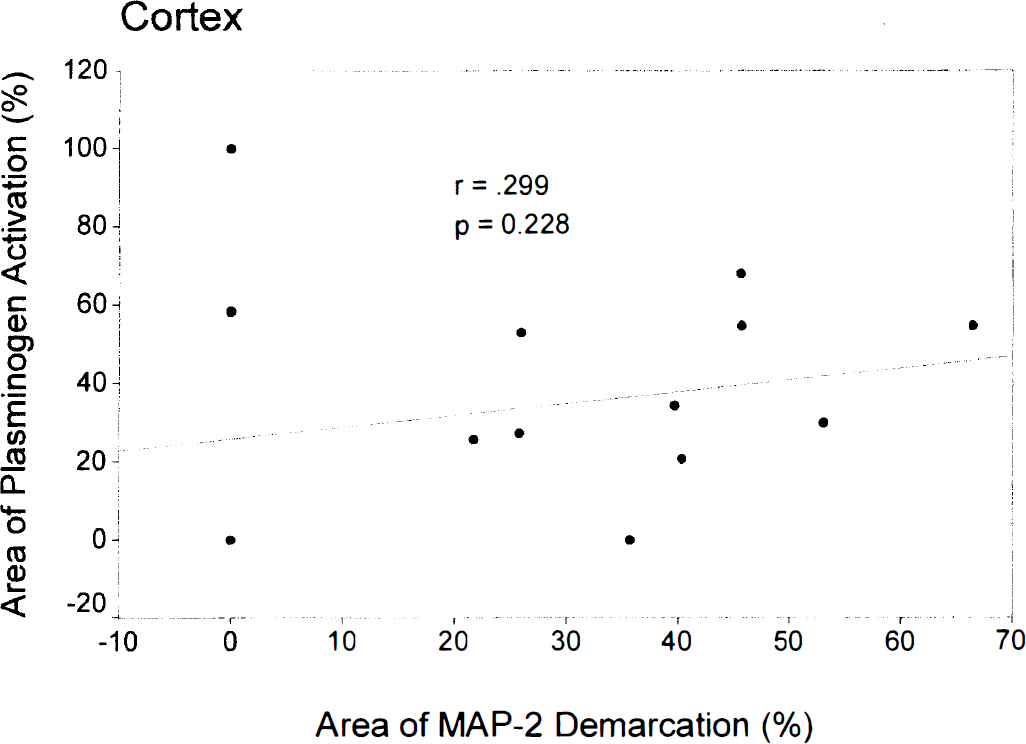
Lack of correlation of plasminogen activation and MAP-2 demarcation in the cortex (Spearman correlation). Notice the several cases of extensive plasminogen activation in the absence of MAP-2 demarcation.
DISCUSSION
Our results demonstrate an increased plasminogen activation after 3 hours of focal ischemia. This increase was limited to the ischemic hemisphere. In the control hemisphere, which represented tissue with unrestricted perfusion, plasminogen activation beyond background activation (as seen in sham-operated animals) was not detected. Plasminogen activation was not limited to certain ischemic brain regions but was observed in both the basal ganglia and in the cortex of the affected hemisphere.
The influence of reperfusion on plasminogen activation remains unclear. The duration of reperfusion in our groups had inconsistent effects on the extent of plasminogen activation. Although reperfused animals showed an increase in basal ganglia plasminogen activation compared with animals exposed to ischemia only, a comparison of different intervals of reperfusion (9 versus 24 hours) showed no significant change. In the cortex, reperfusion had no further effect on plasminogen activation. To further elucidate the role of reperfusion on plasminogen activation, future studies should include control groups with permanent ischemia.
Regions of increased plasminogen activation in the basal ganglia correlated to regions of structural injury detected by MAP-2 demarcation. This was not true for the cortex. In the basal ganglia, the area of detectable structural injury frequently exceeded the area of increased plasminogen activation, whereas the opposite was not seen. In contrast, there were several cases of markedly increased cortical plasminogen activation in the absence of detectable structural injury. The high variability of increased cortical plasminogen activation remains unclear but may explain the lack of correlation with cortical MAP-2 demarcation. Obviously, MAP-2 demarcation and plasminogen activation do not depend on each other in a cause-and-effect relationship.
In light of these results, it is necessary to address the limitations of our study. Because there were few animals per group, significant reperfusion effects might have been missed. Nonetheless, the early increase of plasminogen activation in focal cerebral ischemia is unquestionable. Standard deviations regarding the areas of increased plasminogen activation and MAP-2 demarcation were high. This might result from a high variability of infarct size after proximal middle cerebral artery occlusion (Herz et al., 1996).
The enzymatically detected plasminogen activation revealed no histologic information about the involved anatomic substructures. Potential sources of plasmin activators include endothelial cells of precapillary arterioles and postcapillary venules (Levine and del Zoppo 1994; Schreiber et al., 1998), cerebral capillaries (Zlokovic et al., 1995), and microglial cells (Tsirka et al., 1997).
The mechanisms of plasminogen activation remain unclear. Candidate enzymes are the known physiologic plasminogen activators uPA and tPA. Previous studies investigating their role in focal ischemia have yielded controversial results. In experiments with mice, plasminogen activation was observed at the site of infarction (Wang et al., 1998). In this study, uPA was blocked by amiloride, a selective uPA inhibitor (Vassalli and Belin, 1987), suggesting tPA to be the responsible plasminogen activator. Increased plasminogen activator activity also was seen in spontaneously hypertensive stroke-prone rats. Enzymatic analysis revealed a plasminogen activator with a molecular weight of 72 kD, which is similar to that of tPA (Matsuo et al., 1992). In contrast, gel electrophoresis in experiments on focal ischemia in rats revealed an increase in uPA but a decrease in tPA in the affected hemisphere at different times (Rosenberg et al., 1996). Furthermore, the role of other, yet unknown, enzymes with relevant potency in plasminogen activation cannot be excluded.
The PA inhibitors also may interfere with plasminogen activation in focal cerebral ischemia. In human cell line experiments, PA inhibitor 1 levels decreased after exposure to tPA (Shatos et al., 1996), possibly exacerbating the microvascular injury. Even if stable PA quantities are assumed, the upregulation or downregulation of PA inhibitors may influence overall plasminogen activation. Regardless of the possible mechanisms, we clearly showed that focal ischemia influences the plasminogen-plasmin system toward increased plasmin activity early in focal cerebral ischemia.
Recent experimental studies demonstrate that tPA plays a role in neuronal damage provoked by excitatory amino acids (Tsirka et al., 1995) or focal ischemia (Wang et al., 1998). As in PA/plasmin-mediated microvascular injury, neuronal damage also seems to depend on ECM disruption. In experiments with tPA-deficient mice, the blocking of the ECM component laminin with specific antibodies restored excitotoxic neuronal sensitivity. This suggests that the disruption of the neuron-ECM interaction (by PA/plasmin-mediated proteolysis or blocking of laminin) substantially contributes to tPA-mediated neuronal vulnerability (Chen and Strickland 1997).
In conclusion, our experiments showed an increased plasminogen activation after ischemia and ischemia with reperfusion. This plasminogen activation may substantially contribute to previously described microvascular injury and also may be relevant in patients with acute stroke.
Footnotes
Abbreviations used
Acknowledgements
The authors thank Mrs. Judy Benson for editing the language of this manuscript. The authors also thank G.J. del Zoppo (The Scripps Research Institute, La Jolla, CA, U.S.A.) for helpful comments and suggestions.