
Abstract
Splicing factors (SFs) coordinate nuclear intron/exon splicing of RNA. Splicing factor disturbances can cause cell death. RNA binding motif 5 (RBM5) and 10 (RBM10) promote apoptosis in cancer cells by activating detrimental alternative splicing of key death/survival genes. The role(s) of RBM5/10 in neurons has not been established. Here, we report that RBM5 knockdown in human neuronal cells decreases caspase activation by staurosporine. In contrast, RBM10 knockdown augments caspase activation. To determine whether brain injury alters RBM signaling, we measured RBM5/10 protein in mouse cortical/hippocampus homogenates after controlled cortical impact (CCI) traumatic brain injury (TBI) plus hemorrhagic shock (CCI+HS). The RBM5/10 staining was higher 48 to 72 hours after injury and appeared to be increased in neuronal nuclei of the hippocampus. We also measured levels of other nuclear SFs known to be essential for cellular viability and report that splicing factor 1 (SF1) but not splicing factor 3A (SF3A) decreased 4 to 72 hours after injury. Finally, we confirm that RBM5/10 regulate protein expression of several target genes including caspase-2, cellular FLICE-like inhibitory protein (c-FLIP), LETM1 Domain-Containing Protein 1 (LETMD1), and amyloid precursor-like protein 2 (APLP2) in neuronal cells. Knockdown of RBM5 appeared to increase expression of c-FLIP(s), LETMD1, and APLP2 but decrease caspase-2.
Introduction
Precursor messenger RNA (pre-mRNA) consists of exons (protein-coding RNA) and introns (noncoding RNA). Enzymatic intron removal by splicing mechanisms processes pre-mRNAs into mature mRNAs, which are then translated into protein. Genes that regulate cell death/survival are modified by alternative splicing. 1 Inclusion or exclusion of key coding exons generates different protein variants with altered function(s)—such as converting pro-death mediators into survival molecules or vice versa.1, 2 Alternative splicing and variant selection are regulated by hundreds of nuclear splicing factors (SFs) in the spliceosome. 3 Identifying SFs that promote expression of detrimental splice variants in neurons may lead to new treatments for brain injury.
The nuclear SFs RNA binding motif 5 (RBM5) and 10 (RBM10) regulate alternative splicing of cell death genes and promote apoptosis in some cancer cells.4, 5, 6, 7 RNA binding motif gene targets include caspase-2 and cellular FLICE-like inhibitory protein (c-FLIP). The protease caspase-2 has a long variant caspase-2(L) that is pro-death but a short variant caspase-2(s) that is protective.2, 8 Cellular FLICE-like inhibitory proteins are mostly protective but the long variant c-FLIP(L) is less potent than shorter c-FLIP(s).9, 10, 11 RNA binding motif 5 increases caspase-2(L) and c-FLIP(L) splicing, which favors cell death.4, 12 Overexpression of RBM5/10 greatly sensitizes cancer cells to die by death ligands such as tumor necrosis factor alpha (TNF-α) that induce caspase-mediated apoptosis.13, 14 It is unclear whether RBM5/10 regulate maladaptive splicing and caspase activation after injury in neurons.
Few studies have investigated intron/exon splicing changes after acute central nervous system injury like traumatic brain injury (TBI). Traumatic brain injury is a major cause of death and disability. Secondary insults such as hypotension from hemorrhage (H) or hemorrhagic shock (HS) greatly increase mortality and morbidity in TBI patients. 15 It also increases neuronal death by exacerbating secondary hypoxia-ischemia in injured brain regions in animal models. 16 To the best of our knowledge, SF changes have not been investigated in TBI. Confirmation of SF disturbances in TBI could help to clarify the relevance of this understudied signaling axis in central nervous system therapeutics. We measured protein levels of RBM5, RBM10, splicing factor 1 (SF1), and splicing factor 3A (SF3A) in brain tissue homogenates 4, 24, 48, and 72 hours after combined controlled cortical impact plus HS (CCI+HS) in mice. We included SF1 and SF3A in those studies because they are well-known master regulators of intron and exon splicing.
Here, we show that isolated neurons primarily express a 120-kDa RBM5 protein. Knockdown of neuronal RBM5 inhibited staurosporine (STS)-induced caspase activation, and altered protein expression of survival/death genes. A 90- kDa RBM5 protein was additionally observed in whole brain tissue extracts. The 90-kDa RBM5 protein decreased after CCI+HS. In contrast, RBM5 staining appeared greater in nuclei of hippocampal neurons after CCI+HS—suggesting that neuronal RBM5 is upregulated in select brain regions after injury. Anti-RBM10 antibody detected a 100/120-kDa double band in isolated neurons and whole brain tissue extracts, and increased after CCI+HS. Neuronal RBM10 knockdown exacerbated STS-induced caspase activation revealing potential antiapoptotic functions. Finally, SF1 protein decreased after CCI+HS.
Materials and methods
Chemicals and Reagents
Retinoic acid (RA) was purchased from SIGMA (St Louis, MO, USA). Dimethyl sulfoxide (DMSO) was purchased from SIGMA. Hematoxylin and Eosin Y (H&E) stains were purchased from Thermo Fisher-Shandon (Pittsburgh, PA, USA). Fluorojade B (FJB) was purchased from Chemicon (Temecula, CA, USA). Primary antibodies were purchased from: Anti-RBM5 (Atlas Antibodies, Stockholm, Sweden; Cat#HPA018011, Lot#R07119), Anti-RBM10 (purchased via Sigma but generated by Atlas Antibodies; Cat# HPA034972, Lost#R32333), Anti-SF1 (Atlas Antibodies; Cat#HPA018883, Lot#R07869), Anti-SF3A subunit 1 (purchased via Sigma but generated by Atlas Antibodies; Cat#HPA000690, Lot#R00090) Anti-c-FLIPL/s (H-202) (Santa Cruz, Dallas, TX, USA; Cat#sc-8347, Lot#J2010), Caspase-2L/s (BD Biosciences/Pharmingen, San Jose, CA, USA; Cat#51-1395GR), Anti-TNF-α (TNF-α; Cell Signaling Technology, Danvers, MA, USA; Cat#3707), Anti-Poly ADP ribose polymerase (PARP) Total/cleaved (Cell Signaling Technology; Cat#9532), Anti-Caspase-9 Total/cleaved (Cell Signaling Technology; Cat#9502), Anti-Caspase Cleavage Products (Cell Signaling Technology; Cat#8698), Anti-α-Fodrin (Enzo Life Sciences, Farmingdale, NY, USA; Cat#BML-FG6090-0500, Lot#08231207), Anti-LETM1 Domain-Containing Protein 1 (LETMD1; Lifespan Biosciences, Seattle, WA, USA; Cat# LS-C116277), Anti-amyloid precursor-like protein 2 (APLP2; purchased via Sigma but generated by Atlas Antibodies; Cat# HPA039319, Lost#R326082), Anti-α-Tubulin (Cell Signaling Technology; Cat#2144). Goat anti-rabbit and goat anti-mouse secondary antibodies were purchased from Life Technologies (Grand Island, NY, USA).
Animals
All animal work was approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh. Protocols follow recommendations established by the American Medical Veterinary Association Guideline for Euthanasia. Studies were designed to minimize pain and suffering, and to use the minimum number of animals required to achieve significance. Animals were granted ab libitum accesses to food and water. All animals were maintained on 12 hours light/dark cycle.
