
Abstract
In the infant brain, ischemia-induced ionic and enzyme mechanisms may independently lead to cell death by energy depletion: resequestration of calcium mobilized from intracellular stores consumes ATP, and activated poly(ADP-ribose) polymerase (PARP) uses oxidized nicotinamide adenine dinucleotide to form polyADP-ribosyl nuclear proteins associated with DNA damage, Using 31P nuclear magnetic resonance spectroscopy, we have monitored intracellular pH and cellular energy metabolites in ex vivo neonatal rat cerebral cortex before, during, and after substrate and oxygen deprivation, In an insult that exhibited secondary energy failure and apoptosis we identified a relative 25% augmentation of high-energy phosphates at the end of recovery when the ryanodine-receptor antagonist, dantrolene, was introduced in the early (0- to 40-minute) but not late (40- to 120-minute) stage of recovery (P < 0,05). In contrast to the absence of a late dantrolene-sensitive effect, inhibition of PARP with 3-methoxybenzamide was as effective (P < 0.05) as early dantrolene, even when introduced after a 40-minute delay. The dantrolene and 3-methoxybenzamide effects on high-energy phosphates were not additive, rather the early dantrolene-sensitive effect nullified the potential 3-methoxybenzamide effect. Therefore, in this vascular-independent neonatal preparation, postischemic mobilization of calcium from intracellular stores is associated with PARP-related energy depletion. Inhibition of either of these processes confers improved postischemic bioenergetic recovery in the developing brain.
Keywords
In infants and young children with critical illness, treatment of acute cerebral ischemic insult, in many instances, is only practical after the event (Tasker et al., 1991). Therefore, an understanding of proximate postischemic neurotoxic mechanisms such as dysfunctional ion homeostasis (Erecinska and Silver, 1989; Silver and Erecinska, 1992) and neuron-derived oxidant production (Huang et al., 1994; Dalkara and Moskowitz, 1994) is pertinent to the development of useful therapeutic interventions. However, such mechanisms with their possible interactions are complex (Siesjö, 1992a, 1992b) and produce disparate terminal effects on the neuron, such as cell death associated with swelling (Rothman and Olney, 1986) and cell death induced by gene expression (MacManus and Linnik, 1997).
Alterations in cytosolic calcium (Ca2+) concentration are considered central to the process of ischemia-induced neurotoxicity (Siesjö, 1992a, 1992b). Such changes may result from influx of Ca2+ through voltage-sensitive and agonist-operated calcium channels, as well as release of Ca2+ from intracellular stores. In the immature rat there is evidence that neurotoxicity mediated by the N-methyl-
Oxidants generated during the postischemic period also have a role in neuronal injury. Activation of NMDA receptors can generate nitric oxide (NO) and peroxynitrite (OONO−), which is formed from superoxide anion (O2−) and NO, and these are able to mediate neurotoxicity either directly or indirectly through their effects on nuclear DNA and cellular enzymes (Dawson, et al., 1991, 1993, 1996; Zhang et al., 1994; Bolaños et al., 1997; Eliasson et al., 1997). Taken together with the NMDA-related ionic effects, there is, theoretically, a bioenergetic cost that may contribute to toxicity: sequestration of Ca2+ into endoplasmic reticulum occurs at the expense of ATP (Siesjö, 1992a); accumulation of Ca2+ into mitochondria is driven by the potential across the inner mitochondrial membrane, which is only upheld in the presence of O2 or ATP (Siesjö, 1992a); and poly-(ADP-ribose) polymerase (PARP)—a nuclear DNA repair enzyme that functions upstream of p53 to identify DNA strand breaks—uses oxidized nicotinamide adenine dinucleotide (NAD+) to form polyADP-ribosyl nuclear proteins associated with DNA damage (Berger, 1985; Ueda and Hayaishi, 1985; Lautier et al., 1993; Zhang et al., 1994). (It takes four molecules of ATP to resynthesize one molecule of NAD+ from nicotinamide).
In this context, we have previously undertaken studies using 31P nuclear magnetic resonance (NMR) spectroscopy in the superfused brain slice preparation (Bachelard et al., 1985) to look at energy metabolism after substrate and oxygen deprivation, a form of in vitro “ischemia.” The stage of postnatal cortical development influenced resistance to a hypoxic-ischemic insult and the postischemic response to depolarization and NO, with amelioration rather than exacerbation of bioenergetic state in immature cortex (Tasker et al., 1996). We now describe further infant brain slice studies that examine the hypothesis that both postischemic ionic- and enzyme-induced phenomena have significant bearing on cell bioenergetics. Our findings indicate that postischemic mobilization of Ca2+ from dantrolene-sensitive stores is associated with PARP-related energy depletion: inhibition of either of these processes confers improved postischemic bioenergetic recovery in the developing brain.
MATERIALS AND METHODS
Preparation of cortical brain slices and incubations
Cerebral cortical slices of 350-µm thickness from 7-day-old rats (3 to 7 animals per experiment) were prepared using a McIlwain tissue chopper. Each brain was removed from the cranial cavity within 30 seconds, and slices were rinsed in oxygenated Krebs-Henseleit bicarbonate (KHB) buffer before transferring them to a 20-mm NMR tube. Once in the tube, slices were perfused with KHB buffer containing (in mmol/L) 124 NaCl, 5 KCI, 1.2 MgSO4, 1.2 CaCl2, 26 NaHCO3, 1.2 KH2PO4, and 10
Nuclear magnetic resonance techniques
In these experiments, the metabolic status of the tissue was assessed from determinations of pH; and the relative concentrations of ATP and phosphocreatine (PCr). 31P NMR spectroscopy was used to follow changes in pH; and metabolic recovery of high-energy phosphates. Spectra were recorded using a 20-mm NMR probe in an 8.5 T vertical magnet interfaced with a SMIS console operating at 145.7 MHz. Magnetic field homogeneity was optimized by adjusting room-temperature shim currents until the water proton linewidth from the preparation was less than 30 Hz. Throughout the experiments 31P spectra were acquired (45° pulse; relaxation time, 1.1 seconds; sweep width, 10 kHz; 4,000 points; 280 or 500 scans) and processed as described previously (Kauppinen and Williams, 1990) with a 1/4 sine filter over the first 2 ms of the free induction decay (to suppress broad phospholipid resonances) followed by 15 Hz line-broadening. These pulsing conditions do not allow for full relaxation of all signals (particularly the PCr) between pulses; therefore, peak area ratios do not correspond directly to concentration ratios. The data have not been corrected for this effect, but from a series of experiments previously reported (Tasker et al., 1996) comparing the above acquisition with spectra acquired with an 8-second relaxation time the β-ATP signal is fully relaxed for all conditions. The saturation factor for the PCr signal was 64.4% ± 3.2% (mean ± SD, n = 12) and it was not influenced by condition or age. Therefore, because the acquisition conditions in the experiments described were the same for all 31P spectra and we were interested in time-dependent changes in individual metabolites during and after ischemia, the changes in peak heights of the PCr, γ-ATP, and β-ATP relative to their control values were used (Kauppinen and Williams, 1990).
The pHi was calculated from the chemical shift (σ) of Pi relative to PCr at 0 ppm using the following titration curve (Taylor et al., 1983): pHi = 6.75 + log [(σ – 3.27)/(5.69 – σ)]. It is generally accepted that absolute pH determinations by NMR are accurate to within ±0.1 pH units, but that changes can be measured with a precision of ±0.03 pH units.
Drug interventions
In some experiments, slices were superfused with dantrolene or 3-methoxybenzamide, or both, in the recovery period after in vitro ischemia. Dantrolene (sodium salt) was used at a concentration of 20 µmol/L as a similar concentration (10 to 30 µmol/L) had been reported to protect against glutamate- and NMDA-induced toxicity in cortical neuronal cultures (Frandsen and Schousboe, 1991, 1992), protect against glutamate-and NMDA-induced rises in Ca2+ in cultured hippocampal neurons, and depolarize CA1 neurons in hippocampal brain slices (Krnjevic and Xu, 1996). 3-Methoxybenzamide was used at a concentration of 500 µmol/L as this order of drug concentration significantly inhibits PARP activity (Kofler et al., 1993). Also, similar agents at this concentration limit NMDA-induced toxicity in cortical neuronal cultures (Zhang et al., 1994). Confirmation of the effectiveness of dantrolene and 3-methoxybenzamide at these concentrations in our brain slice preparation was demonstrated in a series of experiments of glutamate-induced energy failure using concentrations of 1, 1.5, and 2 mmol/L (Fig. 1). Briefly, after a control period, cortical slices were perfused serially with ascending concentrations of glutamate, each for 20 minutes followed by a recovery period of 40 minutes. The %ATP at the end of each recovery period relative to control showed that using dantrolene (20 µmol/L) or 3-methoxybenzamide (500 µmol/L) afforded significant amelioration of glutamate-induced bioenergetic failure (P < 0.05). These concentrations were therefore used in the in vitro ischemia experiments described below. All compounds were purchased from Sigma, Poole, Dorset, U.K.
