
Abstract
Traumatic brain injury evokes multiple axonal pathologies that contribute to the ultimate disconnection of injured axons. In severe traumatic brain injury, the axolemma is perturbed focally, presumably allowing for the influx of Ca2+ and initiation of Ca2+-sensitive, proaxotomy processes. Mitochondria in foci of axolemmal failure may act as Ca2+ sinks that sequester Ca2+ to preserve low cytoplasmic calcium concentrations. This Ca2+ load within mitochondria, however, may cause colloid osmotic swelling and loss of function by a Ca2+-induced opening of the permeability transition pore. Local failure of mitochondria, in turn, can decrease production of high-energy phosphates necessary to maintain membrane pumps and restore ionic balance in foci of axolemmal permeability change. The authors evaluated the ability of the permeability transition pore inhibitor cyclosporin A (CsA) to prevent mitochondrial swelling in injured axonal segments demonstrating altered axolemmal permeability after impact acceleration injury in rat. At the electron microscopic level, statistically fewer abnormal mitochondria were seen in traumatically injured axons from CsA-pretreated injured animals. Further, this mitochondrial protection translated into axonal protection in a second group of injured rats, whose brains were reacted with antibodies against amyloid precursor protein, a known marker of injured axons. Pretreatment with CsA significantly reduced the number of axons undergoing delayed axotomy, as evidenced by a decrease in the density of amyloid precursor protein-immunoreactive axons. Collectively, these studies demonstrate that CsA protects both mitochondria and the related axonal shaft, suggesting that this agent may be of therapeutic use in traumatic brain injury.
Keywords
Axotomy after traumatic brain injury (TBI) is not the result of overt mechanical shearing of the axon. Rather, axotomy represents the end stage of a complex process wherein the mechanical insult induces a sequence of events culminating in failure of axoplasmic transport, pooling of intra-axonal contents, and pinching off of the axon from its distal segment. Disconnection develops within hours after the traumatic episode and is termed delayed or secondary axotomy, since the primary mechanical insult provokes secondary biochemical processes that culminate in axotomy. This process of traumatic axonal injury (TAI) has been described in both animal models and the human clinical situation (Povlishock and Jenkins, 1995; Adams et al., 1982).
The precise mechanisms responsible for this delayed/secondary axonal injury are not well characterized, but experimental studies have elucidated several focal axonal abnormalities in TAI including altered axolemmal permeability, cytoskeletal perturbation, ionic dysregulation, and mitochondrial alteration (Povlishock and Pettus, 1996; Pettus and Povlishock, 1996; Gallant and Galbraith, 1997, Maxwell et al., 1997). In this process, it has been posited that, in the case of severe injury, the axolemma fails first, allowing for the influx of extracellular molecules, with particular emphasis on the flux of calcium (Ca2+). High intra-axonal calcium is presumed to activate Ca2+-dependent phosphatases and proteases and initiate the cytoskeletal collapse and mitochondrial swelling observed in injured axons (Maxwell et al., 1997; Xiong, et al., 1997a).
The mitochondrial abnormalities in TAI have been associated with altered Ca2+ compartmentalization. It has been assumed that mitochondria in foci of axolemmal failure act as Ca2+ sinks that sequester Ca2+ within the matrix to preserve low cytoplasmic Ca2+ concentrations. This Ca2+ load within the mitochondrial matrix, however, may lead to colloid osmotic swelling of the organelle and collapse of organelle function (Gunter and Pfeiffer, 1990). Local failure of mitochondria, in turn, could decrease production of high-energy phosphates necessary to maintain membrane pumps and prevent further ionic imbalance in foci of axolemmal permeability change. Thus, mitochondria may be involved in a vicious cycle whereby traumatic axolemmal perturbation leads to Ca2+ influx, which causes mitochondrial failure and loss of energy substrate production, which, in turn, negates the ability of the cell to restore membrane pumps, thereby leading to further Ca2+ entry.
Previous studies document both morphologic (swelling, membrane blebbing) and physiologic (decreased oxidative phosphorylation, altered membrane potential) alterations associated with traumatically induced mitochondrial impairment (Colicos et al., 1996; Colicos and Dash, 1996; Xiong et al., 1997b). Further, we have reported that one of the earliest markers of focal intraaxonal change is mitochondrial swelling, occurring in the first minutes after injury (Pettus and Povlishock, 1996). Also, in vitro work suggests that this mitochondrial swelling may be a factor in the development of the pathobiology of axonal injury (George et al., 1995).
In this process of axonal failure, previous work has focused on discerning the initiating events in axonal Ca2+ accumulation and modulating the downstream consequences of Ca2+ influx in terms of protease and phosphatase activation. Yet, little consideration has been directed toward the more intermediate events such as mitochondrial failure and its overall influence on the fate of the injured axon. It is plausible that preservation of mitochondrial functioning may interrupt the damaging cascade from Ca2+ accumulation and allow the axon to restore cation homeostasis and prevent axotomy.
Mitochondrial integrity depends on the maintenance of the membrane potential across the inner mitochondrial membrane, termed ΔΨM (delta psi mu). Since the inner mitochondrial membrane is the seat of oxidative phosphorylation and energy production for the organelle and the cell, its potential is carefully regulated. The major regulator of ΔΨM under adverse conditions is the permeability transition (PT) pore, a multimeric transmembrane protein on the inner mitochondrial membrane that assembles during times of oxidative stress and Ca2+ accumulation. Opening of the PT pore, loss of ΔΨM, and the resultant mitochondrial permeability transition (MPT) have permanent, deleterious consequences for the organelle and the cell (Petit et al., 1997; Ankacrona et al., 1995). The PT pore has regulatory sites on its cytoplasmic face that are activated by multiple effectors, notably pro-oxidants, uncoupling agents, and divalent cations (Ca2+). The potent immunosuppressant cyclosporin A (CsA) binds and inhibits opening of the PT pore and, thus, inhibits MPT (Zoratti and Szabo, 1995). Cyclosporin A has been shown to prevent pore opening, MPT, and mitochondrial swelling in isolated liver and brain mitochondria exposed to high Ca2+ concentrations (Halestrap and Davidson, 1990).