Traumatic Brain Injury Mouse Model
In all, 12- to 15-week-old male C57BL/6 mice (Jackson Laboratories, Bar Harbor, ME, USA) were used for CCI+HS. Mice were injured by CCI+HS as described by our group. 16 CCI injury: Briefly, anesthesia was initiated by 4% isoflurane in 70% N2O/30% O2. Animals securely placed inside a stereotaxic frame were positioned under isoflurane nose-cone. Isoflurane was reduced to 2% for maintenance anesthesia. Body temperature recorded via a rectal probe, and catheters inserted into the left femoral artery and vein. The head was shaved, sterilized with betadine, and skin opened. A craniotomy over the left parietal cortex was introduced via a dental drill. A pressure-driven pneumatic impactor, with a 3-mm steel flat tip, was applied to the brain for CCI. The magnitude of injury is controlled by impact velocity and depth of tip penetration into the brain. Mild-moderate CCI (velocity 5/ms; depth 1.0 mm) was employed for combined injury experiments. Combined hemorrhagic shock: 5 minutes after CCI, isoflurane was reduced to 0.5%, and blood removed via femoral vein connected to a syringe containing citrate anticoagulant. Blood was withdrawn over 15 minutes until mean arterial blood pressure (MABP) reached 25 to 27 mm Hg in mice. Shock was maintained 35 minutes by either removing or readministering shed blood in 50 μL aliquots (to clamp target MABP). After HS, mice were volume resuscitated using 20 mL/kg lactated ringers. Target MABP during the 90-minutes ‘Pre-Hospital Care Phase’ was 70 mm Hg. Additional 10 mL/kg aliquots of lactated ringers were administered over 90 minutes if required to maintain target MABP. Finally, a ‘Hospital Phase’ was initiated by returning shed blood to mice over 15 minutes. Blood pressure normally returns to baseline levels (∼90 mm Hg). Mice were allowed to recover after CCI+HS injury (weaned from isoflurane and catheters removed). Finally, mice were given supplemental oxygen 30 minutes before return to animal housing for stability. Naïve mice were not exposed to experimental manipulations. Sham mice received craniotomy and catheterization but neither CCI nor HS. Mice were randomized to sham/injury and killing timepoint. Naïve mice were not randomized. For biochemistry studies, mice were killed 4, 24, 48, and 72 hours after injury, tissues snap-frozen in liquid nitrogen, and stored at −80°C until homogenization in radioimmunoprecipitation assay (RIPA) buffer. For immunohistochemistry/staining studies, mice were anesthetized, killed at 48 hours by transcardially perfusing with saline followed by 10% formalin, and brains collected for subsequent processing with antibodies, FJB staining, or H&E staining as previously described by our group. 16 Investigators were not blinded to injury groups.
Primary Cortical Neuron Culture
Cortical neuron cultures were prepared as reported by our group.
17
Briefly, embryos were isolated from timed pregnant (E16 to 18) Sprague Dawley (SD; Charles River, Wilmington, MA, USA) rats. Minced cortical tissue transferred into a 15-mL tube and spun at 5 minutes/200 g/4°C. Tissue was trypsinized 8 minutes at 37°C, triturated (1% DNAse in Hanks Balanced Salt Solution) 10 times through a fire-polished glass Pasteur pipette, and resuspended in Neurobasal/B27 (Life Technologies). Neurobasal Media/B27 plating media was supplemented with 25 μ
Lentivirus Transduction and Differentiation of Human Neuronal SHSY5Y Cells
Human SHSY5Y cells were purchased from ATCC (Manassas, VA, USA). Lentivirus work was approved by the Institutional Biosafety Committee of the University of Pittsburgh. Pre-made BSL-2 lentivirus aliquots containing 200 μL of 1 × 106 infectious particles were purchased from Santa Cruz Biotechnology (Dallas, TX, USA), including nontargeting (NT) shRNAs; Cat#sc-108080, RBM5 targeting shRNAs; Cat#sc-10674-V1, and RBM10 targeting shRNAs; Cat#sc-76362-V. Undifferentiated SHSY5Ys were maintained in Opti-MEM culture media (Life Technologies) supplemented with 10% FBS (GE Healthcare Life Sciences, HyClone Laboratories, Logan, UT, USA). Cells were seeded onto small T-12.5 cm flasks. Twenty-four hours later at ∼50% confluence SHSY5Ys were transduced with lentivirus (1 flask per each group). Transduction media consisted of 50 μL virus+2 μg/mL Polybrene (Santa Cruz) in 2 mL of growth media. Seventy-two hours after start of transduction, cells were washed twice with basal Opti-MEM (no FBS), trypsinized for ∼2 minutes (0.05% Trypsin/EDTA; Life Technologies), protease activity quenched in growth media, centrifuged 5 minutes/200 g/4°C, resuspended in fresh growth media, and seeded onto 75 cm2 sterile flasks (same protocol used for all subsequent propagations). Forty-eight hours later, cultures received fresh growth media containing 1 μg/mL puromycin dihydrochloride (Santa Cruz). Lentiviral expression plasmids contain a puromycin resistance gene. Cells not transduced by lentivirus are killed by puromycin. Pilot dose-death experiments found that 1 μg/mL puromycin killed >90% nontransduced SHSY5Ys by 48 to 72 hours. For the first 3 selection days, media was replaced each day to remove debris. At confluence, they were again propagated into new 75 cm2 and 225 cm2 sterile flasks and selected 3 more days (6 days total puromycin selection). Thawed cryogenically stored NT and RBM knockdown cells were subjected to an additional 3 to 4 extra days of puromycin selection in preparation for different downstream experiments. Puromycin selection was discontinued a minimum of 3 days before experimentation in all studies. Cells were counted using a Cellometer (Nexcelom Bioscience, Lawrence, MA, USA). In all, 1 × 106 cells/well were seeded onto four individual 6-well plates (n=2 replicates in each group per plate). Undifferentiated cells were harvested in RIPA buffer 24 hours later (two plates or n=4 replicates/group). The remaining two plates were differentiated. Growth media was replaced with differentiation media (Opti-MEM/1%FBS/10 μ
Western Blot
As reported by our group, 17 tissues and cells were sonicated in RIPA buffer containing EDTA, protease inhibitors, and phosphatase inhibitors (Thermo Scientific-PIERCE, Rockford, IL, USA). Homogenized material was spun 10 minutes/16,000 g/4°C. Supernatants were collected for downstream SDS-PAGE. Protein quantification was determined by BCA assay (Thermo Scientific-PIERCE). In all, 10 to 20 μg protein was mixed with 2 × or 4 × Laemmli loading buffer (Bio-Rad, Hercules, CA, USA), heated to 95°C/5 minutes, and loaded onto 10- or 15-well pre-cast TGX 1% to 15% gradient gels (Bio-Rad). Gels were run at 150 V. Protein was transferred onto polyvinylidene fluoride membranes (GE Healthcare, Pittsburgh, PA, USA). Precision Plus Kaleidoscope protein standards (Bio-Rad) were included for molecular weight (MW) estimation. Membranes were washed 10 minutes in 1 × Tris-Buffered saline (TBS; Bio-Rad). Traditional housekeeping gene loading controls showed variable changes after injury (Supplementary Figure 1). Staining is a more accurate method to control for protein loading in injured brain.18, 19 Therefore, densitometry of RBM5, RBM10, SF1, and SF3A was normalized to total protein stain in CCI+HS injury experiments (Supplementary Figure 2). Membranes were stained with reversible Swift Membrane Stain (Fisher Scientific, Waltham, MA, USA). Stained blots were saved to file using a flatbed scanner and analyzed by densitometry. Stain was quickly removed, membranes washed in TBS, and blocked 1 hour in TBS-Tween-20 (TBST)+7.5% blotting grade milk (TBS-T/milk). Primary antibodies were prepared in TBS-T/milk, and blots incubated overnight on a slow rocker at 4°C. Blots were washed 3 × /5 minutes in TBS. Secondary antibodies were prepared in TBS-T/milk and incubated 2 hours on an orbital shaker. Blots were washed 3 × /5 minutes in TBS, incubated ∼2 minutes with ECL-2 HRP-detection reagent (Thermo Fisher-PIERECE), and membranes exposed to film in a dark room. Developed films were saved to file using a flatbed scanner, images compiled in Photoshop, and densitometry analyzed by UN-SCAN-IT software (Silk Scientific, Orem, UT, USA).
