
Abstract
Mitochondrial dysfunction may underlie both acute and delayed neuronal cell death resulting from cerebral ischemia. Specifically, postischemic release of mitochondrial constituents such as the pro-apoptotic respiratory chain component cytochrome c could contribute acutely to further mitochondrial dysfunction and to promote delayed neuronal death. Experiments reported here tested the hypothesis that ischemia or severe hypoxia results in release of cytochrome c from mitochondria. Cytochrome c was measured spectrophotometrically from either the cytosolic fraction of cortical brain homogenates after global ischemia plus reperfusion, or from brain slices subjected to severe hypoxia plus reoxygenation. Cytochrome c content in cytosol derived from cerebral cortex was increased after ischemia and reperfusion. In intact hippocampal slices, there was a loss of reducible cytochrome c after hypoxia/reoxygenation, which is consistent with a decrease of this redox carrier in the mitochondrial pool. These results suggest that cytochrome c is lost to the cytosol after cerebral ischemia in a manner that may contribute to postischemic mitochondrial dysfunction and to delayed neuronal death.
Mitochondrial dysfunction remains prominent in hypotheses defining mechanisms of cerebral ischemic injury. For example, postischemic brain shows prominent changes in redox activity of mitochondrial respiratory chain components (Welsh et al., 1982; Welsh et al., 1991; Rosenthal et al., 1995; Pérez-Pinzón et al., 1997; Pérez-Pinzón et al., 1998a,b). Also, mitochondria isolated from postischemic brain showed limitations of “state 3” or activity-dependent respiration (Sims and Pulsinelli, 1987; Sciamanna and Lee, 1993; Bogaert et al., 1994). Postischemic mitochondria also may be a major source of reactive oxygen species. Reactive oxygen species production and free radical-mediated damage were linked to reperfusion injury after brain ischemia (Flamm et al., 1978; Fridovich, 1979; Siesjo et al., 1985; Kontos, 1989; Vlessis et al., 1990; Hall et al., 1993).
Postischemic mitochondrial redox state has been characterized by shifts toward oxidation (hyperoxidation) respiratory chain components (Rosenthal et al., 1995; Pérez-Pinzón et al., 1997; Pérez-Pinzón et al., 1998a,b). This hyperoxidation of electron carriers has been interpreted either as decreased substrate availability (Rosenthal et al., 1995) or to reaction of mitochondrial complexes to reactive oxygen species (Pérez-Pinzón et al., 1997). An alternative explanation is that this hyperoxidation may result from loss of electron carriers, such as cytochrome c and NADH from mitochondria after ischemia. Thus, it is important to determine whether some electron carriers are lost from mitochondria, both because such loss might affect respiratory chain activity and because it may trigger the apoptotic cascade.
Mitochondrial dysfunction appears to underlie delayed neuronal death (apoptotic or necrotic) after cerebral ischemia (Nitatori et al., 1995; Charriaut-Marlangue et al., 1996). This is suggested by findings that apoptosis (programmed cell death) may be linked to mitochondria (Ankarcrona et al., 1995; Schinder et al., 1996; Kluck et al., 1997; Yang et al., 1997) and by reports implicating cytochrome c as a trigger activator of DEVD-specific caspases, and consequently as an inducer of apoptosis (Kluck et al., 1997; Yang et al., 1997). Defining the relation between mitochondrial dysfunction and cell death is complicated. For example, after excitotoxic insults, cells that failed to recover mitochondrial membrane potential appeared to die from necrosis, whereas cells in which mitochondrial membrane potential recovered, showed improved energy status, and survived the necrotic phase still underwent apoptosis (Ankarcrona et al., 1995).
Current research sought to determine whether cytochrome c is released from mitochondria into the cytosol after anoxic or ischemic cerebral insults. Goals were to test whether levels of cytochrome c in the cytosol were increased by global cerebral ischemia in rat brain in situ and to determine whether reducible cytochrome c (presumably the intramitochondrial cytochrome c within the functioning electron transport chain) was decreased after anoxia in hippocampal slices.