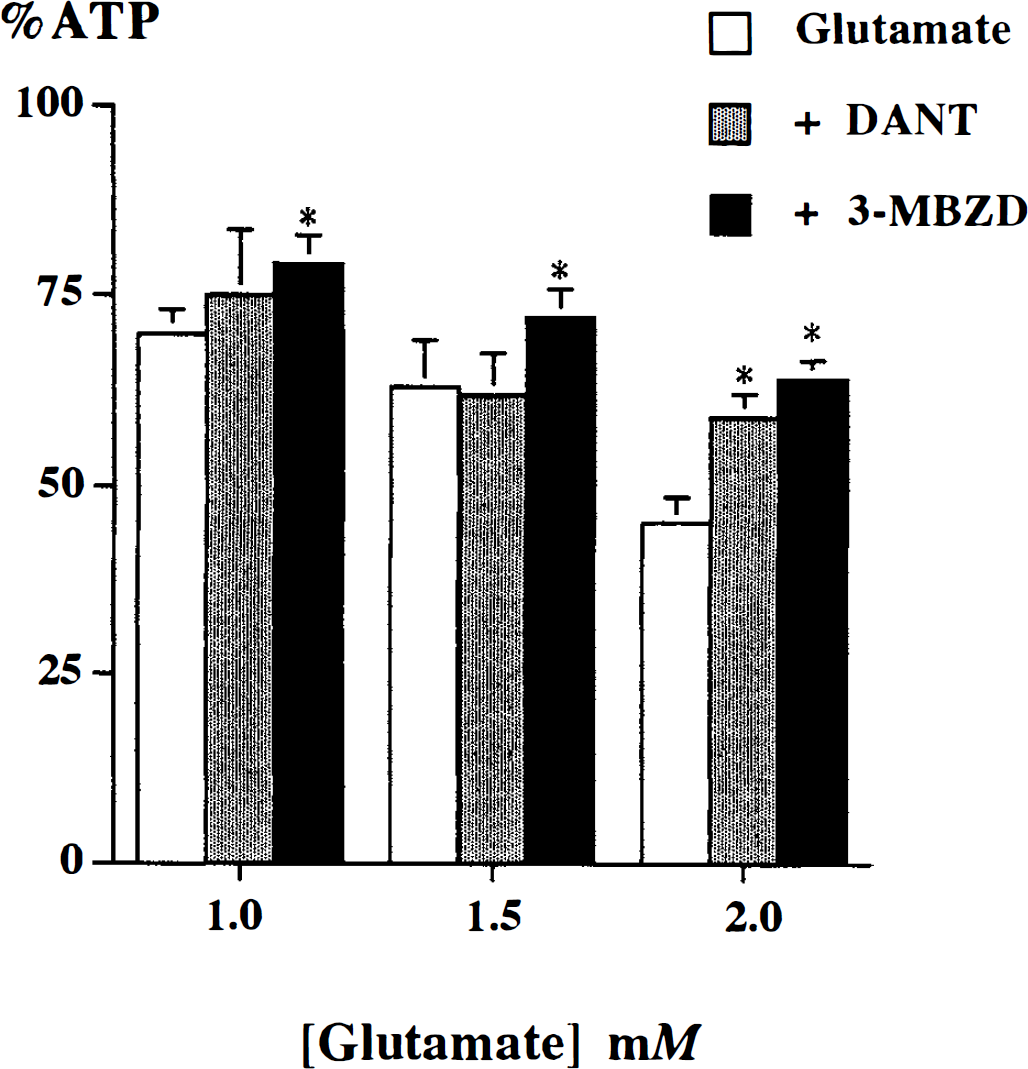
The effect of 20 µmol/L dantrolene (DANT) and 500 µmol/L 3-methoxybenzamide (3-MBZO) on glutamate-induced decrease in β-ATP (%ATP relative to control). Values of mean ± SD are shown. * indicates a significant increase in β-ATP (P < 0.05) compared with glutamate group (analysis of variance followed by post-hoc Tukey-Kramer HSD; n = 9 experiments).
Preparation of DNA from acute cortical brain slices for electrophoresis
Initial experiments were carried out to assess the effect of substrate deprivation (no oxygen, no glucose) on high-energy phosphate metabolites in cortical slices, metabolic stability on early recovery (up to 120 minutes), and DNA fragmentation. At the end of the experiments cortical brain slices were filtered from perfusion buffer and snap frozen in liquid nitrogen. Samples were then stored at –70°C until used for DNA extraction. At that time frozen tissue was shaved and rapidly mixed with cell lysis buffer consisting of 0.1 mol/L EDTA (pH 8.0), 0.01 mol/L Tris-HCl (pH 7.6), 0.02 mol/L NaCl, 7% sodium dodecyl sulfate, and 50 µ/100 mL proteinase K. This material was then incubated at 55°C for 3 hours. The DNA was then extracted with an equal mixture of phenol and chloroform and then precipitated with a 1/10 volume of sodium acetate (3 mol/L, pH 7.0) and 2 volumes of ice-cold ethanol, air dried, and resuspended in TE buffer (10 mmol/L Tris-HCl, pH 7.6, 1 mmol/L EDTA) before being quantified and labeled with fluorescein. Labeling of DNA fragments for automated sequencing of size of fluorescent DNA fragments was carried out by using 1 µg DNA mixed with 5 µL of dNTP mix (10×: 0.2 mmol/L each of dCTP, dGTP, dTTP; 0.1 mmol/L fluorescein-dATP; 0.1 mmol/L dATP; 500 mmol/L Tris-HCl, pH 7.8; 50 mmol/L MgCl2; 100 mmol/L β-mercaptoethanol; and 100 µg/ml nuclease-free bovine serum albumin), and 5 µL enzyme mix (10 ×: BioNick Labelling System, Life Technologies Inc., Rockville, MD, U.S.A.). This mixture, made up to 45 µL, was allowed to react at 16°C for 60 minutes before stopping with 5 µL of stop buffer. The DNA was then reprecipitated in ethanol and sodium acetate as described above and resuspended. Aliquots of 2 µL were then electrophoresed in 4% acrylamide gels together with a standard DNA size marker using the ALF (Pharmacia, Amersham Pharmacia Biotech, Piscataway, NJ, U.S.A.) automated sequencer with the fragment analysis software.
In vitro ischemia studies
A 40-minute ischemic insult was used for subsequent studies investigating the early recovery period with pharmacologic intervention. The effects on changes in pHi and high-energy phosphate metabolites were then compared using statistical measures described below. After recovery from dissection, perfusion of slices with well-oxygenated KHB buffer was continued for 45 to 60 minutes (Fig. 2). After this period, the perfusion medium was changed to KHB buffer containing a reduced concentration of phosphate (100 µmol/L). The washout period for medium changes in our apparatus is less than 5 minutes. Over the next 50 minutes six control 31P spectra were acquired (3 × 500 scans, 3 × 280 scans), and the mean values of peak heights were used as intra-experiment controls for the two methods of acquiring 31P spectra. Next followed four 20-minute intervals, during each of which three 31P spectra were acquired (3 × 280 scans). In the first two intervals the slices were perfused with either KHB buffer lacking oxygen and glucose or normal control buffer. In the second two intervals (recovery after ischemia, 0 to 40 minutes) slices were perfused with either control or dantrolene- (20 µmol/L) containing KHB buffer. Recovery over the next 80 minutes (recovery after ischemia, 40 to 120 minutes) in control KHB buffer with or without 3-methoxybenzamide (500 µmol/L) was also studied, during which time further 31P spectra were acquired (3 × 500 scans).
