
Abstract
Poly(adenosine 5′-diphosphoribose) synthetase (PARS) has been described as an important candidate for mediation of neurotoxicity by nitric oxide. In the current study, we demonstrate for the first time that in vivo administration of a potent PARS inhibitor, 3,4-dihydro 5-[4-1(1-piperidinyl) butoxy]-1(2H)-isoquinolinone, leads to a significant reduction of infarct volume in a focal cerebral ischemia model in the rat. Focal cerebral ischemia was produced by cauterization of the right distal middle cerebral artery (MCA) with bilateral temporary common carotid artery occlusion for 90 minutes. 3,4-Dihydro 5[4-(1-piperidinyl) butoxy]-1(2H)-isoquinolinone was dissolved in dimethyl sulfoxide and injected intraperitoneally. Animals were treated 2 hours before MCA occlusion (control, n = 14; 5 mg/kg, n = 7; 10 mg/kg, n = 7; 20 mg/kg, n = 7; 40 mg/kg, n = 7), and 2 hours after MCA occlusion (same doses as before treatment). Twenty-four hours after MCA occlusion, the total infarct volume was measured using 2,3,5-triphenyltetrazolium chloride. Inhibition of PARS leads to a significant decrease in the damaged volume in the 5 mg/kg–treated group (106.7 ± 23.2 mm3; mean ± SD, P < 0.002), the 10 mg/kg–treated group (76.4 ± 16.8 mm3, P < 0.001), and the 20 mg/kg–treated group (110.2 ± 42.0 mm3, P < 0.02) compared with the control group (165.2 ± 34.0 mm3). The substantial reduction in infarct volume indicates that the activation of PARS may play an important role in the pathogenesis of brain damage in cerebral ischemia through intracellular energy depletion.
Keywords
Nitric oxide (NO) originally was identified as the endothelium-derived relaxing factor (Ignarro et al., 1987; Palmer et al., 1987). It was subsequently found to be a messenger molecule in the nervous system (Garthwaite et al., 1988; Bredt and Snyder, 1989). Excessive NO production has been implicated in playing a significant role in the N-methyl-
Also called PARP [for poly(ADP-ribose) polymerase (EC 2.4.2.30)], PARS is a tightly bound chromosomal enzyme located in the nuclei of cells of various organs including brain (Ikai et al., 1980; Ogura et al., 1990). PARS plays a physiologic role in the repair of strand breaks in DNA (Satoh and Lindahl, 1992). Once activated by damaged DNA fragments, PARS catalyzes the attachment of ADP-ribose units to nuclear proteins, including histones and PARS itself. The PARS activation enhances DNA repair by relaxing chromosomal structure through poly(ADP-ribosyl)ation of histones and other nuclear proteins. The extensive activation of PARS, however, can rapidly lead to cell death through depletion of energy stores, since four molecules of ATP are consumed in NAD (the source of ADP-ribose) regeneration (Berger, 1985). Previously published data show that a series of PARS inhibitors protects various cell cultures from NO- and glutamate-mediated neurotoxicity (Miller et al., 1993; Cosi et al., 1994; Radons et al., 1994) and reactive oxygen radical–induced injury (Thies and Autor, 1991; Aalto and Raivio, 1993). Despite many in vitro studies using inhibitors of PARS, there have been no investigation on the effect of in vivo administration of PARS inhibitors in cerebral ischemia.
A new series of highly potent inhibitors of PARS, the dihydroisoquinolinones, recently has been reported (Suto et al., 1993). 3,4-Dihydro 5-[4-(1-piperidinyl) butoxy]-1(2H)-isoquinolinone is one member in this potent series of PARS inhibitors. 3,4-Dihydro 5-[4-(1-piperidinyl) butoxy]-1(2H)-isoquinolinone is mimicking nicotinamide to competitively inhibit PARS at the NAD binding site. The current study examines the effect of in vivo administration of 3,4-dihydro 5-[4-(1-piperidinyl) butoxy]-1(2H)-isoquinolinone on focal cerebral ischemia produced by middle cerebral artery (MCA) occlusion in the rat.
MATERIALS AND METHODS
Animal preparation
All procedures performed on animals were approved by the University Institutional Animal Care and Use Committee of the University of Pennsylvania. A total of 42 male Long-Evans rats (weights 230 to 340 g) obtained from Charles River Laboratory (Wilmington, MA, U.S.A.) were used in this study. The animals were fasted overnight with free access to water before the surgical preparation. They were anesthetized with halothane (4% for induction and 0.8% to 1.2% for surgical procedure) in a mixture of 70% nitrous oxide and 30% oxygen. The body temperature was monitored by a rectal probe and maintained at 37.5° ± 0.5°C with a heating blanket regulated by a homeothermic blanket control unit (Harvard Apparatus Limited, Kent, U.K.). A catheter (PE-50) was placed into the tail artery, and arterial pressure was continuously monitored and recorded on a Grass polygraph recorder (Model 7D, Grass Instruments, Quincy, MA, U.S.A.). Samples for blood gas analysis (arterial pH, Pao2 and Paco2) also were taken from the tail artery catheter and measured with a blood gas analyzer (ABL 30, Radiometer, Copenhagen, Denmark).