Activation of the PT pore by Ca2+ results in loss of ΔΨM and colloid osmotic swelling within the mitochondrial matrix, a process that is prevented by CsA (Gunter and Pfeiffer, 1990). Based on this information, in the current communication we evaluate the fate of mitochondria in axonal disease. We examine the ability of the PT pore-inhibitor CsA to inhibit mitochondrial swelling after impact acceleration TBI in injured axons undergoing traumatically induced axolemmal change.
In addition to evaluating the potential preservation by CsA of mitochondrial morphologic features, the current communication also addresses whether this protection translates into protection for the axon itself reflected in a reduction in the number of damaged axons. To this end, antibodies to amyloid precursor protein (APP) were used, since axonal accumulations of APP are recognized as a sensitive marker of traumatically induced axonal insult (Sherriff et al., 1994; Gentleman et al., 1993). We examined whether preservation of mitochondrial integrity can limit axonal disturbances that produce accumulations of APP.
MATERIALS AND METHODS
Electron microscopic analysis of mitochondrial status in cyclosporin A-treated and untreated injured animals
Drug delivery and injury protocol. We investigated whether pretreatment with the MPT inhibitor CsA could prevent mitochondrial abnormalities in axons demonstrating traumatically induced axolemmal perturbations. To identify injured axons, we examined the potential for focal axolemmal permeability change that previously has been associated with related intraaxonal cytoskeletal and mitochondrial abnormality. For this purpose, we used a large molecular weight species, horseradish peroxidase (HRP), normally excluded by the intact, uninjured axolemma. After traumatically induced axolemmal change, the membrane is sufficiently disrupted to permit the local influx of HRP. Subsequent histochemical visualization of HRP specifically identifies this target subpopulation of injured axons.
Ten male Sprague Dawley rats (375 to 393 g) were subjected to an impact acceleration model of TBI in a manner consistent with that previously described (Marmarou et al., 1994). Animals were provided food and water ad libidum before surgery. Animals were anesthetized for 5 minutes under 4% isoflurane and a 2:1 mixture of N2O:O2. Rats were endotracheally intubated and maintained under 1.5% to 2% isoflurane. Rats were secured in a stereotactic frame, and a dorsal midline neck incision was made over the cervical vertebrae and occiput. The atlanto-occipital membrane was exposed with blunt dissection. A 25-gauge spinal needle was stereotactically lowered into cisterna magna, and 0.1 mL of CSF was withdrawn into a 1-mL tuberculin syringe. One hour before injury, 15 mg of HRP was dissolved in the withdrawn CSF and the solution was reinfused slowly into cisterna magna. In five rats at 30 minutes before injury, 10 mg/kg of CsA (Sandoz Sandimmune Injection Prep; Novartis, Basel, Switzerland) in polyethylene glycol/sterile saline/cremophor oil solution (250 mg CsA/10 mL vehicle) was infused through the spinal needle after an equal volume of CSF (∼0.12 mL) was withdrawn. These animals constituted the treatment group, CsA treated. In the other five rats, no drug was given, thereby generating the control group, untreated. The incision was sutured, and rats were removed from the stereotactic frame.
Ten minutes before injury, parietal bones were exposed, cleaned, and dried. A steel helmet was secured in the midline between bregma and lambda with dental acrylic. Just before injury, anesthesia was withdrawn. Rats were secured on a foam bed beneath the injury tower and injured from 2 m with a 450-g weight. Thus, injury was induced 60 minutes after HRP injection in untreated animals, whereas in CsA-treated animals, injury was induced 60 minutes after HRP injection and 30 minutes after CsA delivery. After injury, rats were ventilated with oxygen and monitored for recovery of respiration and reflexes.
In four of the CsA-treated animals, the femoral artery was cannulated before exposure of the atlanto-occipital membrane and blood withdrawn at specific time intervals to monitor any change in arterial blood gas parameters as a result of CsA administration. In addition, since some pharmacologic agents have been shown to exert protection through the induction of hypothermia rather than their implied mechanism of action (Buchan and Pulsinelli, 1990), we placed a temperature probe in the temporalis muscle of two animals as a window to possible alterations in brain temperature resulting from CsA administration.
Electron microscopic preparation. After 1-hour survival time, all animals received an intraperitoneal overdose of sodium pentobarbital and were transcardially perfused with 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 mol/L Millonig's buffer. Brains were harvested and sagittally blocked to within 1 mm of the midline by placing the brain in a sagittal specimen holder, cutting just lateral to the pyramids, and including the cervicomedullary junction. Blocked brains were postfixed overnight in the same fixative. Fifty-micrometer vibratome sections were removed into 0.1 mol/L Millonig's buffer and reacted with cobalt-glucose oxidase for HRP visualization. Sections then were processed for light microscopic analysis to detect peroxidase-containing axonal segments. In all cases, analysis was confined to the corticospinal tract (CSpT) in the region of the pontomedullary junction, a region of known vulnerability to traumatic axonal change in this animal model (Povlishock et al., 1997).
Sections serially adjacent to those containing HRP-flooded axonal segments within the pontomedullary CSpT identified at the light microscopic level then were prepared for electron microscopic investigation. These sections were postfixed in 1% OsO4 in 0.1 mol/L sodium phosphate buffer for 1 hour in darkness at 4°C. The sections were rinsed in 0.1 mol/L sodium phosphate buffer, serially dehydrated in ascending grades of alcohols, and flat-embedded on plastic slides in medcast resin (Ted Pella). Axonal segments demonstrating HRP uptake, a marker of local axonal injury, in the plastic-embedded sections were cut out, mounted on beam capsules, and sectioned at 2 μm on a LKB microtome. Thin sections (75 nm) were taken with a Diatome diamond knife, mounted on Formvar-coated slot grids, and stained in 5% uranyl acetate in 50% methanol and 0.5% lead citrate for electron microscopic investigation using a Zeiss EM 10CA electron microscope. Micrographs of HRP-containing axons were photographed at magnifications of either 5100× or 10,000×.