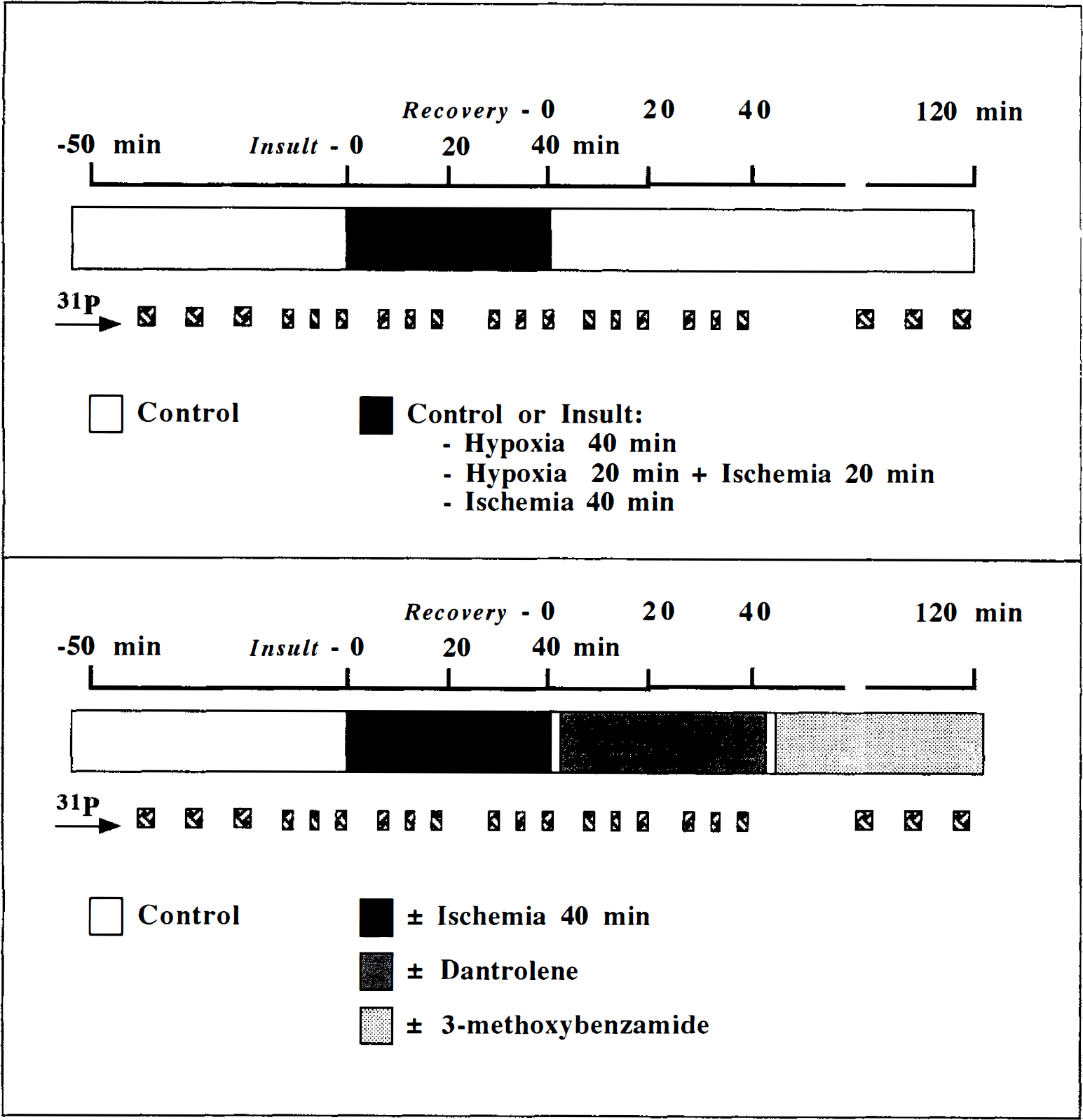
Diagrams illustrating the time course of experiments after an initial 45- to 60-minute equilibration period in Krebs-Henseleit bicarbonate buffer. The horizontal bar represents a series of experiments. 31P indicates times when 31P NMR spectra were acquired. The upper panel outlines the substrate- and oxygen-deprivation experiments. The lower panel outlines the postischemic pharmacologic experiments.
Thirty-nine separate brain slice NMR spectroscopy experiments were conducted: (1) n = 12 to evaluate the effect of insult on postischemic recovery and DNA fragmentation (3 experiments × 4 conditions: control; hypoxia, 40 minutes; hypoxia, 20 minutes then ischemia, 20 minutes; ischemia, 40 minutes); (2) n = 9 to study the effect of dantrolene and 3-methoxybenzamide on glutamate toxicity (3 experiments × 3 conditions); (3) n = 18 drug experiments during the postischemic period.
Statistical analysis
Throughout this article quantitative results are presented as means ± SD. We have used a nonparametric test to calculate the rank correlation coefficient (Kendall's tauB) when considering the relation between increasing severity of insult and consequence on relative NMR metabolite values and change in pHi at a priori chosen time points (Fig. 2) in the protocol (end of insult, 40 minutes; recovery, 40 and 120 minutes). In all other experiments, multiway analysis of variance (ANOVA) was carried out to examine for significant main effects and interactions between factors. Significant differences in NMR metabolites and change in pHi among groups was determined with ANOVA followed by the Tukey-Kramer HSD or Dunnett's multiple comparison tests for differences between mean values. Significance was accepted with P < 0.05. Analysis of variance was performed on the logarithms of percent changes to overcome the nonnormal distribution of ratios.
RESULTS
The effect of substrate deprivation (no oxygen, no glucose) on bioenergetic failure and DNA fragmentation
Phosphorus 31 NMR spectra from a superfused brain slice preparation before, during, and after a 40-minute in vitro insult are shown in Fig. 3. In these studies, before insult the pHi was 7.20 ± 0.02, and the PCr to β-ATP ratio in 500- and 280-scan spectra was 1.70 ± 0.33 and 1.59 ± 0.31, respectively (not significantly different). All insults resulted in significant changes in pHi (ΔpHi) by the end of the insult of approximately -0.18 pH units (Table 1). The energy metabolites in control experiments were well conserved (PCr, 100% ± 6.6%; β-ATP, 100% ± 2.7%). However, PCr and β-ATP were particularly sensitive to changes in the availability of oxygen and glucose, with the effect dependent on the type of insult. Cortical slices deprived of oxygen for 40 minutes (in vitro hypoxia) retained at the end of the insult 52% ± 10.4% PCr and 73% ± 9.5% β-ATP relative to preischemic levels, whereas slices deprived of oxygen and glucose for 40 minutes (in vitro ischemia) had lost all detectable PCr and β-ATP. An intermediate insult—20 minutes of hypoxia followed by 20 minutes of ischemia—had a highly variable and intermediate effect (PCr, 16% ± 27.1%; β-ATP, 40% ± 34.7%). These changes exhibited a trend with increasing severity of insult (control; 40 minutes of hypoxia; 20 minutes of hypoxia then 20 minutes of ischemia; 40 minutes of ischemia) resulting in significantly worsened energetic state (τ for PCr and β-ATP: –0.84 and –0.90, respectively; P < 0.001).