MATERIALS AND METHODS
All animal procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health.
Procedure for studies in vivo
Male Wistar rats weighing 300 to 350 g were fasted overnight and then anesthetized with 3% halothane and 70% nitrous oxide (balance oxygen). The femoral arteries were cannulated for blood pressure measurements and for arterial sampling of blood gases. The rats underwent endotracheal intubation and were artificially ventilated with 0.5% halothane and 70% nitrous oxide (balance oxygen). Both common carotid arteries were exposed by a midline ventral incision and gently dissected free of surrounding nerve fibers. Ligatures of polyethylene (PE-10) tubing, contained within a double-lumen silastic tubing, were passed around each carotid artery. Brain temperature was monitored with a 33-gauge thermocouple implanted on the temporalis muscle and maintained at 36.5° to 37°C throughout the experiment.
Before each ischemic insult, blood was gradually withdrawn from the femoral artery into a heparinized syringe to reduce systemic blood pressure to 40 to 50 mm Hg. Cerebral ischemia then was produced by tightening the carotid ligatures bilaterally. To allow postischemic reperfusion, the carotid ligatures were removed, and the shed blood was reinjected into the femoral artery.
Cytochrome c measurements from in vivo brain. To measure cytosolic concentrations of cytochrome c from intact brains, the rats were perfused for 1 minute with cold physiologic saline to remove all blood and hemoglobin contamination. The cortex was removed and homogenized in a ratio of 5 to 1 (mL/mg) in an extraction buffer with the following composition (in millimolar amounts): 220 mannitol, 68 sucrose, 20 Hepes-KOH, pH 7.4, 50 KCl, 5 EGTA, 2 MgCl2, 1 dithiothreitol, 1 phenylmethylsulfonyl fluoride, and 10 μmol/L cytochalasin B. Dithiothreitol was used to fully reduce cytochrome c. The homogenate was centrifuged for 10 minutes at 500 g (4°C). The supernatant was centrifuged at 17,500 g for 15 minutes at 4°C. The new supernatant was centrifuged in an ultracentrifuge (Beckman Model TL-100, Palo Alto, CA, U.S.A.) for 20 minutes at 106,000 g at 4°C. The final supernatant then was transferred to a spectrophotometer cuvette, and the absorption peak was measured at 419 nm (Fig. 1) with a Beckman DU 640 spectrophotometer. The final cytochrome c concentration was obtained from a standard curve expressed over milligrams of protein. Protein concentration was determined with the Bio-Rad copper sulfate reagent by spectrophotometry. All drugs were purchased at Sigma Chemical Co. (St. Louis, MO, U.S.A.).