Immunohistochemistry and Tissue Staining
At 48 hours after injury, anesthetized mice were flushed with heparinized ice-cold saline by transcardial perfusion into the left ventricle. Tissues were immediately flushed with 10% buffered formalin. Brains removed and postfixed in 10% buffered formalin for 72 hours. Brains were cut into 3 mm coronal sections, dehydrated by serial alcohol preparations, cleared with Xylene, and embedded into paraffin wax. Coronal blocks were cut into 5 μmol/L sections via a microtome, and mounted onto glass slides. Avidin-biotin complex/3′,3′-Diaminobenzidine was used for visualization of RBMs by immunodetection. Briefly, deparaffinized tissue slides were subjected to antigen retrieval by heat-microwave in Antigen Decloaker solution (Biocare Medical, Concord, CA, USA). Slides were incubated 30 minutes in 3% H2O2/methanol to quench endogenous peroxidase activity. Slides were blocked 20 minutes in automation buffer containing 3% normal goat serum. Primary antibodies were diluted in 3% goat serum/buffer and incubated overnight at 4°C. Slides were washed and incubated 1 hour with biotinylated goat anti-rabbit IgG diluted in buffer (Vector Laboratories, Burlingame, CA, USA). Slides were washed, incubated 1 hour with Avidin-biotin complex reagent (Vector Laboratories), and treated with 3′,3′-Diaminobenzidine reagent (Vector Laboratories) for 5 to 10 minutes. Sections were counterstained with hematoxylin for 30 seconds. Slides were mounted with a coverslip and images collected on a Nikon Eclipse 90i microscope (Nikon). Hematoxylin and Eosin and FJB staining procedures were performed in CCI+H mice as reported by our group. 16 Sections were deparaffinized, treated with hematoxylin stain (Thermo Scientific, Waltham, MA, USA), washed, treated with Bluing reagent (Thermo Scientific), washed, and treated with Eosin Y (Thermo Scientific). Alternatively, sections were stained 30 minutes with FJB, washed, mounted, and imaged.
Statistical Analysis
Data were analyzed using the GraphPad PRISM software (GraphPad Software Inc., La Jolla, CA, USA). Multiple comparisons were analyzed by ANOVA and Newman-Keuls Multiple Comparison post-hoc analysis. Data were considered as significant at P<0.05 using two-tailed tests. All graphs were created in GraphPad PRISM software and show mean+s.e.m. Experiments in which raw data of group comparisons were acquired as relative units (i.e., densitometry/pixel intensity), data were standardized by expressing group differences as values between 0 and 1 (i.e., on the y axis). In Figure 7E, graphed densitometry data of caspase cleavage products normalized to α-tubulin were transformed to log(Y) values for statistical comparisons to correct for nonnormality in data distribution.
Results
RNA Binding Motif 5 Regulates Caspase Activation and Protein Expression/Splicing in Neurons
Human neuronal SHSY5Ys were transduced with lentivirus to deliver NT control, RBM5 targeting, or RBM10 targeting shRNAs. RNA binding motif 5 is a 92-kDA protein and reportedly migrates on SDS-PAGE at ∼90 kDa but also commonly observed at ∼120 kDa. 20 The ∼120-kDa form of RBM5 was detected in SHSY5Ys, and decreased by knockdown (Supplementary Figure 3A). RNA binding motif 10 is a 103-kDa protein. Both an ∼100-kDa and ∼120-kDa RBM10 signal were detected in SHSY5Ys and decreased by knockdown (Supplementary Figure 3D).
Stably expressing/transduced SHSY5Ys cells were differentiated to neuronal phenotype by 7 days RA treatment. Neurons were administered the caspase activator STS for 8 hours. Knockdown of RBM5 decreased PARP cleavage (Figures 1A and 1D), decreased caspase-9 activation (Figure 1C), and blocked cleavage of a major ∼70 kDa caspase substrate (Figures 1B and 1E). Knockdown of RBM10 increased STS-induced caspase activation. It increased PARP cleavage (Figures 1A and 1D), increased caspase-9 activation (Figure1C), and increased cleavage of the ∼70-kDa caspase substrate (Figures 1B and 1E).
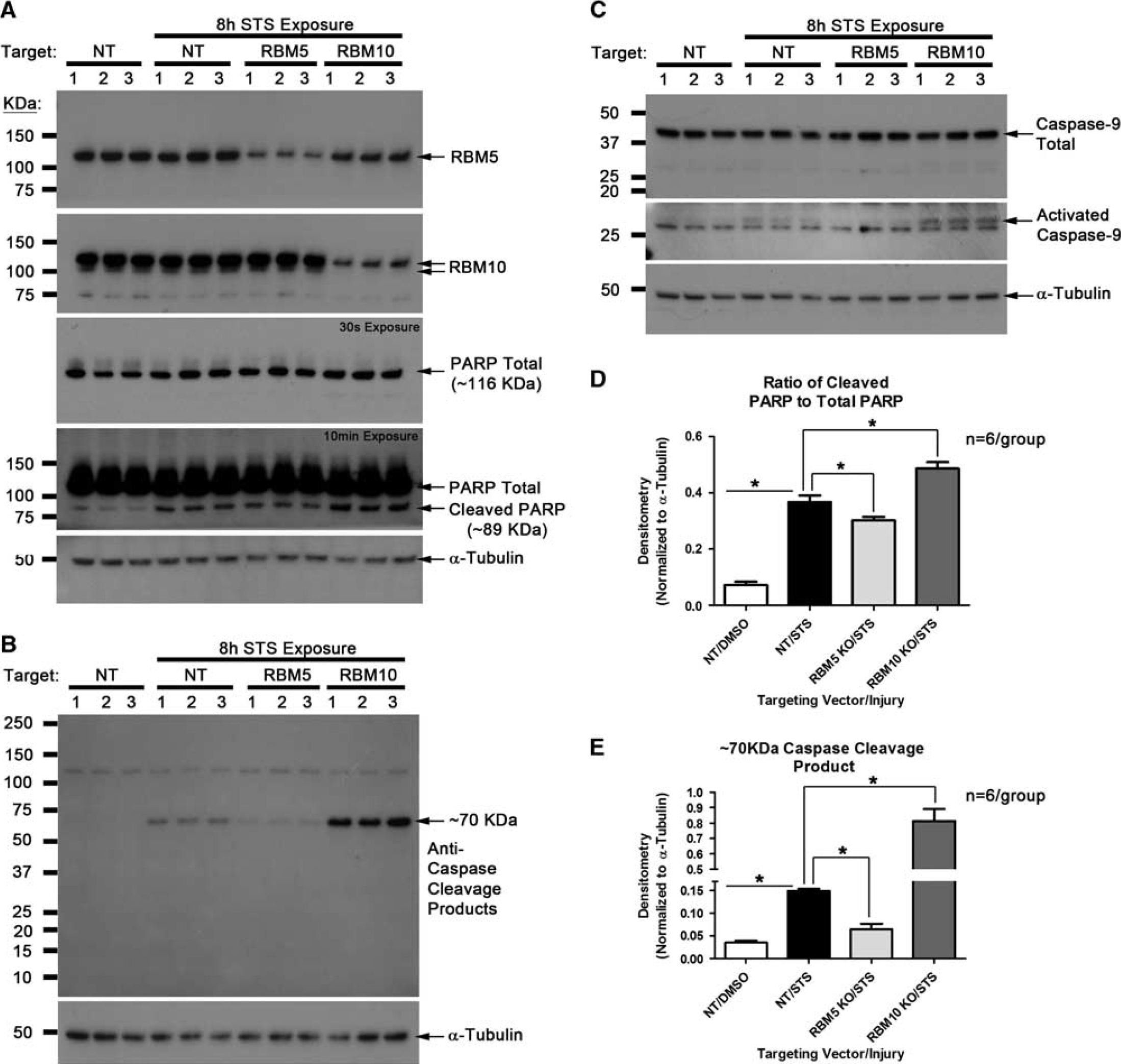
RNA binding motif 5 (RBM5) knockdown decreases staurosporine (STS)-induced caspase activation. Differentiated human neurons were subjected to 150 nmol/L STS injury for 8 hours. (
Cellular FLICE-like inhibitory protein and caspase-2 are known RBM5 splicing targets. We first measured whether the neuronal differentiation process affected protein variant expression of c-FLIP (L/s) and caspase-2 (L/s). Also, the c-FLIP and caspase-2 antibodies used in this experiment were reported to detect (L/s) splice variants in human cells.8, 21 Neuronal differentiation appeared to increase ∼55 kDa c-FLIP(L) as well as ∼26 kDa c-FLIP(s) variant (Figure 2A). Long exposure times required to observe c-FLIP(s) protein are consistent with its high degradation rate relative to stable c-FLIP(L). 22 Only the caspase-2(L) variant was present in neurons (Figure 2A).