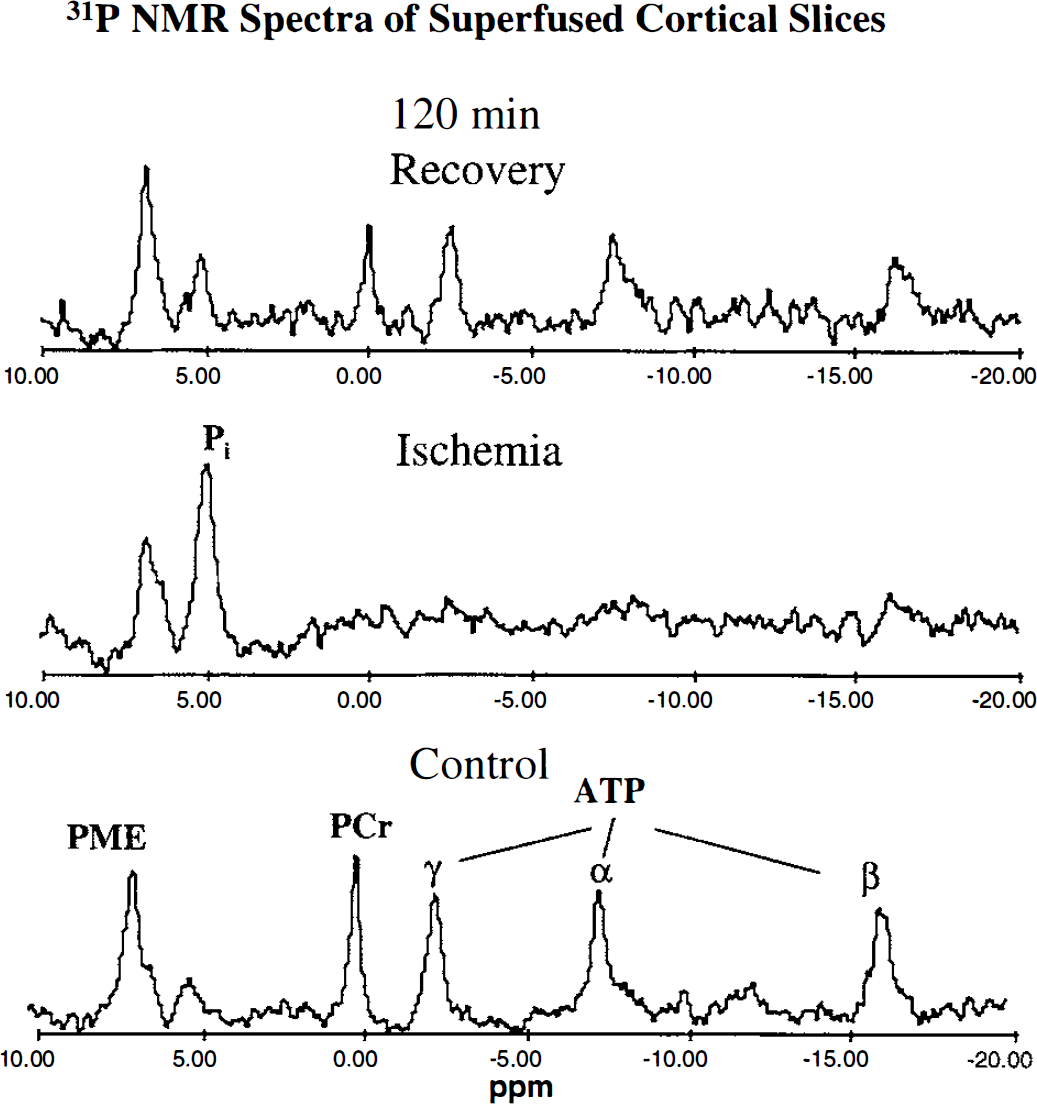
Phosphorus 31 NMR spectra from neonatal rat superfused cortical slices recorded before ischemia (control), at the end of the ischemic period, and at the end of a 120-minute recovery period. Acquisition and processing conditions are given in the methods. PME, phosphomonoesters (mainly phosphorylethanolamine); ppm, parts per million chemical shift scale expressed relative to PCr at 0 ppm.
A summary of changes relative to preinsult values induced by substrate and oxygen deprivation for 40 minutes observed at the end of insult, and after 40- and 120-minute recovery
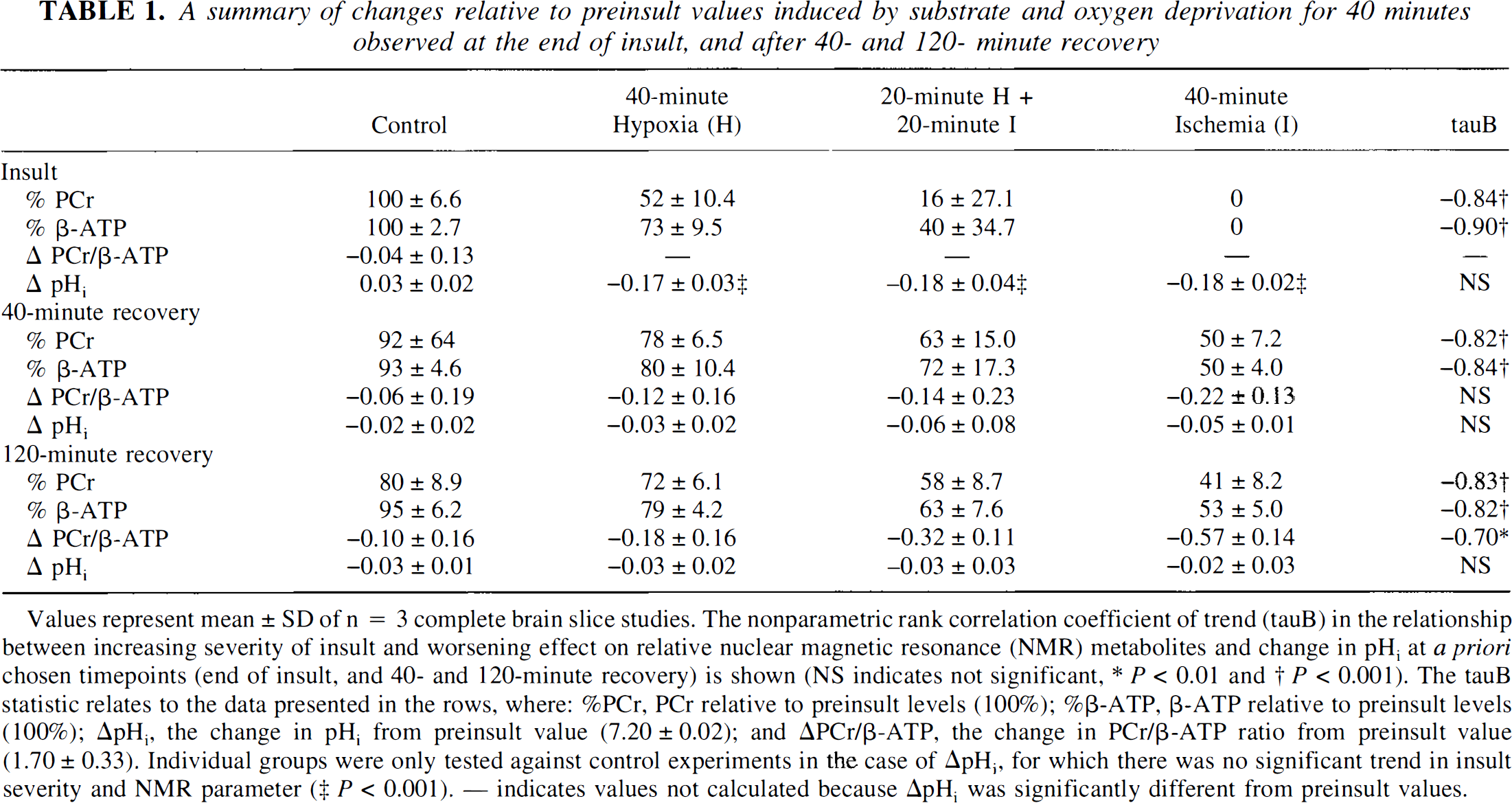
Values represent mean ±SD of n = 3 complete brain slice studies. The nonparametric rank correlation coefficient of trend (tauB) in the relationship between increasing severity of insult and worsening effect on relative nuclear magnetic resonance (NMR) metabolites and change in pHi at a priori chosen timepoints (end of insult, and 40- and 120-minute recovery) is shown (NS indicates not significant, * P < 0.01 and † P < 0.001). The tauB statistic relates to the data presented in the rows, where: %PCr, PCr relative to preinsult levels (100%); %β-ATP, β-ATP relative to preinsult levels (100%); ΔpHi, the change in pHi from preinsult value (7.20 ± 0.02); and ΔPCr/β-ATP, the change in PCr/β-ATP ratio from preinsult value (1.70 ± 0.33). Individual groups were only tested against control experiments in the case of ΔpHi, for which there was no significant trend in insult severity and NMR parameter (‡ P < 0.001). — indicates values not calculated because ΔpHi was significantly different from preinsult values.