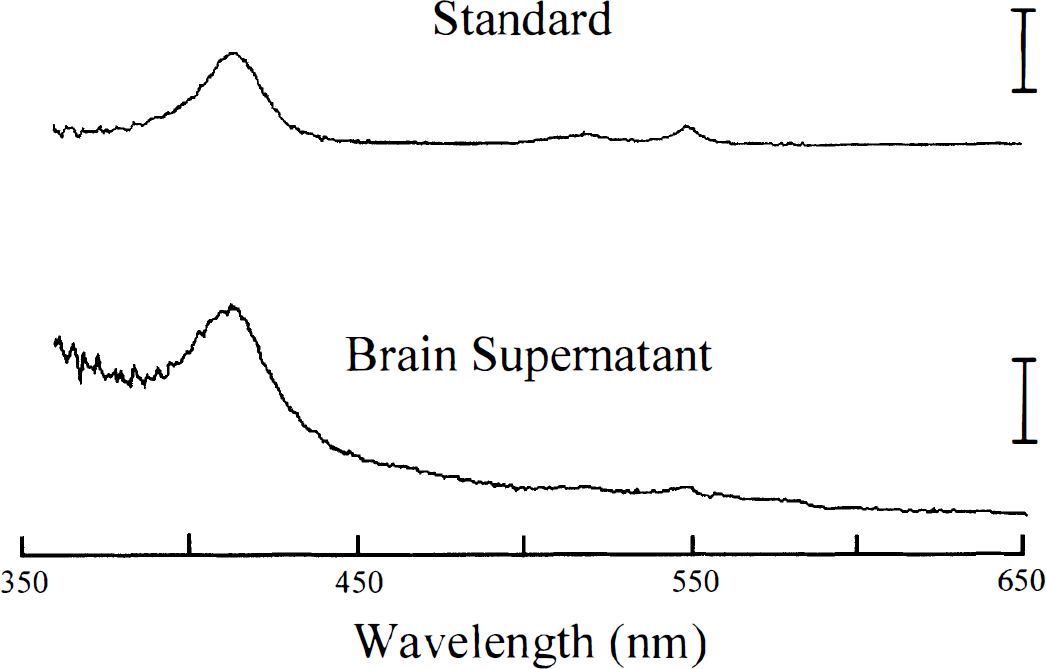
Absorption spectra from a 35 nmol/L cytochrome c standard solution (upper panel, scale 0.02 log units) and cytosolic fraction derived from the cerebral cortex of a rat that underwent 20 minutes of ischemia followed by 30 minutes of reperfusion (lower panel, scale 0.007 log units). Brain supernatant spectra clearly shows that no other cytochrome is present and that the largest peak is found at 419 nm.
Study groups and experimental paradigms in vivo. There were two in vivo study groups:
In vivo control group: Rats underwent all surgical procedures except ischemia. The length of time was similar to that of the ischemia group (see later). In vivo ischemia group: Rats underwent 20 minutes of ischemia followed by 30 minutes of reperfusion.
Procedure for studies in hippocampal slices
Slice preparation has been previously described (Pérez-Pinzón et al., 1997). Briefly, male Wistar rats (250 to 300 g) from Charles River Laboratories (Cambridge, MA, U.S.A.) were deeply anesthetized with pentobarbital sodium (60 mg/kg) and cooled down. The rats were perfused for 1 minute with cold physiologic saline to remove all blood and hemoglobin contamination. After decapitation, the cranium was opened and the brain was superfused with cold artificial cerebrospinal fluid with the following composition (in millimolar amounts): 126 NaCl, 3.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 2 MgSO4, and 10 glucose equilibrated with 95% O2 and 5% CO2.
Slices of 400-μm thickness were prepared from the hippocampal region of the hemissected brain with a motorized Vibroslice microtome from Campden Instruments, Ltd (Sileby, U.K.). Each slice was incubated in artificial cerebrospinal fluid oxygenated with 95% O2/5% CO2 and stored at 28°C for at least 1 hour before they were transferred to the recording chamber.
Absorption spectra were obtained by rapid scanning spectrophotometry (LaManna et al., 1985) from the control slices in normoxia and again at the final 10 minutes of anoxic insult. Absorption differences between normoxic and anoxic control slices at 550 nm (with reference to 535 and 575 nm), attributable to cytochrome c, were compared with absorption differences measured in experimental slices.