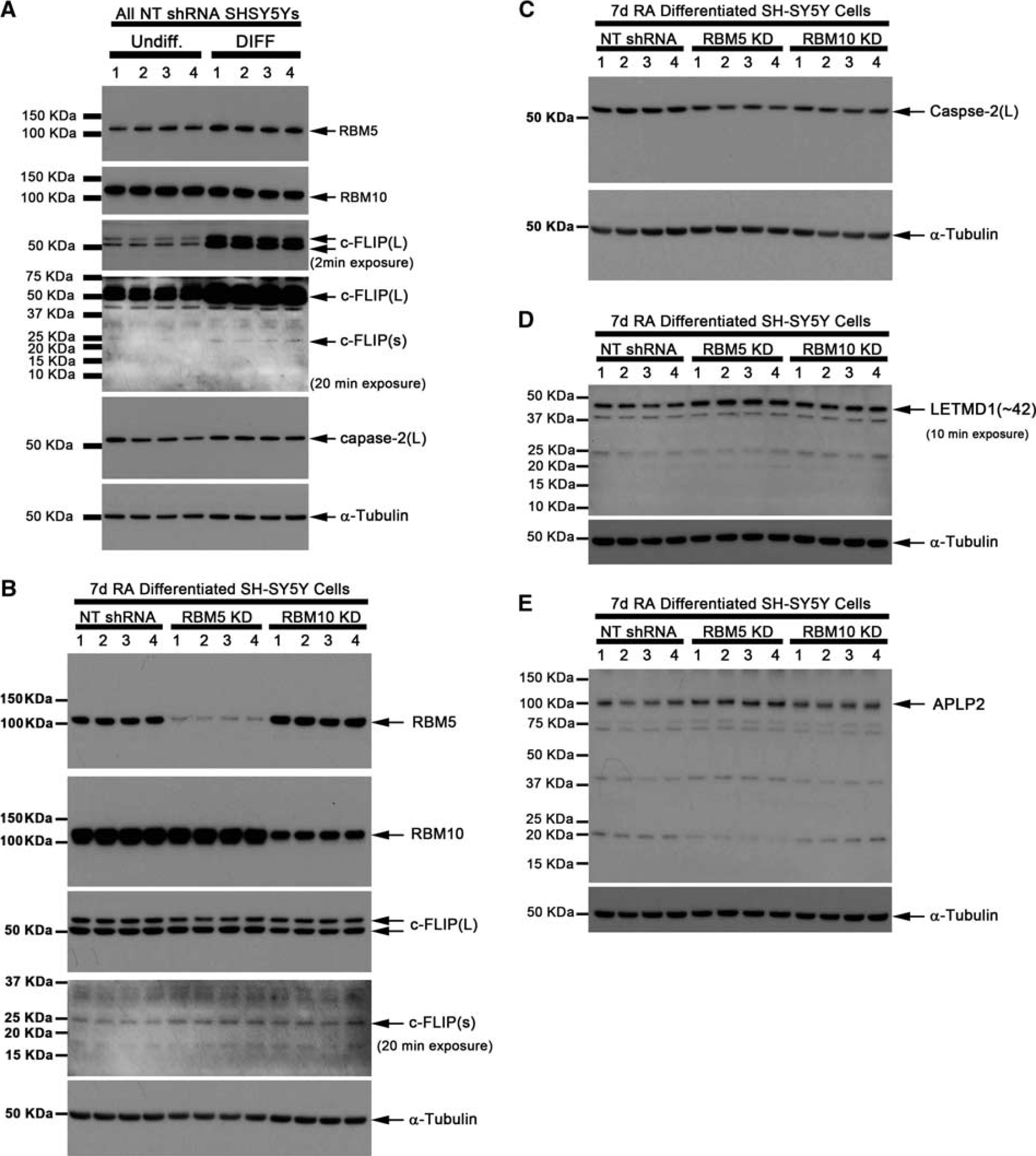
RNA binding motif 5 (RBM5) knockdown increases c-FLIP(s), LETM1 Domain-Containing Protein 1 (LETMD1), and APLP2 but decreases caspase-2(L) in human neuronal SHSY5Y cells. (
The effect of RBM5 or RBM10 knockdown on c-FLIP and caspase-2 expression/splicing in differentiated neurons was studied. Knockdown of RBM5 appeared to increase c-FLIP(s) but decrease caspase-2(L) protein levels (Figures 2B and 2C). Caspase-2(s) was not detected in any groups/treatments. We also measured levels of anti-apoptotic protein LETMD1, and levels of neuron development regulator APLP2—both reportedly RBM10 splicing targets. Knockdown of RBM5 and RBM10 appeared to increase LETMD1 protein in neurons (Figure 2D). Knockdown of RBM5 appeared to increase APLP2 (Figure 2E).
Splicing Factor Disturbances after Traumatic Brain Injury in Mice
Figure 3A illustrates SFs of interest (RBM5, RBM10, SF1, and SF3A) and summarizes their known roles in RNA splicing. We first characterized specificity of antibodies to detect SFs in brain. Consistent with prior reports using custom generated antibodies to detect RBM5, 20 we observed multiple signals across the MW range including an ∼90-kDa and ∼120-kDa product (Figure 3B). Of note, we tested several commercially available antibodies and only the anti-RBM5 antibody made by Atlas Antibodies detected RBM5 in our hands. We observed ∼100 kDa (i.e., RBM10 variant 1; RBM10-V1) and ∼120 kDa bands across the MW range in brain (Figure 3C). Splicing factor 1 encodes an ∼75 kDa protein and the only signal detected in brain (Figure 3D). The large subunit of the SF3A complex is ∼120 kDa and the only signal observed in brain (Figure 3E).