Image and statistical analysis. Sixty-two micrographs of HRP-containing CSpT axonal segments in CsA-treated injured animals and 51 micrographs of HRP-containing axonal segments in untreated injured animals thus were generated. Numbered micrographs were coded to blind the assessors to experimental condition. Coded micrographs then were videographically captured, digitized, and enlarged on an image analysis unit (Imaging Research, St. Catherine's, Ontario, Canada) to permit quantitation of axoplasmic area, counting of total mitochondrial number, and assessment of mitochondrial integrity. Mitochondria with evidence of swelling or blebbing were scored “damaged.” For this purpose, a mitochondrion was considered swollen if its matrix was expanded with loss of discernible cristae. Blebbing was considered present when one or more of the cristae were grossly expanded.
Numbered micrographs were decoded, the percentage of damaged mitochondria in axons within each group was calculated, and mean percent damaged mitochondria in each group (CsA-treated versus untreated) were compared with independent samples t tests. Also, the total damaged mitochondrial density in axons within each group was calculated and mean density damaged mitochondria (per μm2) in each group was compared with independent samples t tests.
Amyloid precursor protein immunoreactivity in cyclosporin A-treated, untreated, and vehicle-treated injured animals
Injury protocol. Because APP immunoreactivity is widely recognized as a marker for traumatically induced axonal change (Gentleman et al., 1993), we also explored the potential for any change in the number of APP-immunopositive axons in another sample of CsA-treated injured animals versus untreated and vehicle-treated injured animals.
In this case, 12 additional male Sprague Dawley rats (375 to 400 g) were anesthetized, intubated, and secured in a stereotactic frame as described earlier. Animals were provided food and water ad libidum before surgery. A dorsal midline neck incision was made over the cervical vertebrae and occiput, and the atlanto-occipital membrane was exposed with blunt dissection. In four of the animals, 30 minutes before injury, a 25-gauge spinal needle was stereotactically lowered to tap cisterna magna, and 10 mg/kg of CsA in polyethylene glycol/cremophor oil (25 mg/mL of solution) solution were infused through the spinal needle after an equal volume of CSF was withdrawn. In the four vehicle-treated animals, 0.12 mL of CSF was withdrawn and an equal volume of polyethylene glycol/sterile saline/cremophor oil vehicle was infused. In the four untreated animals, 0.12 mL CSF was withdrawn and reinfused.
Ten minutes before injury, parietal bones were exposed, cleaned, and dried. A steel helmet was secured midway between bregma and lambda with dental acrylic, and the animals were injured in a manner consistent with that described earlier.
Immunohistochemistry. In all 12 cases, after 24 hours of survival, rats were administered an intraperitoneal overdose of sodium pentobarbital and transcardially perfused with 4% paraformaldehyde and 0.1% glutaraldehyde in 0.1 mol/L Millonig's buffer. Brains were harvested, sagittally blocked just lateral to the pyramids (consistent with the methods previously described for our mitochondrial studies), and postfixed overnight in the same fixative. Fifty-micrometer vibratome sections were cut and placed in phosphate-buffered saline. Sections were reacted for immunohistochemical visualization of APP using a protocol adapted in our laboratory (Stone et al., 1997) modified from Gentleman and others (1993). Briefly, sections were reacted with 0.5% H2O2 to block endogenous peroxidase activity and microwaved in citric acid while maintaining a 45°C maximum temperature for 10 minutes. The tissue was incubated for 1 hour in 10% Triton X:10% NHS and then 16 to 18 hours in 1:200 APP antibody in 1% NHS. On the following day, sections were incubated for 1 hour in 1:200 biotinylated rat-adsorbed anti-mouse IgG and for 1 hour in a Vectastatin Elite ABC kit, then reacted with diamino benzidene/H2O2 with imidazole enhancement for visualization of secondary antibody. Sections were mounted on gelatin-coated glass slides and serially dehydrated, and coverslips were applied.
Image and statistical analysis. For image analysis of the CsA-treated, untreated, and vehicle-treated cases, slides were examined with an Olympus BH-2 microscope at 10×, and areas of high-density APP immunoreactivity within the CSpT were videographically captured and digitized. Scanning was restricted in all cases to the CSpT in the region of the pontomedullary junction, a known vulnerable site to traumatic axonal change (Povlishock et al., 1997). The digitized images were imported into an image analysis unit (Imaging Research, St. Catherine's, Ontario, Canada), and the area of the CSpT captured was measured. Total number of APP-positive axons greater than 10 μm in diameter within this area were marked, counted, and expressed as density of APP-positive axons per unit area. Investigators were blinded in the data analysis of the CsA-treated and untreated cases.
Mean densities of APP-immunopositive CSpT axons were compared among CsA-treated, untreated, and vehicle-treated injured animals with a one-way analysis of variance (ANOVA) statistical test.
RESULTS
Physiologic response to cyclosporin A administration and injury
CsA administration did not produce physiologically significant changes in arterial blood gas parameters (Table 1). Temporalis temperature monitoring indicated that CsA administration produced a slight reduction in brain temperature from 37.5° to 36.5°C over 1 hour after injection. Brain temperature returned to baseline within 90 minutes of administration. Such a decline in brain temperature, however, is not consistent with temperature ranges necessary for any therapeutic effect of hypothermia (Buchan and Pulsinelli, 1990).