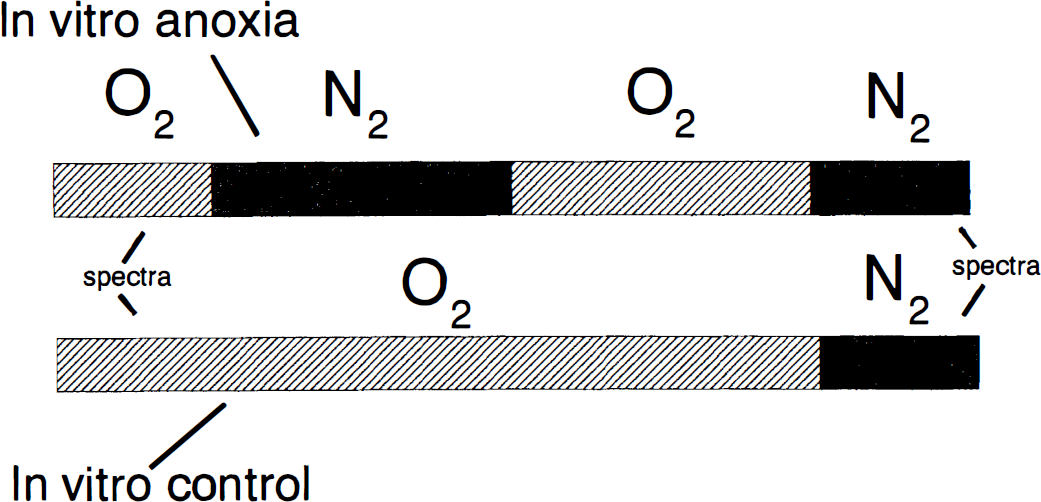
Diagram of the two experimental groups in hippocampal slices as explained in Materials and Methods. First panel represents anoxic experiments (group 2). Second panel represents control experiments (group 1). The first anoxic insult (top panel) represents 20 minutes of anoxia. The final anoxic insult represents 10 minutes of anoxia.
Study groups and experimental paradigms in hippocampal slices. There were two in vitro study groups:
In vitro control: Slices underwent a single anoxic insult of 10 minutes' duration after 50 minutes in normal oxygenated artificial cerebrospinal fluid. Spectra of cytochromes were collected after 10 minutes of normoxia and at 10 minutes of anoxia (for fully reduced spectra). Slices obtained for this group came from the same rat as those used for the next group; thus, comparisons were made between cytochrome c levels in both groups. Paradigms used for this group and that of group 2 are depicted in Fig. 2. In vitro anoxia: After 10 minutes of normoxia, slices underwent a 20-minute anoxic insult followed by 20 minutes of reoxygenation, after which 10 minutes of anoxia was induced. Spectra of cytochromes were collected at 10 minutes of normoxia before the first anoxic insult, at 20 minutes of anoxia, at reoxygenation, and after 10 minutes of final anoxia (Fig. 2).
Statistical significance between groups was determined by one-tailed Student's t test. Significance was accepted with P < 0.05. All data were expressed as mean ± SD.
RESULTS
Fig. 1 shows a typical absorption spectra obtained from the cytosolic fraction from postischemic rat neocortex. This spectrum is nearly identical to that from a 35-nmol/L cytochrome c standard with absorption maxima near 419 and 550 nm. A key finding is that the brain spectrum does not exhibit absorption peaks that are characteristic of other mitochondrial pigments (e.g., Fig. 3, top panel). This confirms that the supernatant was devoid of mitochondria or mitochondrial pigments other than cytochrome c.