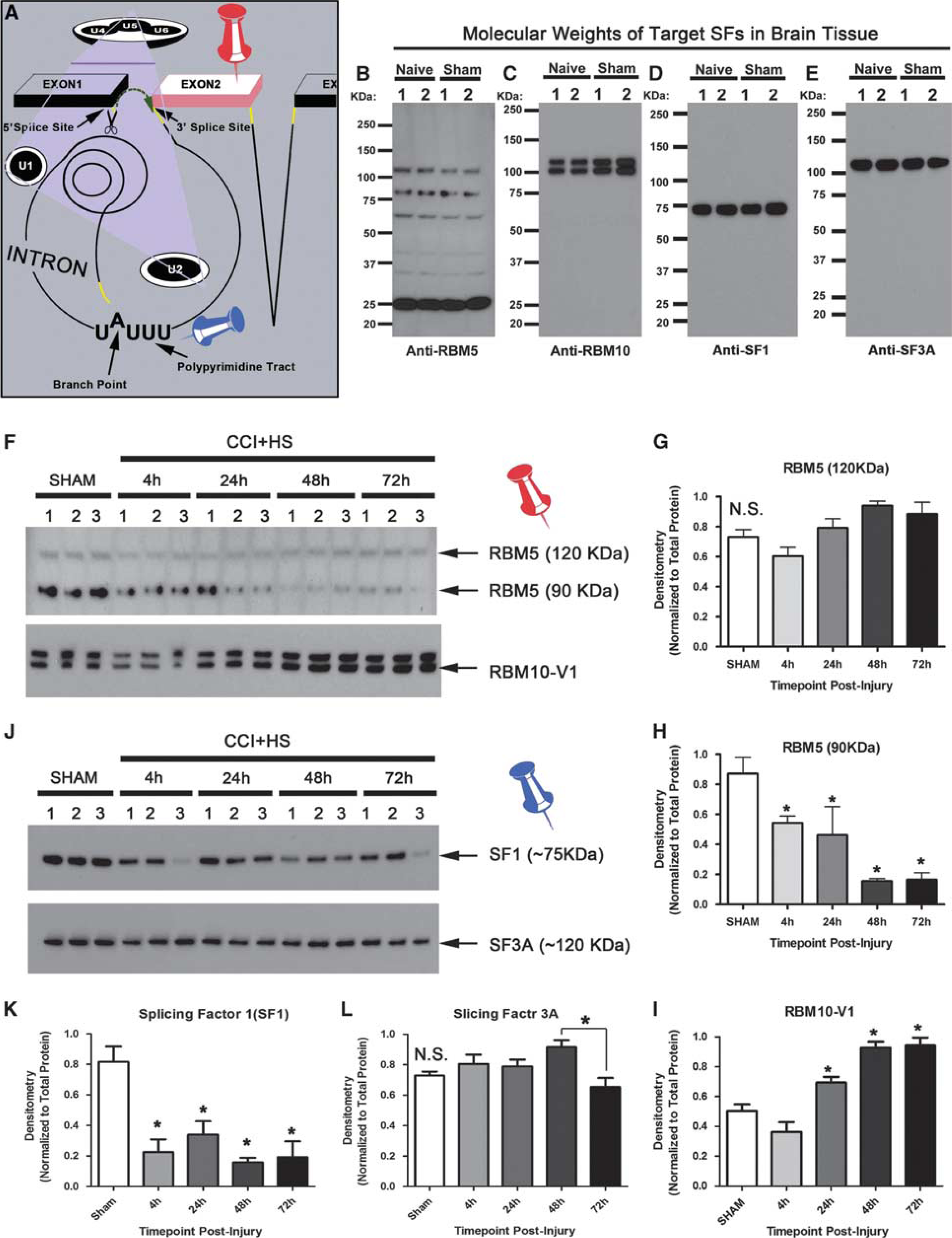
Brain disturbances in splicing factor (SF) levels after CCI+HS. (
We next tested whether SFs are disturbed in the acute/subacute phase after CCI+HS—a brain injury model to maximize secondary neuronal death in TBI. 16 Splicing factors dynamically altered after brain injury. In all, ∼120 kD RBM5 did not increase after injury compared with sham levels (Figures 3F and 3G) but was a larger component of the total RBM5 protein pool at later time points (Supplementary Figure 4B). In contrast, ∼90 kDa RBM5 decreased after injury (Figures 3F and 3H). RBM10-V1 significantly increased by 24 hours after injury compared with shams (Figure 3F), and doubled by 48 to 72 hours after injury (Figure 3I). The higher migrating ∼120 kDa RBM10 band was significantly higher by 48 hours after injury (Supplementary Figure 4C). Caspase cleavage products also increased by 48 hours after injury (Supplementary Figures 5G and 5H). Splicing factor 1 levels were significantly low at each time point postinjury compared with sham (Figures 3J and 3K). Splicing factor 3A levels did not differ from shams after injury but 48-hour levels were slightly increased relative to the 72-hour time point (Figure 3L).
We focused immunohistochemistry studies on RBM5 and RBM10 at 48 hours after injury because both increased by this time point. Levels of RBM5 appeared to increase in subfield neurons of the hippocampus 48 hours after injury (CA1, CA3, and DG; Figure 4). RNA binding motif 10 staining showed similar changes in the hippocampus (Figure 5). RNA binding motifs also appeared to increase in some cortical neuron populations but not striatal or cerebellum neurons (Figure 6). Circled cells in cortex/striatum show magnified fields of view from background images—the speckled pattern of RBM staining in nuclei of shams is consistent with spliceosomal regulators. 23 Also, RBM10 staining in some Purkinje neurons of the cerebellum seemed to favor cytoplasmic versus nuclear localization (Figure 6—black arrows in RBM10 cerebellum panels).
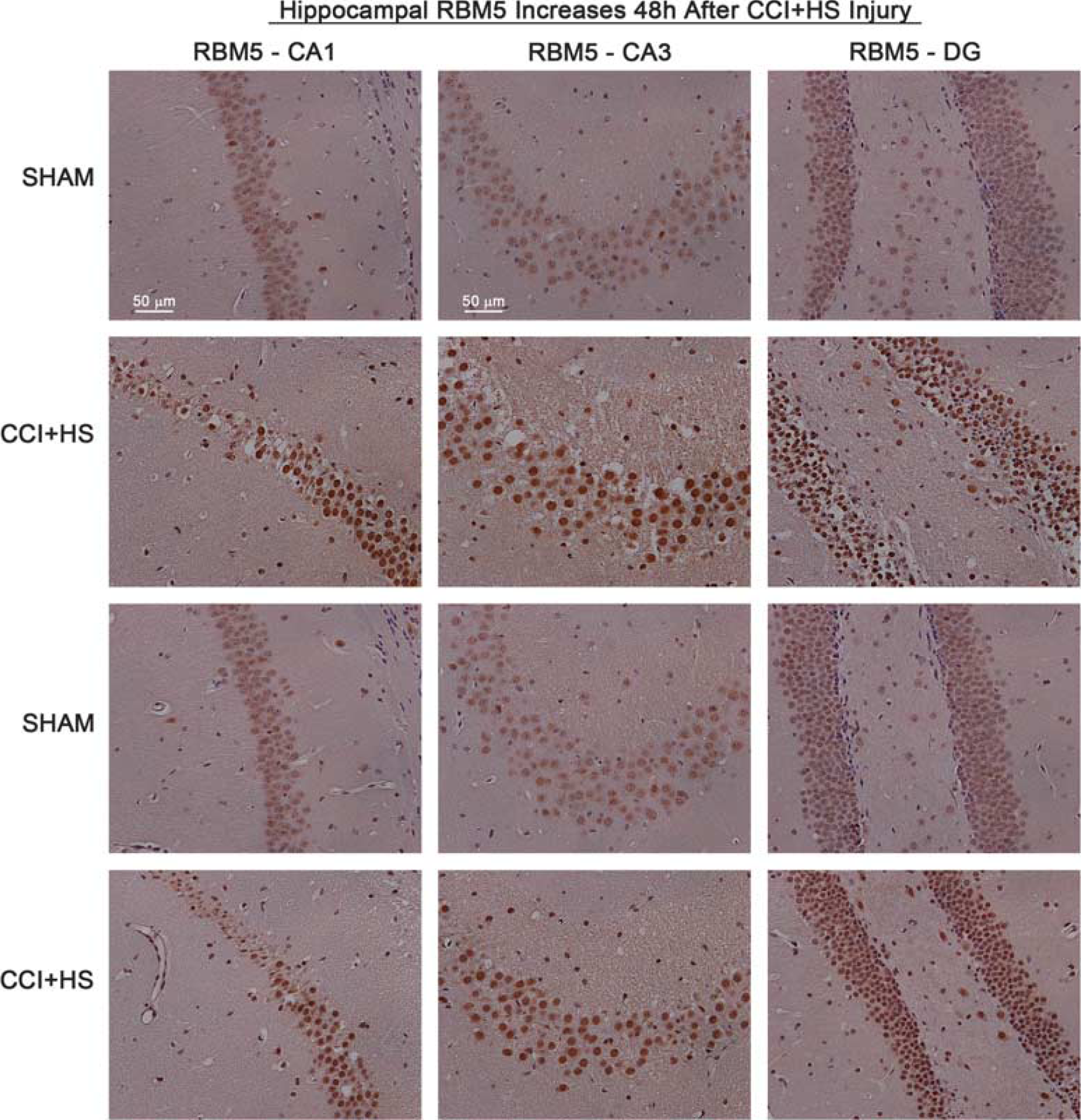
Increased RNA binding motif 5 (RBM5) staining in injured hippocampus. Brain tissue sections from sham (n=2) and CCI+HS injured mice (n=2) were collected 48 hours after injury and probed with antibody against RBM5. Immunohistochemistry shows × 20 magnification of CA1, CA3, and DG subregions. RBM5 staining (brown) is increased in all hippocampal regions of CCI+HS mice. CCI, controlled cortical impact; HS, hemorrhagic shock.