Arterial blood gas parameters in CsA-treated animals
Preservation of mitochondrial morphology in axons undergoing axolemmal permeability change in cyclosporin A-treated injured animals
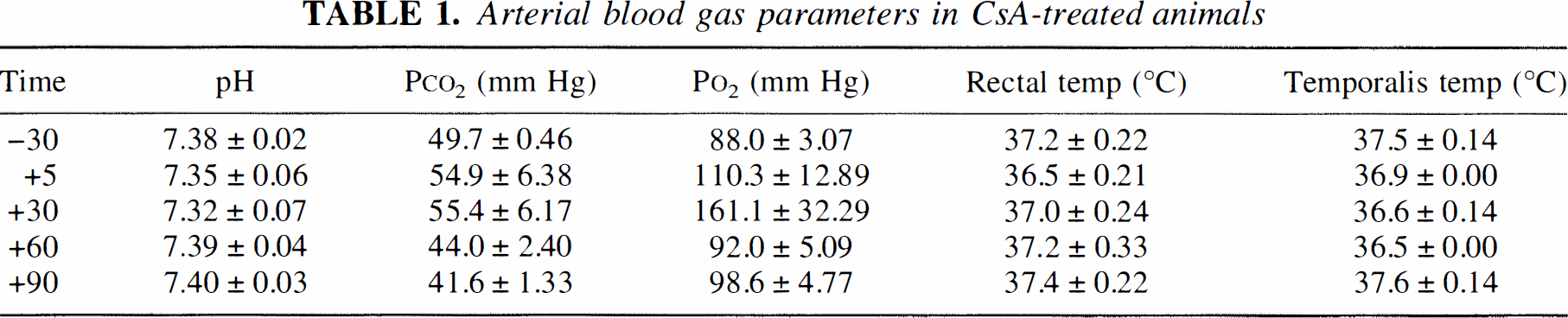
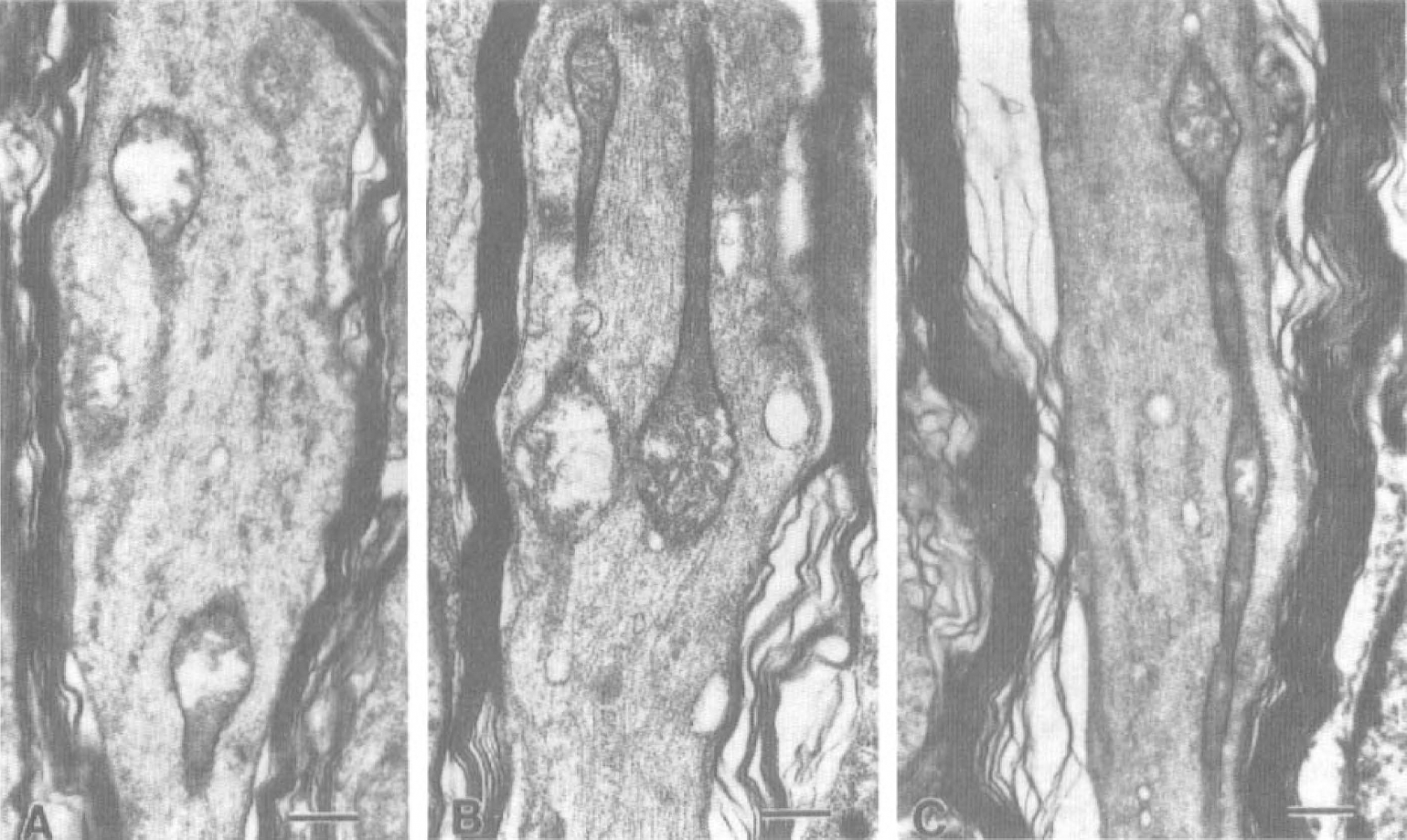
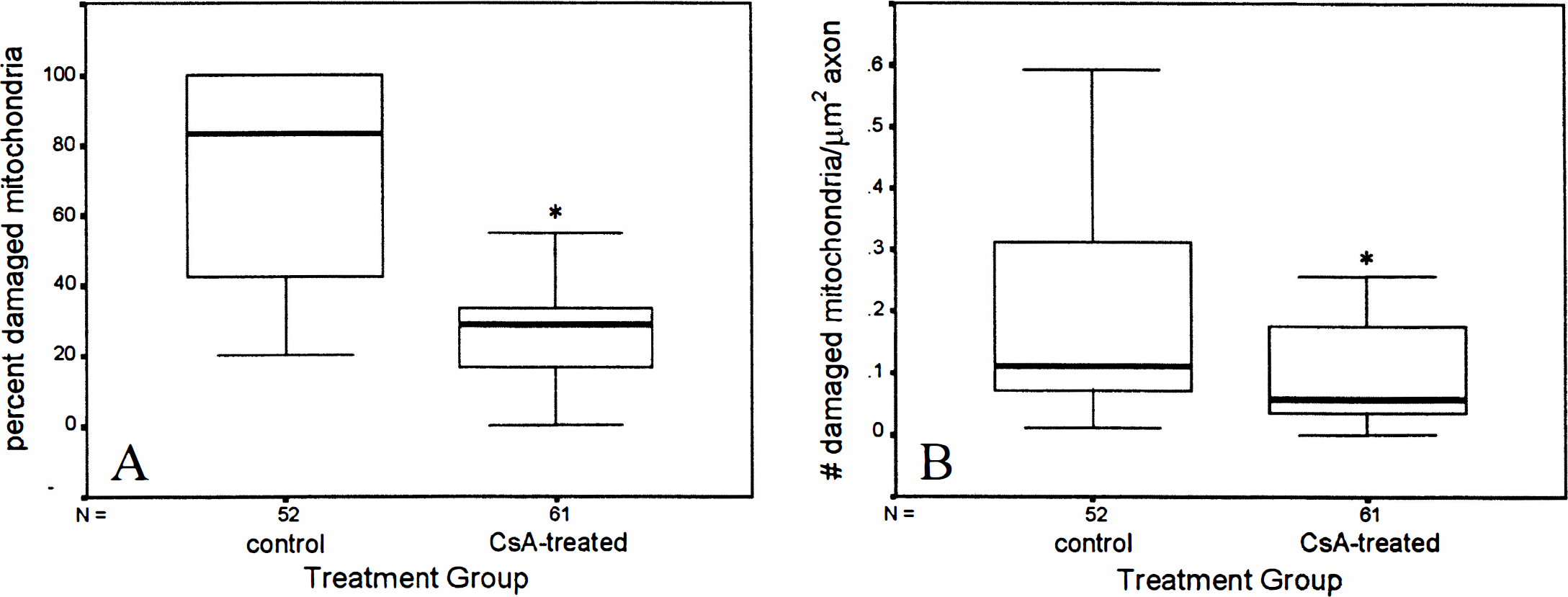
Pretreatment with CsA protected mitochondrial morphologic features in injured axons undergoing axolemmal permeability change at 1 hour after injury. Micrographs of HRP-containing axonal segments harvested from untreated injured animals revealed heavy focal mitochondrial damage as evidenced by high percentages of swollen or “blown-out” mitochondria (Fig. 1), whereas HRP-containing axonal segments from CsA-treated injured animals had significantly fewer swollen mitochondria (Fig. 2). The mean percentage of damaged mitochondria in HRP-containing axons of untreated animals was 71.94 ± 31% (mean ± SD (N = 52), whereas