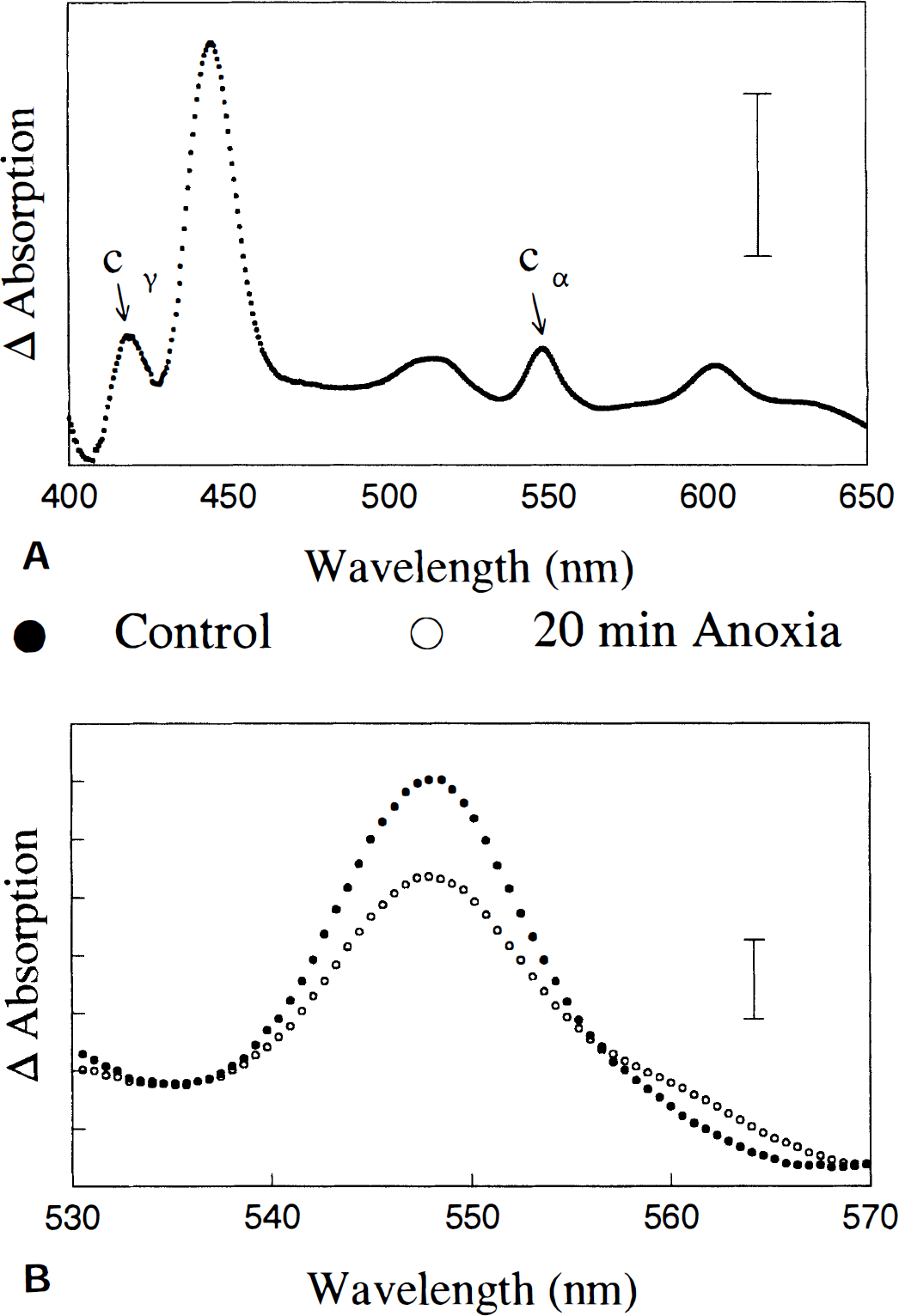
Spectrum of control conditions (normoxia) and anoxic slices (
Fig. 4A shows that concentrations of cytosolic cytochrome c (estimated from a standard curve and protein content) increased significantly after cerebral ischemia by 42% (*P < 0.05). Rats that underwent 20 minutes of ischemia followed by 30 minutes of reperfusion had 5.44 ± 1.8 × 10−3 μmol/L/mg of protein (n = 7) of cytochrome c in the cytosolic fraction. Sham-operated rats showed 3.83 ± 0.8 × 10−3 μmol/L/mg of protein of cytochrome c (n = 7).