The severity of energy failure at insult influenced recovery between 40 and 120 minutes. The data in Table 1 summarize the changes in PCr and β-ATP relative to preischemic levels, and the overall change in pHi (preischemia—recovery ΔpHi) in the recovery period. There was no significant effect of insult on pHi with a return to control values by 40 minutes of recovery. Increasing severity of insult, however, resulted in significantly worsened recovery (PCr and β-ATP: after 40 minutes, τ –0.82 and –0.84, respectively; P < 0.001; after 120 minutes, τ –0.83 and –0.82, respectively; P < 0.001). In addition, the most severe insult, 40 minutes of ischemia, resulted in a significant reduction in the PCr to β-ATP ratio at 120 minutes of recovery (ΔPCr to β-ATP, control and ischemia: –0.10 ± 0.16 and –0.57 ± 0.14, respectively; P = 0.01). In the absence of a change in pHi, which would have resulted in a decrease in the amount of PCr because of an acid shift in the creatine kinase equilibrium, this observation indicates a greater fall in PCr than ATP—or alternatively less recovery of PCr than ATP. It also suggests an increase in free ADP. Thus between 40 and 120 minutes of recovery from severe insult there is either a PCr-consuming state, presumably to replenish ongoing, mismatched ATP consumption, or compromised mitochondrial function requiring an increase in ADP to maintain respiration, or both.
Fragment analysis of low-molecular weight DNA from control, 40 minutes of hypoxia, and 40 minutes of ischemia brain slices (after 120 minutes of recovery) revealed consistent nonrandom fragmentation (∼1,800, ∼900, and ∼600 bp) in the postischemia tissue (lanes 5 through 7, Fig. 4), but not the posthypoxia or control tissue (lanes 1 through 4, Fig. 4). This is consistent with apoptosis-associated fragmentation postischemia because the DNA bands represent integer multiples of the internucleosomal DNA length of about 180 bp. Based on these findings, i.e., severe energy failure with reproducible bioenergetic recovery, secondary deterioration, and execution of apoptosis, we chose the 40-minute in vitro ischemia and 120-minute recovery protocol for further study of postischemic ionic and apoptotic interactions.
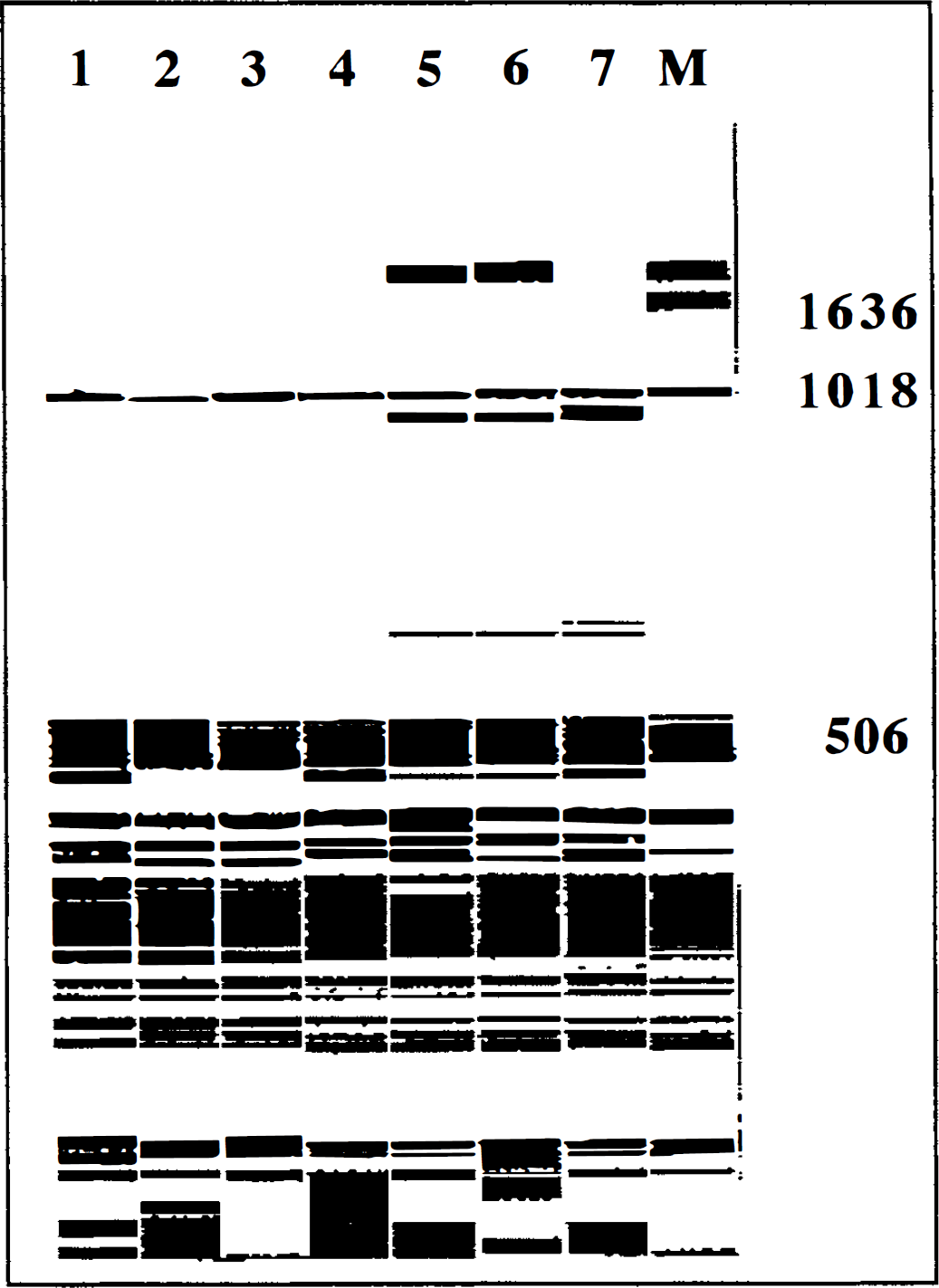
Fragment analysis of fluorescein-labeled low-molecular weight DNA electrophoresed in 4% acrylamide and analyzed on the ALF fragment analysis program. Eight lanes (1–7 and M) are shown: 1, control; 2 to 4, 40 minutes of hypoxia; 5 to 7, 40 minutes of ischemia; and M, standard DNA size marker. Lanes 5 and 6 contain fragments ∼1,800, ∼900 and ∼600 bp, and lane 7 contains fragments ∼900 and ∼600 bp, consistent with DNA laddering associated with apoptosis.
Recovery from in vitro ischemia and effect of dantrolene
After the 40-minute ischemic insult, the brain slices were allowed to recover for a further 40 minutes in either control or dantrolene-containing buffer (“early” dantrolene). In some experiments, recovery in dantrolene-containing buffer was delayed by 40 minutes (“late” dantrolene). The data in Figs. 5 and 6 and Table 2 summarize the changes in NMR metabolites relative to preischemic levels, and the overall change in pHi (preischemia-recovery, ΔpHi) during the recovery period. There was no significant change in pHi at 40 and 120 minutes of recovery (Fig. 5), but earlier (20 minutes) recovery in pHi was delayed in slices exposed to dantrolene (P < 0.05) with the ΔpHi approximately –0.12 in the dantrolene groups. In the high-energy phosphates early dantrolene yielded a significant (P < 0.05) improvement in recovery of PCr and β-ATP by 40 minutes: PCr and β-ATP for control ischemia and ischemia with dantrolene were, respectively, PCr, 54% ± 2.6% and 68% ± 3.2%; β-ATP, 55% ± 2.4% and 66% ± 2.9%. This degree of improvement with early dantrolene was still evident after 120 minutes (Fig. 6 and Table 2). Later postischemic introduction of dantrolene did not ameliorate bioenergetic state. The early dantrolene-induced recruitment of PCr and ATP indicates that in the neonatal cortical slice preparation mobilization of intracellular calcium pools is involved in bioenergetic stress early after an ischemic insult.