mean percentage of damaged mitochondria in HRP-containing axonal segments of CsA-treated animals was 27.96 ± 16% (N = 61). Independent samples t test revealed a significant difference between the groups with P < 0.001 (Fig. 3A). Furthermore, the density of damaged mitochondria in HRP-containing axonal segments in CsA-treated injured animals (0.107 ± 0.11/μm2 of axoplasm) was significantly less than the density of damaged mitochondria in HRP-containing axonal segments harvested from untreated animals (0.304 ± 0.38/μm2) (Fig. 3B). Damaged mitochondria per unit area were statistically lower in CsA-treated injured animals at P < 0.001.
Morphologic appearance of mitochondria in injured axons undergoing an axolemmal permeability change harvested from untreated animals at 1 hour after injury.
Morphologic appearance of mitochondria in injured axons undergoing an axolemmal permeability change in cyclosporin A (CsA)-treated animals at 1 hour after injury.
Preservation of axonal integrity in cyclosporin A-treated animals
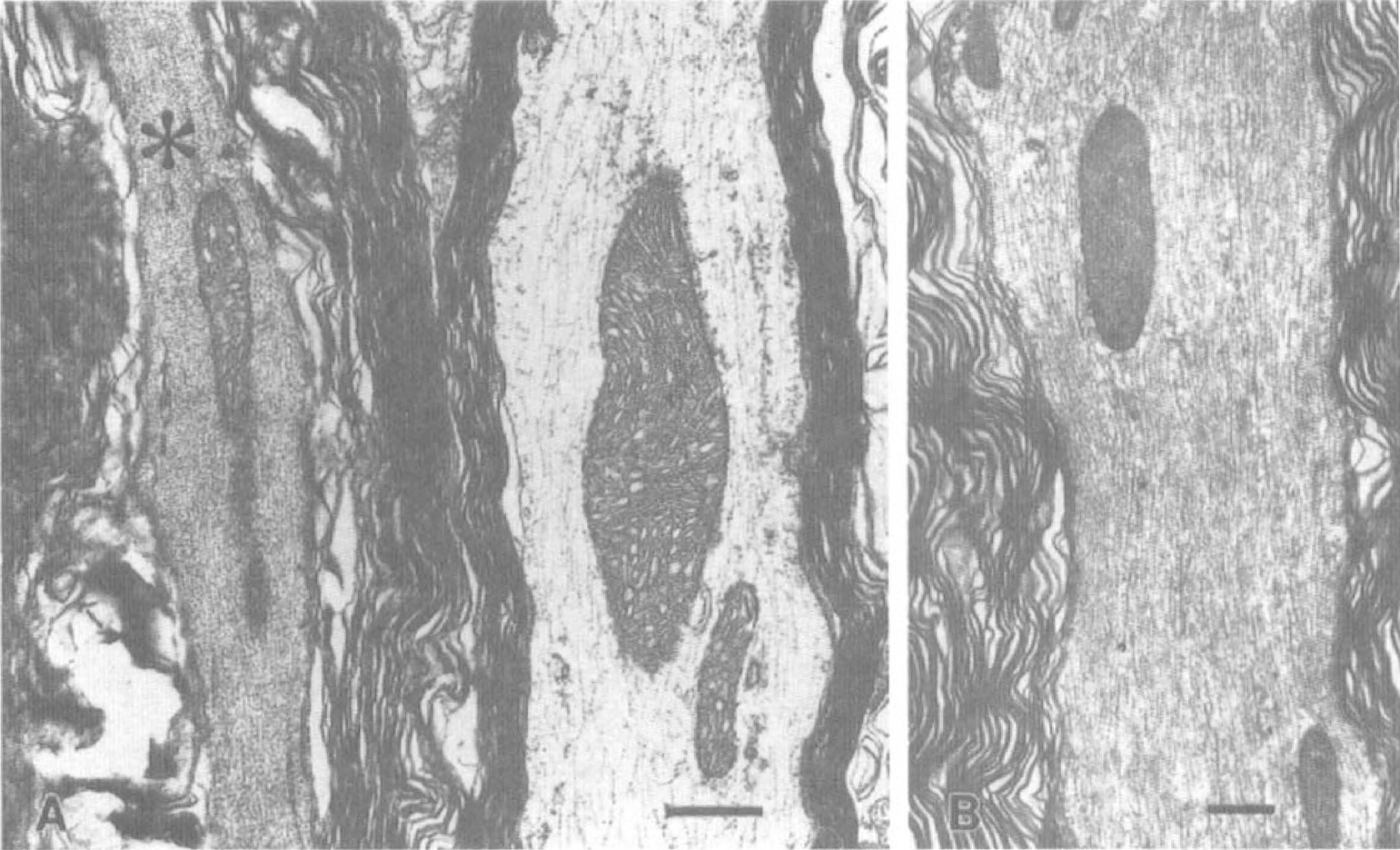
A single bolus of CsA before injury also significantly protected against delayed axotomy after impact acceleration injury. Digitized fields of CSpT in untreated animals showed larger numbers of APP-immunopositive axons when compared with similar fields from CsA-treated animals (Fig. 4). Mean density of APP-immunoreactive axons in CsA-treated injured animals was 247.40 ± 161/mm2 (N = 20), whereas mean density of APP-immunoreactive axons in untreated injured animals was 388.43 ± 65/mm2 (N = 20) (Fig. 5). The difference in means was significant at P < 0.001 (one-way ANOVA).
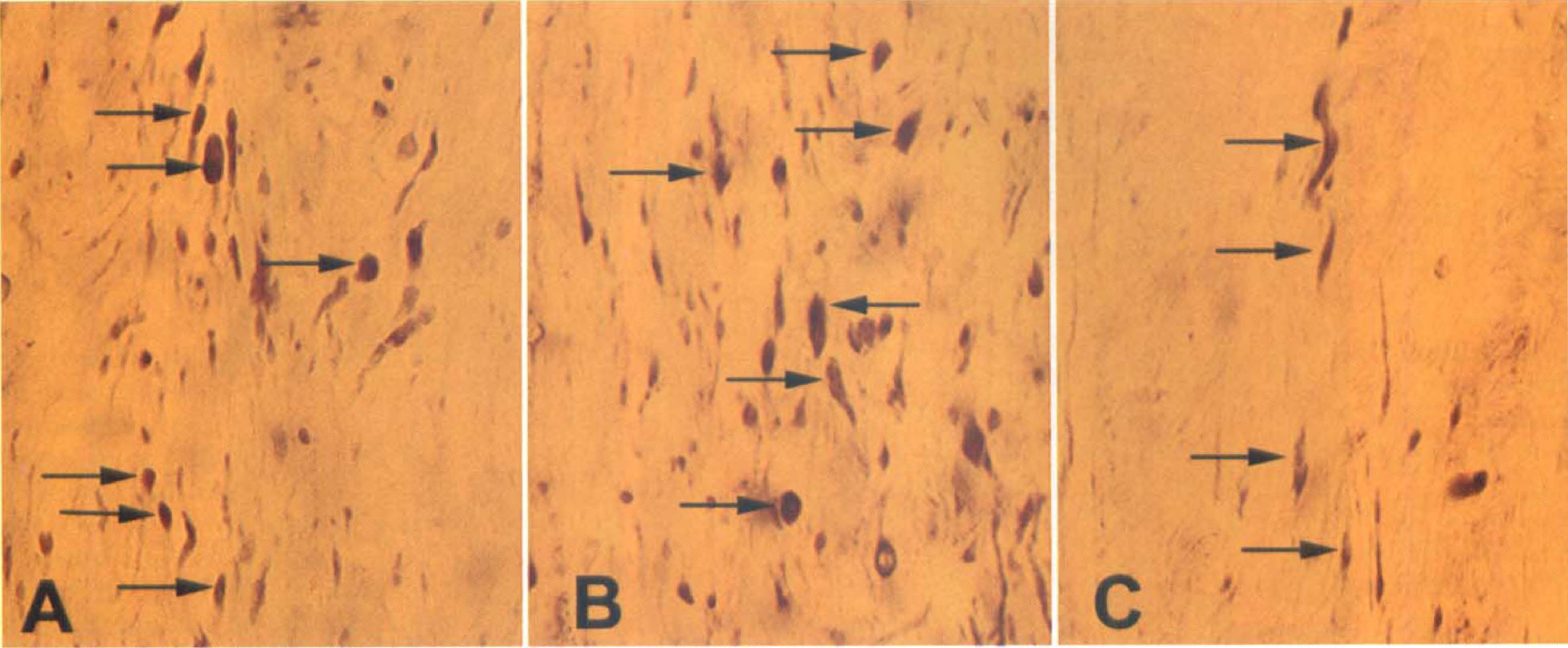
Light micrographs of corticospinal tracts reacted with antibodies against amyloid precursor protein (APP), a marker of injured and axotomized axonal segments.