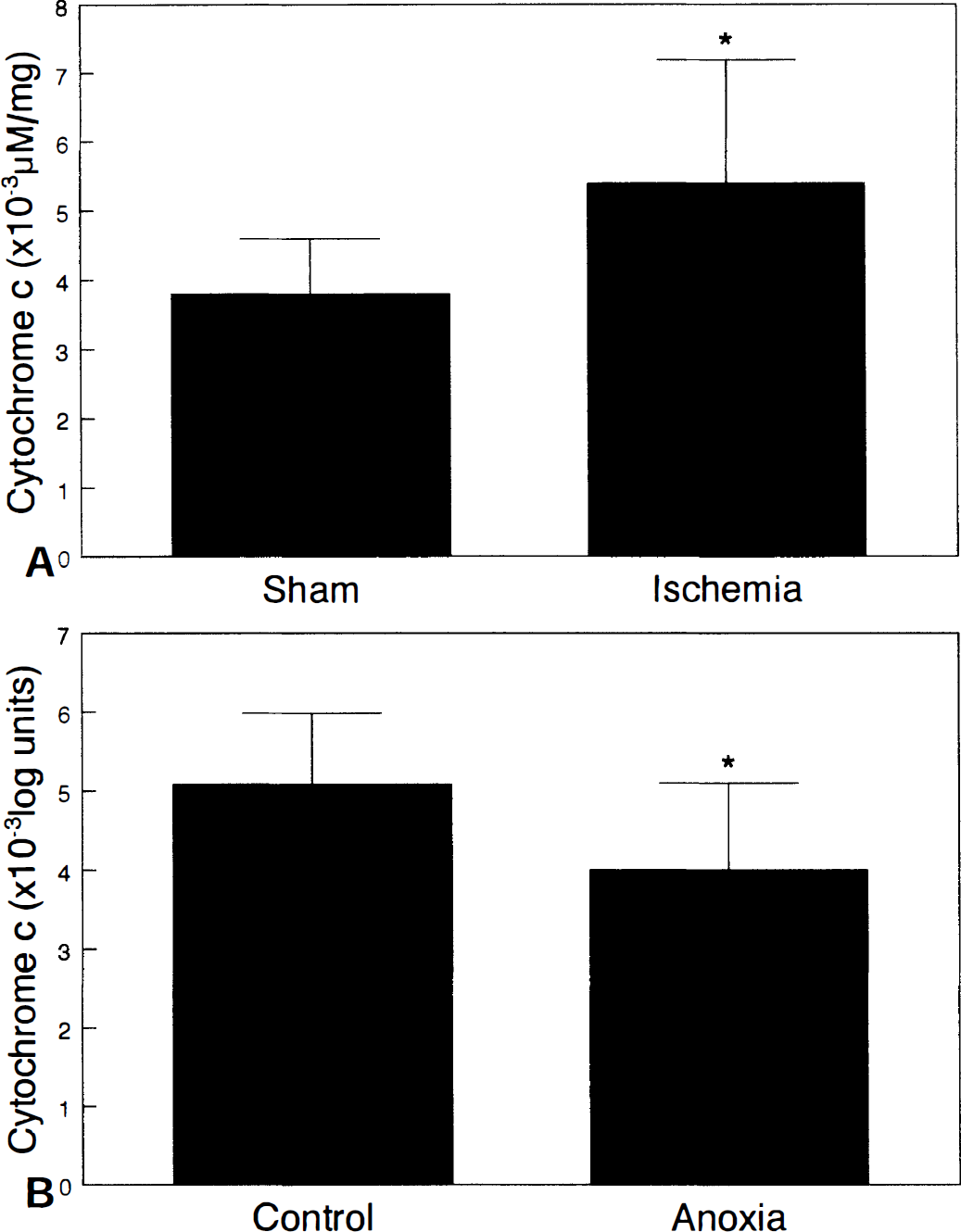
Absorption difference spectra depicting the transition from normoxia to anoxia in control hippocampal slices (c.f., Fig. 3, top panel) contained an absorption maxima for reduced cytochrome c at 550 and 419 nm, respectively. Such spectra also contained peaks at 445 and 605 nm attributable to the absorption maxima of reduced cytochrome a,a3. Fig. 2 (bottom panel) illustrates typical findings that absorption by cytochrome c produced by anoxia was greater in control slices than in slices reoxygenated for 20 minutes after an earlier 20-minute anoxic insult. Fig. 4B confirms that reduction of cytochrome c produced by anoxia in experimental slices after an earlier anoxic insult (4.00 ± 1.1 × 10-3 log units; n = 9) was significantly decreased by 21% when compared with that in control slices (5.08 ± 0.9 × 10−3 log units; n = 9; *P < 0.05).