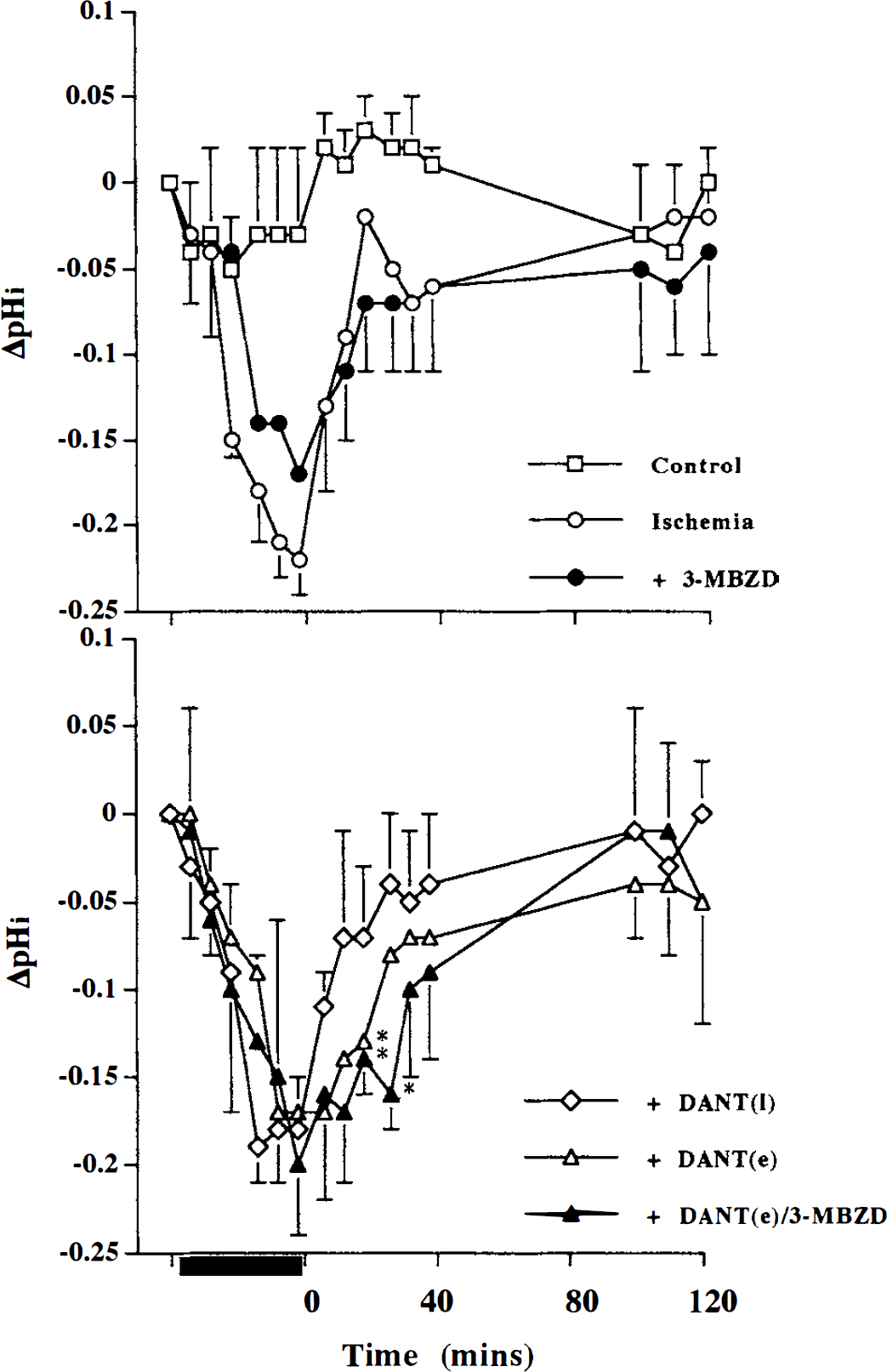
Change in intracellular pH (ΔpHi—relative to preinsult control) during a 40-minute in vitro ischemic insult (black bar on abscissa) and during 120 minutes of recovery (time 0 to 120 minutes). Upper panel: nonischemic control (Control), 40-minute ischemia (Ischemia), and 40-minute ischemia with 500 µmol/L 3-methoxybenzamide during 40- to 120-minute recovery (3-MBZD) groups. Lower panel: 40-minute ischemia with 20 µmol/L dantrolene during 40- to 120-minute recovery (DANT (I)), 40-minute ischemia with 20 µmol/L dantrolene during 0- to 40-minute recovery (DANT (e)), and 40-minute ischemia with DANT (e) and 3-MBzD groups. Values represent mean ± SD and * indicates significantly different from ischemia group (analysis of variance with post-hoc Tukey-Kramer HSD, P < 0.05).
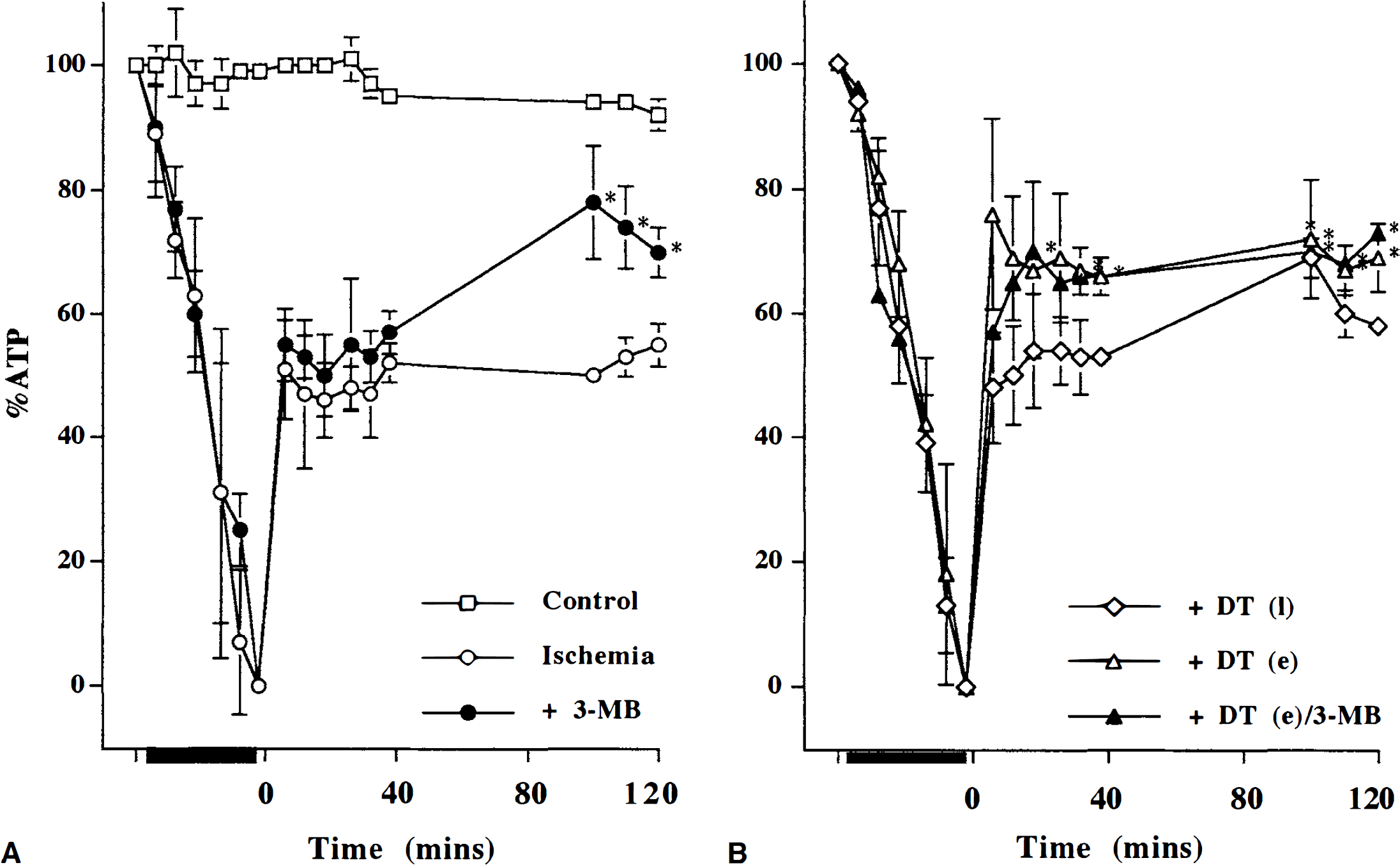
β-ATP relative to preinsult levels (% ATP) during a 40-minute in vitro ischemic insult (black bar on abscissa) and during 120 minutes of recovery (time 0 to 120 minutes).