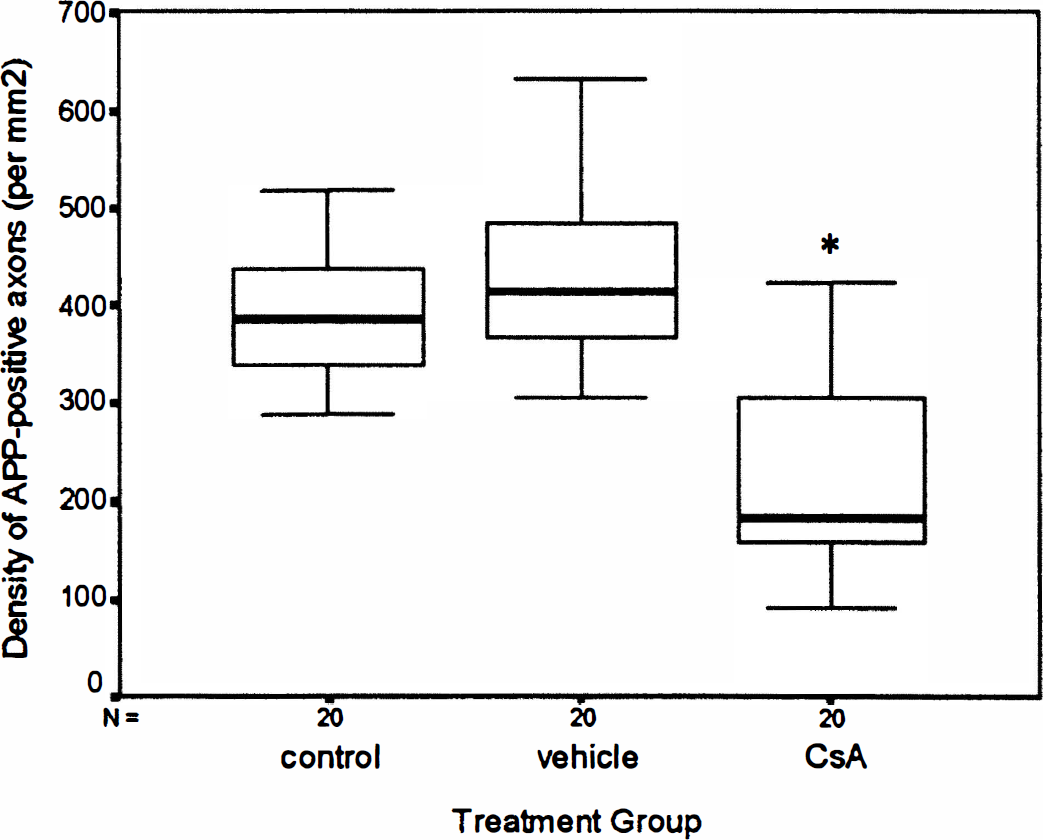
Box plot of density of APP-immunopositive axons per square millimeter of tissue in each group of cases. The number of damaged mitochondria per unit area of corticospinal tract is significantly lower in CsA-treated injured animals than untreated and vehicle-treated injured animals (P < 0.001 and P < 0.0001, respectively).
Vehicle infusion had no effect on the progression to delayed axotomy after impact acceleration injury. Mean density of APP-immunoreactive axons in CSpT of vehicle-treated animals was 427.09 ± 88/mm2 (N = 20), which was significantly higher than mean density in CsA-treated animals (P < 0.0001, one-way ANOVA). Importantly, no difference was noted between mean density of APP-positive profiles in untreated control versus vehicle-treated animals (P > 0.1, one-way ANOVA).
DISCUSSION
In the current communication, we demonstrate two essential facts concerning pretreatment with CsA in TBI. First, a single bolus of CsA before impact acceleration TBI preserves mitochondrial morphologic features in axons undergoing traumatically induced axolemmal change. Secondly, pretreatment with CsA limits delayed axotomy and axonal damage as evidenced by a decrease in APP immunoreactivity, which normally delineates injured and axotomized axons. These findings represent the first direct evidence of in vivo pharmacologic preservation of mitochondrial morphologic features in TBI and provide intriguing insight into the role of mitochondria in the pathogenesis of TAI. Furthermore, these results intimate that interruption of secondary injury processes provides protection to the axon.
The key question in this process, then, centers on how CsA protects mitochondria in injured axons and how this translates into protection for the axon itself. As observed earlier, CsA is known to block the Ca2+-induced collapse of mitochondrial respiration by inhibiting opening of the PT pore, the main route through which Ca2+ crosses the inner mitochondrial membrane under conditions of oxidative stress (Zoratti and Szabo, 1995; Gunter and Pfeiffer, 1990). The mechanism of action of CsA is dependent on binding to its partner molecule cyclophilin, a member of the immunophilin family of proteins, which are present in the mitochondrial matrix and cytosol (Chadhuri and Stephan, 1995). Under conditions of oxidative stress (high Ca2+, low ATP, high Pi), cyclophilins can induce a conformational change in the PT pore that allows nonspecific passage of solutes less than 1.5 kd (Halestrap and, 1990; Halestrap et al., 1997). Binding by CsA prevents cyclophilin from interacting with the PT pore complex.
Additionally, CsA-cyclophilin complexes within the cytosol have other targets including calcineurin, a Ca2+-dependent and calmodulin-dependent serine/threonine phosphatase (Liu et al., 1991). Calcineurin inhibition is believed to be the primary target mediating the main historical on-label use of cyclosporin as a potent immunosuppressant. In lymphocytes, calcineurin dephosphorylates a transcription factor, nuclear factor of activated T cells, thereby decreasing interleukin-2 secretion in antigen-reactive T cells and, thus, T-cell proliferation (Schreiber and Crabtree, 1992).
Whereas the implications for calcineurin inhibition in neurons and in neurotrauma are unclear, the discovery that the immunophilins are expressed in high concentrations in the brain (Steiner et al., 1992; Dawson et al., 1994) led to numerous investigations of the possible neural roles for immunophilins. Immunophilin-immunosuppressant complexes have been implicated in regulation of IP-3-regulated Ca2+ release, nitric oxide-stimulated glutamate release, neurite outgrowth, kindling, hippocampal long-term potentiation, and a host of other processes occurring in both normal and diseased brain (see Sabatini et al., 1997; Gold 1997).