DISCUSSION
These results demonstrate that cytosolic cytochrome c is increased after global cerebral ischemia and that mitochondrial cytochrome c is decreased after anoxia in hippocampal slices. Absorption spectra from the cytosolic fraction of rat neocortex after ischemia showed no evidence that other electron carriers were lost from the mitochondrial matrix. This is not surprising, since cytochrome c is soluble, whereas the other electron carriers are bound to mitochondrial membranes or in the mitochondrial matrix and are more likely to remain associated with the membrane fraction, even after an ischemic insult. These findings support the hypothesis that anoxia or ischemia provokes the release of cytochrome c from cerebral mitochondria into the cytosol. These results also are in agreement with recent preliminary data by Fujimura and others (1997) showing release of cytochrome c into the cytosol after focal cerebral ischemia.
The mechanism by which cytochrome c is released into the cytosol remains undefined. Ouyang and associates (1997) showed that large molecules such as the mitochondrial aspartate aminotransferase were released from mitochondria into the cytosol after transient focal cerebral ischemia, suggesting the opening of the mitochondrial permeability transition pore. Fiskum and colleagues (1997) showed that cytochrome c release could be induced from isolated dog brain mitochondria after 10 minutes of cardiac arrest in beagles by increasing Ca+2 concentration ([Ca+2]). This release occurred in a Ca+2 dose-dependent manner. Interestingly, also hyperoxidation of NADH after anoxia increased with [Ca+2]O in hippocamal slices (Pérez-Pinzón et al., 1998a). Our results and results from these studies suggest that during reperfusion after anoxia/ischemia, mitochondrial permeability transition or osmotic lysis of mitochondria, or both, may occur. Either event could lead to an overall loss in pyridine nucleotides and cytochrome c from mitochondria. It is possible that hyperoxidation of NADH previously reported (Rosenthal et al., 1995; Pérez-Pinzón et al., 1997; Pérez-Pinzón et al., 1998a) results from the loss of NADH from the matrix after mitochondrial permeabilization.
There is another possible explanation for the current results: after global ischemia in vivo, mitochondria may be more susceptible to breakage. As a result, the mechanical fractionation procedure, by which cytosol was isolated, could have a greater influence on these mitochondria than those from control brains. Previous studies using the fractionation method used in the current study also have detected cytochrome c in the cytosolic fractions derived from control cells (Yang et al., 1997), suggesting that this is a normal artifact of this method. This is one reason why complementary experiments were carried out in hippocampal slices.
In slices, prolonged anoxia plus reoxygenation resulted in a decrease in labile reducible cytochrome c signal. An explanation for this result could be that a fraction of mitochondrial cytochrome c was not available for reduction by the respiratory chain during anoxia. We interpret this to mean that a fraction of cytochrome c was released from mitochondria to the cytosol. However, these data only suggest that cytochrome c is released from mitochondria, since differences in spectra were not obtained from maximally oxidized versus maximally reduced conditions. It is also possible that the baseline cytochrome c values between the two groups were different. Additional in vitro studies are required to confirm these findings.
In summary, data here support the hypothesis that cerebral anoxia or ischemia provoked release of cytochrome c from mitochondria to the cytosol. This translocation of cytochrome c may be associated with either apoptotic and/or necrotic pathways after anoxia/ischemia. Cytochrome c release also provides another possible explanation for how postischemic mitochondrial hyperoxidation of electron carriers results. Also, these data suggest that a common mechanism may exist to produce acute and chronic injury after cerebral ischemia.
Footnotes
Acknowledgements
The authors thank Drs. Carlos T. Moraes and Myron D. Ginsberg for their helpful comments and criticisms of the study, and Dr. Ricardo Prado for technical assistance.