A summary of in vitro ischemia-induced changes in bioenergetic state modified by dantrolene and 3-methoxybenzamide administered during recovery, and observed after 120-minute recovery
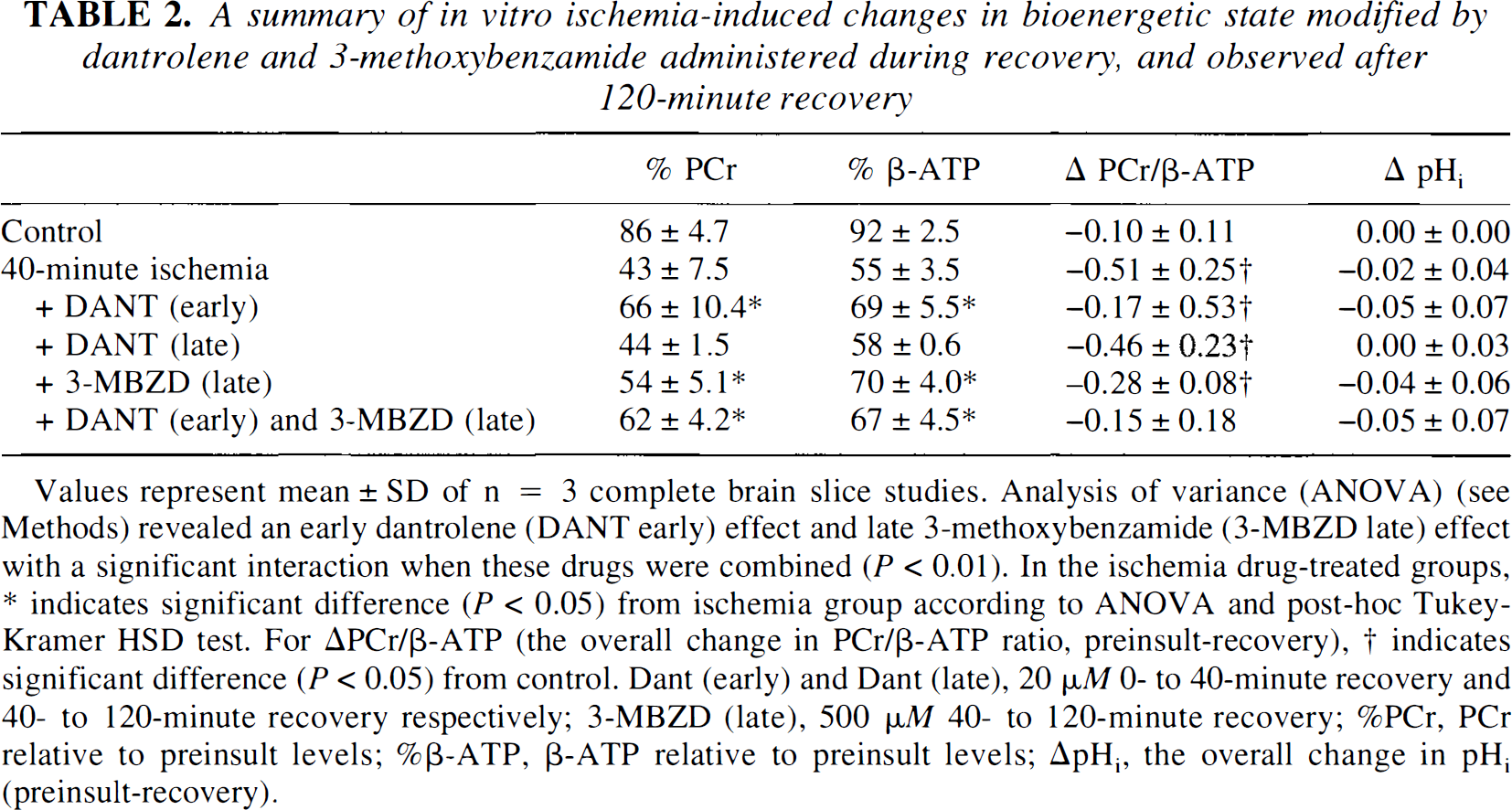
Values represent mean ± SD of n = 3 complete brain slice studies. Analysis of variance (ANOVA) (see Methods) revealed an early dantrolene (DANT early) effect and late 3-methoxybenzamide (3-MBZD late) effect with a significant interaction when these drugs were combined (P < 0.01). In the ischemia drug-treated groups, * indicates significant difference (P < 0.05) from ischemia group according to ANOVA and post-hoc Tukey-Kramer HSD test. For ΔPCr/β-ATP (the overall change in PCr/β-ATP ratio, preinsult-recovery), † indicates significant difference (P < 0.05) from control. Dant (early) and Dant (late), 20 µM 0- to 40-minute recovery and 40- to 120-minute recovery respectively; 3-MBZD (late), 500 µM 40- to 120-minute recovery; %PCr, PCr relative to preinsult levels; %β-ATP, β-ATP relative to preinsult levels; ΔpHi, the overall change in pHi (preinsult-recovery).
Recovery from in vitro ischemia and effect of late postischemic 3-methoxybenzamide
Poly(ADP-ribose) polymerase can be inhibited by benzamide and its derivatives (Ueda and Hayaishi, 1985; Kofler et al., 1993). When PARP has been activated, ATP is consumed by the cell; correspondingly, when the active enzyme is inhibited by benzamide, ATP consumption will be reduced and a measurable increment in ATP will indicate indirectly not only that the enzyme was activated but also the bioenergetic cost to the tissue of PARP activation.
In contrast to the lack of effect of late (40 minutes) postischemic dantrolene, the late postischemic introduction of 3-methoxybenzamide produced a significant recruitment in the level of PCr and β-ATP (P < 0.05), which indicated PARP activation between 40 and 120 minutes of postischemia (Fig. 6 and Table 2). The mean absolute percent increases in PCr and β-ATP were II % and 15%, respectively, which represent 25% relative improvement from the untreated postischemic bioenergetic state. Because early postischemic dantrolene had augmented the bioenergetic state to a similar degree we tested whether the dantrolene and 3-methoxybenzamide effects were additive when added sequentially. Early (0 to 40 minutes) postischemic dantrolene combined with late (40 to 120 minutes) postischemic 3-methoxybenzamide did not result in any additional improvement in the level of PCr and β-ATP (Fig. 6) above that seen with early dantrolene alone: there was a significant dantrolene-3-methoxybenzamide interaction for β-ATP (P < 0.05), indicating that the early dantrolene nullified any later potential 3-methoxybenzamide effect. We also noted that, in contrast to either dantrolene or 3-methoxybenzamide alone, the combination resulted in a PCr to β-ATP ratio that was not significantly different from control values (Table 2). Taken together, postischemic exacerbation of energetic stress can be limited early (0 to 40 minutes) with dantrolene or later (40 to 120 minutes) with 3-methoxybenzamide.
DISCUSSION
The results of these studies show that in the developing brain postischemic ionic- and enzyme-induced phenomena have significant bearing on cell bioenergetics and the mechanism of injury.
Postischemic bioenergetics
The study of energy metabolism using NMR spectroscopy in ex vivo respiring cortical brain slices has a number of methodological advantages over similar in vivo brain studies in live animals (Bachelard et al., 1985; Kauppinen and Williams, 1990; Espanol et al., 1992): NMR signals from extracerebral soft tissue and muscles can be eliminated; specific brain regions can be studied; spectral resolution is greater; pharmacology is more controlled; and variations in blood flow and systemic physiology are not present. However, interpretation of changes in this preparation needs to take into account certain unavoidable facts, which may limit the relevance of any findings to complex in vivo pathophysiology: the tissue being studied has been previously compromised by the process of preparation; ex vivo brain slices are metabolically suppressed, i.e., they show only trivial spontaneous electrical activity; and the milieu when studying slices is nonphysiologic. However, concerning the first of these criticisms, after reoxygenation absolute concentrations of adenylates and PCr in the ex vivo brain slice preparation may only attain approximately 80% of those found in vivo, the PCr to ATP ratio is the same as in vivo (Whittingham et al., 1984).