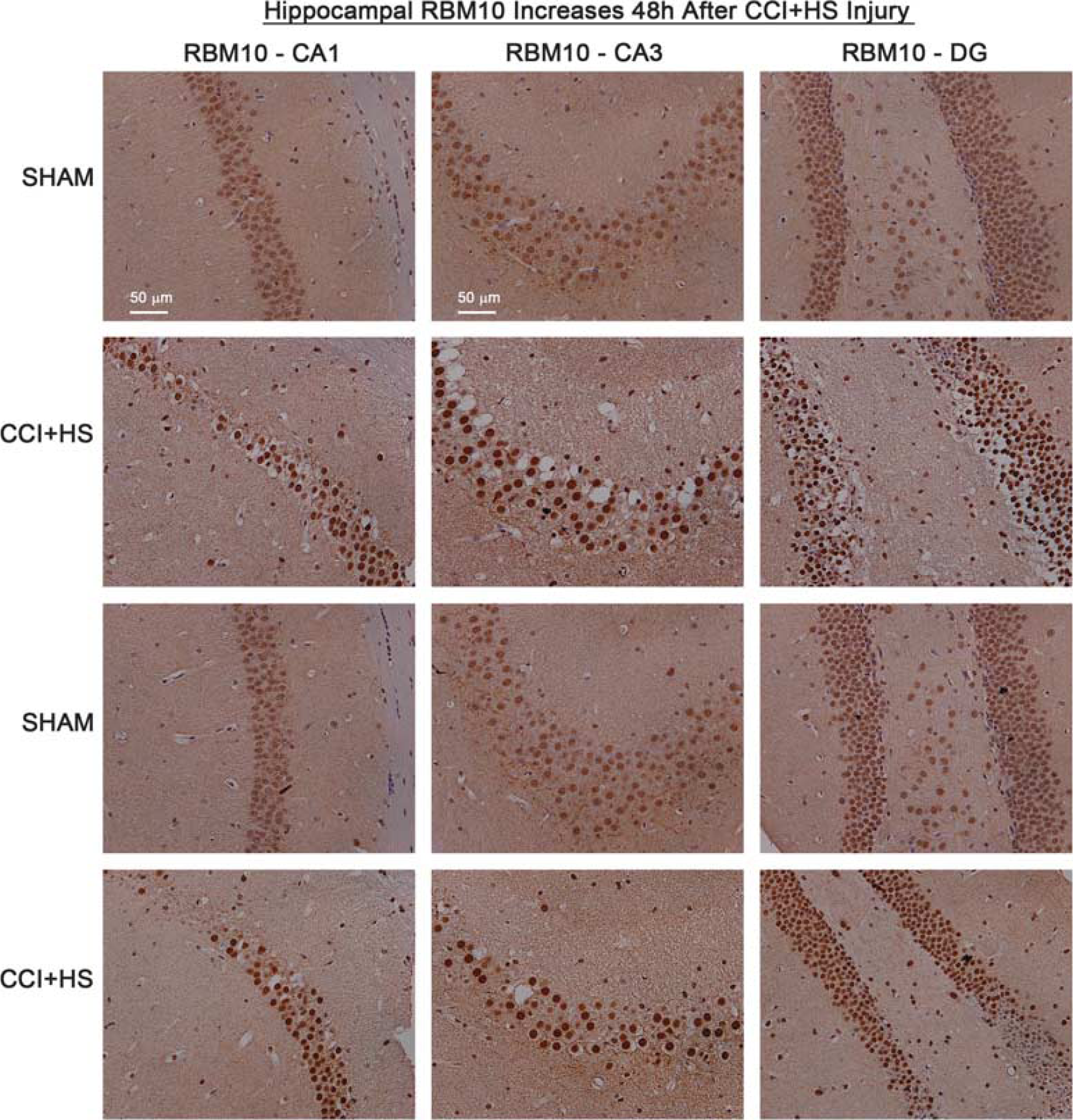
Increased RNA binding motif 10 (RBM10) staining in injured hippocampus. Brain tissue sections from sham (n=2) and CCI+HS injured mice (n=2) were collected 48 hours after injury and probed with antibody against RBM10. Immunohistochemistry shows × 20 magnification of CA1, CA3, and DG subregions. RBM10 staining (brown) is increased in all hippocampal regions of CCI+HS mice. CCI, controlled cortical impact; HS, hemorrhagic shock.