Based on this, it is clear that although the role of calcineurin inhibition is uncertain, CsA-mediated blockade of the PT pore could preserve the supply of energy substrates in injured axons by limiting the extent of mitochondrial damage. Evidence to support this premise derives from multiple sources. In the field of ischemia, there is evidence to support a role for mitochondria in mediating axonal failure as well as evidence for neuroprotection by CsA (Siesjo and Siesjo, 1996). Such studies in ischemia also have pointed to the failure of membrane pumps and loss of ionic homeostasis in the development of axonopathy and to the mitochondrial mediation of axonal failure (LoPachin and Stys, 1995; Stys et al., 1992; Waxman et al., 1992; Waxman et al., 1994). In this process, it has been posited that the axolemma first fails, allowing for membrane depolarization and the ultimate axoplasmic accumulation of Ca2+ through reverse operation of the Na+-Ca2+ exchanger (LoPachin and Lehning, 1997). High axonal Ca2+ induces mitochondrial swelling and initiates Ca2+-sensitive enzymes (proteases, phospholipases, endonucleases), which cause a procession toward irreversible axonal degeneration (Duchen et al., 1993). Interestingly, Ca2+ accumulation within mitochondria precedes subsequent accumulation of Ca2+ within the axoplasm, and this mitochondrial failure contributes to the deficit of energy substrates in these failing neurons (LoPachin and Lehning, 1997). This has led some to contend that mitochondria may be the key determinants of the irreversibility of axonal insult (Duchen et al., 1993; LoPachin and Lehning, 1997).
Although this is the first report of CsA neuroprotection in TBI, several groups have reported preservation of the bioenergetic state of mitochondria and increased survival of ischemic hippocampal neurons after CsA administration (Uchino et al., 1995; Li et al., 1997; Shiga et al., 1992; Folbergrova et al., 1997; Dawson et al., 1993; Steiner et al., 1996, Balakirev, et al., 1997). These studies report lower incidences of delayed hippocampal cell death and smaller areas of cerebral infarction after CsA, and all point to mitochondrial protection as the common mediator of protection. Collectively, their data speak to the ability of CsA to limit ischemic neurotoxicity through direct and indirect inhibition of MPT. In contrast to the current communication, however, these studies of ischemia used relatively gross measures of neuronal cell survival and triphenyl tetrazolium chloride staining to speculate on the protective effects of CsA administration. Our work, on the other hand, directly assesses the ultrastructural fate of mitochondria after CsA treatment, in addition to evaluating its effects on the total number of damaged axons. Thus, our results define the site of protection as the mitochondria themselves rather than only addressing the more global issue of cell or axonal survival.
Our results indicate that, in foci of axonal perturbation in CsA-treated injured animals, roughly three fourths of mitochondria are morphologically intact and, ostensibly, functioning, whereas one fourth undergo an MPT and swell. Perhaps bioenergetic failure of mitochondria diminishes the axon's ability to restore ionic homeostasis. Thus, maintenance of oxidative respiration and ATP supply from the functioning three fourths of mitochondria may permit the continued functioning of ATP-dependent membrane pumps, thereby limiting enduring changes in ionic imbalance. A MPT in the swollen one fourth of mitochondria with consequent Ca2+ accumulation in the matrix could protect the axoplasm from the damaging effects of a large Ca2+ load. This would create a conducive environment for recovery of the injured axon: continued ATP supply and low axoplasmic Ca2+.
A physiologic role of MPT has been proposed whereby a MPT would create large Ca2+ sinks in the matrix that could release Ca2+ later, while still permitting recovery of that mitochondrion. In this scenario, ATP production from the organelle would temporarily cease, but the cytoplasm would be protected from Ca2+ overload. The subsequent recovery from a massive Ca2+ influx into the matrix across permeabilized mitochondrial membranes would require less energy than to have extruded the same load of Ca2+ using membrane pumps. Given the in vivo reversibility of MPT, such a proposal would represent a highly cost-effective protection mechanism for the cell.
Moving on the premise of CsA neuroprotection in TAI, other questions remain concerning the efficacy of CsA if administered in a delayed treatment paradigm or if given through other routes of administration. In the studies reported here, CsA was administered 30 minutes before injury. Therefore, the potential therapeutic benefits of delayed administration must await further study. Also, since the intrathecal administration route used in the current study is not an optimal route of administration, the efficacy of CsA through systemic routes also remains to be explored. These issues are confounded by the fact that CsA does not cross the blood-brain barrier in appreciable quantities (Begley et al., 1990). However, it is known that TBI transiently alters blood-brain barrier properties, so that CsA access to the brain parenchyma across the vascular border may be possible (Povlishock et al., 1978).
Possible criticisms of the current findings could arise on two fronts: first, whether the applied intrathecal CsA actually reached the target sites under evaluation, and, secondly, if the observed axonal protection translated into improved behavioral outcome in the rat. Although because of technical limitations we were unable to radiolabel CsA, our experience using intrathecal tracers strongly argues that the agent did reach the target sites. As we have both in this and past studies demonstrated that intrathecal injection of HRP (44,000 molecular weight [MW]) successfully delivers this macromolecule to known vulnerable sites within the neuraxis (Pettus and Povlishock, 1996; Povlishock and Pettus, 1996), there is no compelling argument to dismiss a similar level of access for CsA, which is only a 1203-MW polypeptide.
Whether CsA improves behavioral outcome after TBI related to the fibers under investigation is an issue of importance in our laboratory. Motor outcome, however, is difficult to assess directly because in animals, as in humans, motor deficits after TBI exhibit spontaneous recovery. Thus, experimental strategies to assess motor recovery have poor sensitivity. Parallel investigations in our laboratory were conducted to examine motor recovery using the same drug delivery protocol and doses as the studies reported here. Unfortunately, no beneficial effect of a single pretreatment with CsA was detected. We believe that this most likely resulted from the spontaneous recovery of motor function, which limited our ability to detect therapeutic efficacy given the small population of inured axons.
These results provide clear evidence that mitochondria are important mediators in the development of TAI. The literature supports that CsA both induces a favorable Ca2+ compartmentalization within the axon and preserves the bioenergetic state of mitochondria. Mediation of mitochondrial functioning in injured foci by CsA has positive effects on the outcome of the traumatically injured axon and thus may have long-range therapeutic implications for use in brain-injured humans.
Footnotes
Acknowledgments
The authors thank Thomas Coburn, Lynn Davis, and Sue Walker for their skilled technical assistance. The authors also thank Dr. Sung Choi for his statistical expertise.