Our previous work in brain slice preparations (Tasker et al., 1992, 1996; Vornov et al., 1994) indicates that ionic-, NMDA-receptor, and endogenous NO-mediated events in the first 30 to 40 minutes of reperfusion are significant energy-consuming factors that warrant the concentration on energetics—reflecting ionic stress (Haddad and Donnelley, 1990; Xia et al., 1992; Bickler et al., 1993)—during the early recovery period. In the experiments reported here we have looked at bioenergetic recovery after a severe in vitro ischemic insult. We chose an insult that exhibited secondary energy failure (the loss in PCr while ATP is spared between 40 and 120 minutes of recovery indicates an increase in free ADP and decrease in phosphorylation potential) and chemical evidence of apoptosis to study interactions between ionic and oxidant mechanisms of injury. In this period we have observed a 25% relative augmentation in bioenergetic state after manipulations either at an early (0 to 40 minutes) or later (40 to 120 minutes) stage. In isolation, such a change may appear inconsequential as disturbances of energy metabolism are not generally considered to be involved in the mechanism of delayed neuronal death (Abe et al., 1995). In these experiments, however, the improvement in compromised postischemic bioenergetic state represents the benefit of not expending high-energy phosphates on sequestering calcium into endoplasmic reticulum or mitochondria (Siesjö, 1992a) or on the process requiring four molecules of ATP to recycle from nicotinamide the NAD+ used in forming polyADP-ribosyl nuclear proteins associated with repairing DNA damage (Berger, 1985; Ueda and Hayaishi, 1985; Lautier et al., 1993; Zhang et al., 1994). Furthermore, the consequence of combined inhibition of these processes was a PCr to β-ATP ratio no different from control tissue, possibly suggesting that energetic flux in the salvaged tissue had reattained normality.
Postischemic physiology and the interaction between dantrolene-sensitive and 3-methoxybenzamide-sensitive changes
In our studies, introducing dantrolene and 3-methoxybenzamide to postischemic cortex at different times in recovery disclosed important insights into the proximate neurotoxic mechanisms in neonatal brain. Changes in high-energy phosphates, as a result of 3-methoxybenzamide, indicated indirectly that PARP-related energy failure occurred between 40 and 120 minutes of recovery from ischemia, but not when earlier mobilization of intracellular Ca2+ had been inhibited by dantrolene.
The specificity of PARP inhibition by 3-aminobenzamide and related derivatives is indicated by the similarity between functional studies of cells containing a cellular PARP inhibitor (subcloned cDNA encoding only the amino-terminal 42- to 46-kDa DNA-binding domain of PARP) and cells treated with 3-aminobenzamide (Lindahl et al., 1995). Other possible metabolic effects of benzamide (an analog of nicotinamide), such as inhibition of cyclic adenosine monophosphate phosphodiesterase and nicotinamide N-methyltransferase (Ueda and Hayaishi, 1985), would compromise postischemic cells; thus, in relation to the findings in the present study they are probably not relevant. Inhibitors of PARP prevent NMDA- and NO-mediated neurotoxicity in dissociated cortical cell cultures (Zhang et al., 1994). Deletion of PARP by gene disruption renders cortical cell cultures from such mice virtually resistant to the effects of oxygen-glucose deprivation (Eliasson et al., 1997), and the animals are also protected against experimental transient focal ischemia (Eliasson et al., 1997; Endres et al., 1997). In vivo, homozygous PARP-deleted mice subjected to 2 hours of middle cerebral artery occlusion (with regional cerebral artery blood flow decreased to ∼30% of baseline) followed by 22 hours of reperfusion have an infarct volume approximately 20% of that seen in wild-type mice (Eliasson et al., 1997). These results provide cogent evidence for a primary involvement of PARP activation in neuronal damage associated with cerebral ischemia. However, studies using gene-disrupted mice can be criticized on the possibility that PARP-deletion influences resistance to, rather than just recovery from, cerebral insult, and therefore the potential benefit of postischemic therapeutic intervention remains open. In the present studies, we have used a severe ischemic insult—defined as complete loss of high-energy phosphates—and demonstrated that delayed (40 minutes) postischemic inhibition of PARP was effective at reducing cellular bioenergetic stress by approximately 25%, which may be sufficient to reduce injury.
Oxidative stress associated with stimulation of NMDA receptors is thought to be mediated by Ca2+ influx, leading to a series of potentially neurotoxic events such as activation of nitric oxide synthase with the subsequent production of NO and OONO− (Bolaños et al., 1995), activation of PARP (Zhang et al., 1994; Eliasson et al., 1997), and initiation of apoptosis (Bonfoco et al., 1995). Alternatively, redistribution of intracellular Ca2+ may be the major signal for NO production (Bhardwaj et al., 1997), mitochondrial generation of O2− (Piantadosi and Zhang, 1996), and activation of Ca2+-dependent apoptotic endonuclease activity (Arends et al., 1990; MacManus et al., 1997). The latter is suggested, first, by the observation in murine fibroblasts that DNA fragmentation is caused by a transient increase of [Ca2+]i and can be inhibited by complexing of intracellular Ca2+ (Penning et al., 1994), and second, by studies in macrophages in which quin-2, an intracellular Ca2+-chelator, inhibits the OONO−-induced suppression of mitochondrial respiration, without preventing PARP activation or NAD+ depletion (Szabó and Salzman, 1996). In the studies of neonatal brain tissue reported here, the data indicate that after an ischemic insult dantrolene augments bioenergetic recovery and ameliorates PARP-related bioenergetic failure only when administered within the initial recovery phase. This observation lends support to the hypothesis that in the infant brain early mobilization of intracellular Ca2+ is of importance after ischemia.
Alternatively, it is also possible that the dantrolene-induced changes in bioenergetics are a consequence of some other indirect effect. For example, we have previously reported that postischemic depolarization in neonatal rat cortex ameliorates postischemic levels of PCr and that this effect is mediated by NO and mimicked by the nitrosonium ion (NO+) donor glyceryl trinitrate (Tasker et al., 1996). (NO+-mediated neuroprotection may result from its reaction with free sulfhydryl groups and alteration of function at the redox modulatory site on the NMDA receptor [Lei et al., 1992; Lipton et al., 1993]). Dantrolene (20 µmol/L) can depolarize brain tissue by approximately 13 mV (Krnjevic and Xu, 1996), and depolarization can reduce the driving force for Ca2+ to enter mitochondria, thereby decreasing uptake (Gunter and Pfeiffer, 1990) and limiting mitochondria-derived reactive oxygen species. Of note, in the present study, is that in the first 20 minutes of postischemic recovery in the presence of dantrolene, recovery in pHi was delayed with a ΔpHi of approximately –0.12, which is equivalent to the change we reported with postischemic depolarization (Tasker et al., 1996). Interestingly, when dantrolene was introduced in later recovery, there was no such change in pHi and the drug had no significant protective effect. Although indirect depolarization with early postischemic dantrolene remains a possible explanation for our current findings, we consider this of secondary importance because, in contrast to the depolarization-induced, postischemic exacerbation of bioenergetic state in mature cortex (Tasker et al., 1996), dantrolene has been reported to be neuroprotective in mature animals (Wei and Perry, 1996). However, it may be that dantrolene is more protective in young animals because of the associated depolarization effect (Krnjevic and Xu, 1996), the lower peak [Ca2+]i achieved with energy failure in immature cortex (Bickler et al., 1993), and the effects described in this report. Finally, we noted that the PCr to β-ATP ratio was similar to control values only when postischemic dantrolene was combined with 3-methoxybenzamide, which raises the possibility that when dantrolene alone is used activation of PARP and secondary energy failure is delayed beyond the bounds of our experimental paradigm.
Taken together, these postischemic ionic- and enzyme-induced phenomena have significant bearing on the complex interaction between cell bioenergetics and mechanism of injury in brain tissue. In the immature brain the beneficial effects of limiting postischemic mobilization of intracellular Ca2+ and inhibition of PARP activation on bioenergetics has been clearly underscored. The intricate link between these phenomena may also explain the “vexatious issue” of neuronal morphology after cerebral ischemia recently reviewed by MacManus and Linnik (1997). Features of both necrosis (mitochondrial swelling) and apoptosis (nuclear margination) in the same cells have been reported in a variety of models (Colicos and Dash, 1996; Portera-Cailliau et al., 1997a, 1997b), and this finding is entirely consistent with Ca2+-dependent effects on intracellular organelles occurring simultaneously with PARP-activation and cellular bioenergetic stress. We have demonstrated in our studies that recovery from an insult known to deplete all high-energy phosphates exhibits secondary energy failure and chemical evidence of apoptosis within 2 hours of reperfusion with oxygen and glucose. The findings suggest that therapies designed toward inhibiting mobilization of intracellular Ca2+ and inhibition of PARP even after the insult may be effective: in lesser insults it is also possible that a slower time course means a longer postinsult therapeutic window.