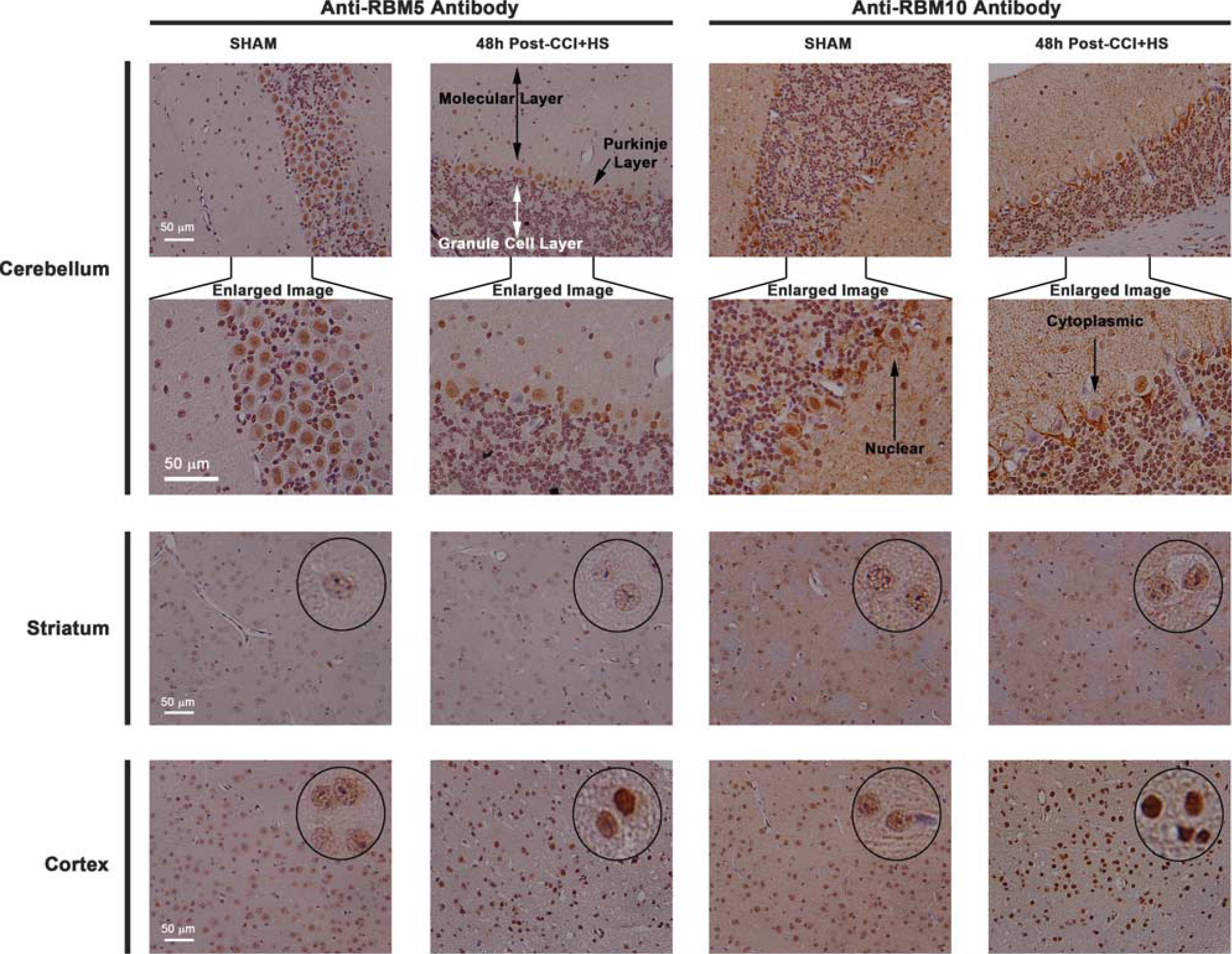
Increased RNA binding motif 5 (RBM5)/10 staining in injured cortex but not in ipsilateral striatum or cerebellum. Brain tissue sections of a sham and CCI+HS injured mouse at 48 hours after injury were probed with antibodies against RBM5 and RBM10 (× 20 magnification). Cerebellar images at top (first row across) show representative pictures for injury group/RBM target. Cerebellar areas of interest were magnified in Photoshop (second row across) to highlight nuclear/cytoplasmic differences of RBM10 staining in some cerebellar neurons. Injury did not affect faint RBM staining in striatal neurons (third row across). Injury increased RBM staining in cortical neurons (fourth row across). Black circles show randomly selected neurons within each image field that were magnified in Photoshop to show speckled staining pattern.
RNA binding motif staining in mouse tissues appeared highly neuronal and restricted to nuclei. To confirm this observation, we performed colocalization studies using primary rat cortical neurons. Day in vitro 12 neurons were probed with antibodies against RBM5/10 and signals colocalized with neurons and astrocytes. RBM5/RBM10 (RED) almost exclusively stained neurons (RBM5; Figures 7A to 7C; RBM10; Figures 7G to 7I). Little staining was observed in astrocytes (RBM5; Figures 7D to 7F; RBM10; Figures 7J to 7L). RNA binding motif 5/10 staining was restricted to nuclei and colocalized with DAPI (RBM5; Figures 7B and 7E; RBM10; Figures 7H and 7K). Protein homogenates were prepared from DIV 10 pure rat cortical neurons (i.e., ARA-c added to inhibit glial proliferation) and probed with antibodies against RBM5/10. The ∼120-kDa RBM5 signal was the only band detected in primary rat cortical neurons (Figure 7M). Both the ∼100-kDa and ∼120-kDa RBM10 bands were detected (Figure 7N).
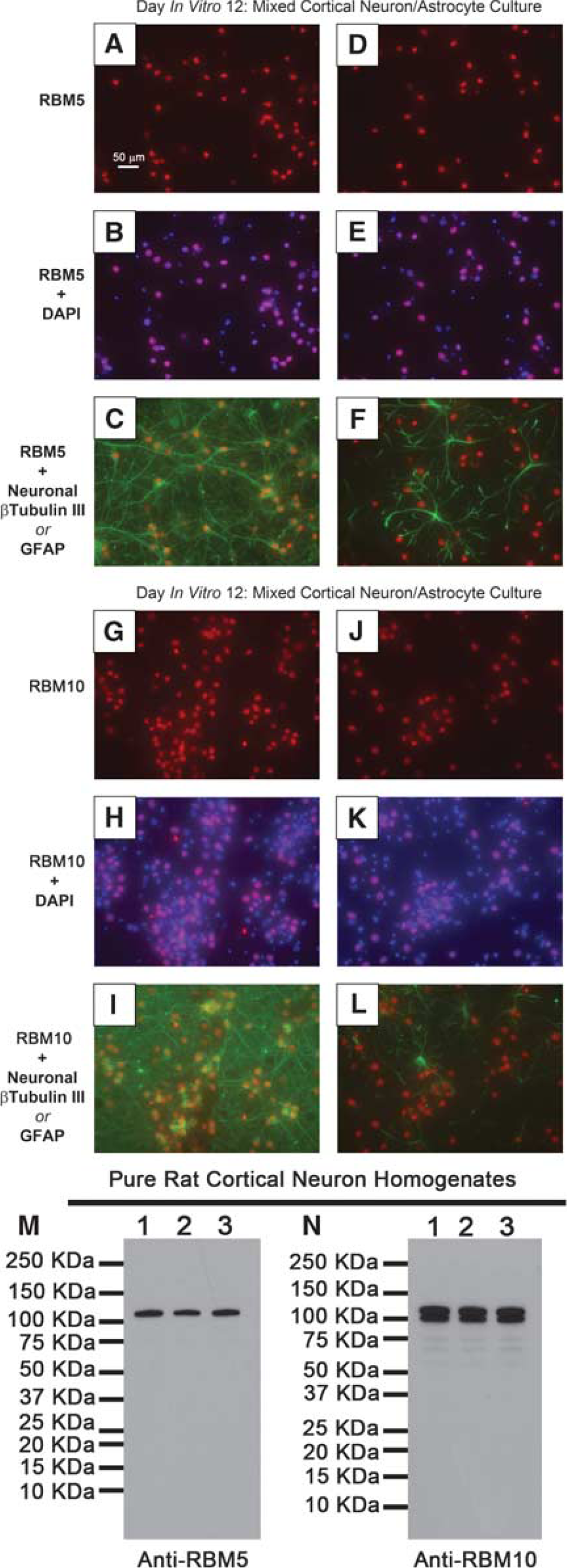
Characterization of RNA binding motif 5 (RBM5)/10 in cultured primary cortical neurons. Day in vitro (DIV) 12 mixed cortical neuron/astrocyte cultures were prepared for immunofluorescence. Images show × 40 magnification of (
Discussion
RNA Binding Motif 5 and RNA Binding Motif 10 Regulate Cell Death Biochemistry in Neurons
Caspase-2 and c-FLIP regulate neuronal death/survival.24, 25, 26 Both genes encode multiple protein splice variants with different effects on cell death. The caspase-2(L) variant is toxic but caspase-2(s) is protective. Cellular FLICE-like inhibitory protein is mostly protective but c-FLIP(s) is more potent than c-FLIP(L). No therapy currently exists to selectively upregulate protective splice variants.
The splicing factor RBM5 is abundant in reproductive organs and brain relative to other tissues. 27 RNA binding motif 5 may be a potential target to enhance pro-survival splicing of select genes including caspase-2 and c-FLIP. In cancer cells, RBM5 inhibition increases protective caspase-2(s) and c-FLIP(s) levels. However, it is unknown whether RBM5 also regulates splicing and/or promotes cell death in neurons. Here, we show that lentivirus/shRNA knockdown of RBM5 appears to increases c-FLIP(s), and reduce caspase-2(L) in human neuronal cells. Our results are consistent with findings by others in cancer cells. We did not observe an increase in caspase-2(s). Caspase-2(s) reportedly exists as a transient mRNA species and subject to rapid nonsense mediated decay. 28 Thus, it may not be a major effector of neuron survival—our results seem to support this idea because caspase-2(s) protein was undetectable in SHSY5Ys. To test the functional effect of protein changes on survival, we treated RBM knockdown neurons with caspase activator STS. As predicted, RBM5 inhibition decreased caspase activation. Surprisingly, RBM10 inhibition increased STS-induced caspase activation. Results confirm that RBM5 promotes caspase activation and pro-death splicing in neurons. Finally, RBM5 knockdown also increased APLP2—a key protein involved in cortical neuron development and synaptic function in adult mice.29, 30, 31 This novel finding suggests that RBM5 may also regulate genes involved in neuronal function.
Inhibition of RBM10 did not increase c-FLIP or caspase-2 proteins in SHSY5Ys. This finding is supported by others. Inoue et al 5 report that RBM10 knockdown did not alter c-FLIP nor caspase-2 splicing in HeLa cells. 5 Furthermore, a recent (2013) study reported that RBM10 knockdown in human embryonic kidney 293 (HEK293) cells caused 304 mRNA exon splicing changes (256 inclusion events and 48 exclusion events). 32 Caspase-2 and c-FLIP were not reported as gene targets in those studies. However, LETMD1 and APLP2 were RBM10 gene targets—prompting us to examine them here. We confirm that inhibition of RBM10 or RBM5 appears to upregulate LETMD1 expression in SHSY5Ys. LETM1 Domain-Containing Protein 1 may be neuroprotective because it protects cells from UV and STS induced death. 33
Prior studies suggest RBM5 and RBM10 have compensatory regulation (Supplementary Tables 3 and 4 in study by Wang et al 32 ). Our findings agree with those observations. Knockdown of RBM10 in SHSY5Ys significantly raised RBM5 levels (Supplementary Figures 3A and C). Furthermore, decreased RBM5 levels correlate with increased RBM10 in sham versus injured mouse brain homogenates (Supplementary Figure 4A).
Exon Splicing Factors RNA Binding Motif 5 and RNA Binding Motif 10 are Disturbed after Traumatic Brain Injury
We predicted that SF disturbances might occur after TBI. Here, we report that RBM5/10 increase in hippocampus and cortex 48 hours after CCI+HS. Increased RBM levels coincide with neuronal degeneration (FJB), and neuronal loss (H&E) in hippocampus (Supplementary Figure 6; Figure 7). In addition, TNF-α, caspase cleaved α-Fodrin, and caspase cleavage products increased after CCI+HS (Supplementary Figure 5). Previous studies in cancer cells show that RBM5/10 overexpression sensitizes cells to TNF-α induced death and caspase activation (as well as the death ligands TNF-related apoptosis-inducing ligand and Fas ligand). Here, we confirm that RBM5 promotes caspase activation in neuronal cells. It is possible that increased RBM5 in brain similarly sensitizes hippocampal/cortical neurons to die by extracellular TNF-α and other death ligands after injury. Future studies should examine whether RBM5 overexpression in primary rat cortical neurons sensitizes them to die by treatment with TNF-α, TNF-related apoptosis-inducing ligand, and Fas ligand. Alternatively, increased RBM10 may represent an endogenous neuroprotective response but again this hypothesis needs to be challenged by overexpression studies in primary neurons.
Western analysis of brain tissue homogenates show temporal changes of RBM5 proteins after injury. The calculated MW of RBM5 is 92 kDa. Approximately 90 kDa RBM5 decreased by 48 hours after injury. Approximately 120 kDa RBM5 levels did not differ in shams versus injury. However, the ratio of ∼120 kDa/∼90 kDa increased postTBI (Supplementary Figure 4B). Also, RBM5 staining appeared greater in neurons of the injured hippocampus—potentially revealing increased expression of neuronal RBM5 in select brain regions. Western analysis failed to detect IHC-like changes. This may be due to nonneuronal sources of ∼120 kDa RBM5 diluting neuron-specific effects in whole tissue extracts. Consistent with the idea that neuronal RBM5 staining is detecting the ∼120-kDa protein, immunofluorescence studies in SHSY5Ys show that differences in nuclear RBM5 staining match protein changes after knockdown (i.e., protein and IF signal appear to be reduced in RBM5 knockdown cells but increased in RBM10 knockdown cells; Supplementary Figure 3). Future studies need to compare RBM5 protein levels in culture homogenates from neurons, astrocytes, oligodendrocytes, microglia, pericytes, and brain endothelial cells. Sutherland et al 20 suggest that both ∼90 kDa and ∼120 kDa forms of RBM5 are linked to cell death. Results from SHSY5Y studies suggest that ∼120 kDa RBM5 promotes cell death in neurons but the role of ∼90 kDa RBM5 is still unclear. The calculated MW of RBM10-V1 is 103 kDa. The ∼100-kDa and ∼120-kDa forms of RBM10 increase after injury in hippocampus and cortex. Both signals are readily detected in primary rat neurons and human neuronal cells.
A limitation of this study is that we could not correlate RBM5/10 changes with c-FLIP(L/s) and caspase-2(L/s) alterations after CCI+HS. Rodents do not express the c-FLIP(s) variant, precluding its detection in rodent tissues here. 34 However, both c-FLIP(L) and c-FLIP(s) increase after TBI in human brain. 25 Our results show that RBM5 inhibition appears to increase c-FLIP(s) and decrease caspase-2(L) in human neuronal cells. Thus, it is possible that RBM5 inhibition in humans could be a way to further augment c-FLIP(s) after TBI. We also measured caspase-2 in mouse brain homogenates but like human neuronal cells caspase-2(s) was undetectable (data not shown). Future exon microarray studies may reveal other major splicing targets of RBM5 in neurons.
Intron Splicing Factor 1 Is Disturbed after Traumatic Brain Injury
The scope of splicing disturbances after TBI is unknown. A few studies report changes in splicing of a few select proteins after TBI.35, 36, 37 Global splicing derangements could exist if key components of intron splicing are also disturbed. We measured levels of SF1 and SF3A—both important facilitators of intron and exon splicing in cells. Splicing factor 1 protein was markedly decreased in brain after CCI+HS. Levels of SF3A did not decrease below sham. Studies in human cells show that SF1 knockdown induces apoptosis. In contrast, knockdown of the 120-kDa subunit of SF3A (i.e., the subunit measured in our study) induces necrosis. 38 One potential explanation for this difference is that SF3A knockdown also markedly decreases total protein levels in human cells because general loss of pre-mRNA intron splicing decreases translatable mRNAs. 38 In contrast, SF1 depletion only slightly decreases total protein levels—suggesting a more restricted and selective set of intron/exon pre-mRNA gene targets. 38 Regardless the mechanism of cell death, both SF3A and SF1 seem essential for cell viability. Decreased SF1 after CCI+HS may cause intron splicing disturbances, which could affect cellular recovery after injury—whether SF1-dependent splicing disturbances exist after TBI deserves future study. Splicing factor 1 changes have been observed after global brain ischemia in gerbil. In those studies SF1 mRNA acutely peaks 4 hours after ischemia in resistant DG regions but vanished by 4 days in dying hippocampal CA1 subregion. 39 Future studies need to test whether SF1 protein is needed for neuronal viability, and thus if it may represent another important target for splicing therapies in brain.
Footnotes
Dr Travis C Jackson and Dr Patrick M Kochanek are inventors on a filed international PCT patent application (#PCT/US2013/040995) titled: ‘Small molecule inhibitors of RNA binding motif (RBM) proteins for the treatment of acute cellular injury.’
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.