Laser Doppler flowmetry
The head was positioned in a stereotaxic frame, and a right parietal incision between the right lateral canthus and the external auditory meatus was made. Using a dental drill constantly cooled with saline, a 3-mm burr hole was prepared over the cortex supplied by the right MCA, 4 mm lateral to the sagittal suture and 5 mm caudal to the coronal suture. The dura mater and a thin inner bone layer were kept, with care being taken to position the probe over a tissue area devoid of large blood vessels. The flow probe (tip diameter of 1 mm, fiber separation of 0.25 mm) was lowered to the bottom of the cranial burr hole using a micromanipulator. The probe was held stationary by a probe holder secured to the skull with dental cement. The microvascular blood flow in the right parietal cortex was continuously monitored with a laser Doppler flowmeter (FloLab, Moor, Devon, U.K.; and PeriFlux 4001, Perimed, Stockholm, Sweden).
Focal cerebral ischemia
Focal cerebral ischemia was produced by cauterization of the distal portion of the right MCA with bilateral temporary common carotid artery (CCA) occlusion (Chen et al., 1986; Liu et al., 1989). Bilateral CCA were isolated, and loops made from polyethylene catheter (PE-10) were carefully passed around the CCA for later remote occlusion. The incision made previously for placement of the laser Doppler probe was extended to allow observation of the rostral end of the zygomatic arch at the point of fusion to the squamous bone. A burr hole was made in the parietal bone 2 to 3 mm rostral to the fusion point using a dental drill, and the dura mater overlying the MCA was cut. The MCA distal to its crossing with the inferior cerebral vein was lifted by a fine stainless steel hook attached to a micromanipulator, and after bilateral CCA occlusion, the MCA was cauterized with an electrocoagulator. The burr hole was covered with a small piece of Gelform, and the wound was sutured to maintain the brain temperature. After 90 minutes of occlusion, the carotid loops were released, the tail arterial catheter was removed, and all of the wounds were sutured. Gentamicin sulfate (10 mg/mL; SoloPak Laboratories, IL, U.S.A.) was topically applied to the wounds to prevent infection. The anesthesia was discontinued, and the animal was returned to its cage after awakening. At the dose we used in the study, 3,4-dihydro 5-[4-(1-piperidinyl) butoxy]-1(2H)-isoquinolinone had no apparent effects on the rats in terms of mortality or recovery from anesthesia. No animals died during the study, and all rats awakened from the halothane anesthesia within 20 minutes after halothane was discontinued. Water and food were allowed ad libitum.
Therapeutic protocol
3,4-Dihydro 5-[4-(1-piperidinyl) butoxy]-1 (2H)-isoquinolinone was dissolved in 100% dimethyl sulfoxide using a sonicator and injected intraperitoneally. Animals were treated with 3,4-dihydro 5-[4-(1-piperidinyl) butoxy]-1(2H)-isoquinolinone 2 hours before MCA occlusion (5 mg/kg, n = 7; 10 mg/kg, n = 7; 20 mg/kg, n = 7; 40 mg/kg, n = 7) and 2 hours after MCA occlusion (same doses as before treatment) in a volume of 1.28 mL of dimethyl sulfoxide per kilogram of body weight per injection. This dosing schedule was used to keep the volume of dimethyl sulfoxide constant across groups. Fourteen animals were injected similarly with equivalent volumes of the vehicle in the same schedule. These studies were designed to demonstrate the principle of in vivo PARS inhibition for reducing neuronal damage during ischemia. Consequently, the PARS inhibitor was administered both before and after the start of ischemia so as to maximize the inhibition during the early ischemic period.
Morphometric measurement of damaged volume
Twenty-four hours after the MCA occlusion, the rats were killed with an intraperitoneal injection of pentobarbital sodium (150 mg/kg). The brain was carefully removed from the skull and cooled in ice-cold artificial CSF for 5 minutes and then sectioned in the coronal plane at 2-mm intervals using a rodent brain matrix (RBM-4000C, ASI Instruments, Warren, MI, U.S.A.) The brain slices were incubated in phosphate-buffered saline containing 2% 2,3,5-triphenyltetrazolium chloride (Sigma, St. Louis, MO, U.S.A.) at 37 °C for 10 minutes. Color slides were obtained from the posterior surface of the stained slices and were used to determine the damaged area at each cross-sectional level using a computer-based image analyzer (NIH Image version 1.59). To avoid artifacts caused by edema, the damaged area was calculated by subtracting the area of the normal tissue in the hemisphere ipsilateral to the stroke from the area of the hemisphere contralateral to the stroke (Swanson et al., 1990). The total volume of infarction was calculated by summation of the damaged volume of the brain slices.
Statistical analysis
The data are expressed as mean ± SD. The significance of differences between groups was determined using an analysis of variance followed by probing for the source of the difference using a Student's t test in which a Bonferroni correction was made for multiple comparisons.
RESULTS
Physiologic variables
The values of arterial blood gases (Pao2, Paco2, and pH) were within the physiologic range in the control and treated groups with no significant differences in these parameters among the five groups (Table 1). There were no significant differences in mean arterial blood pressure before MCA and CCA occlusion among the five groups. Although mean arterial blood pressure was significantly elevated after occlusion in all five groups, there were no significant differences in mean arterial blood pressure during the occlusion period among the groups (Table 1).
Physiologic variables
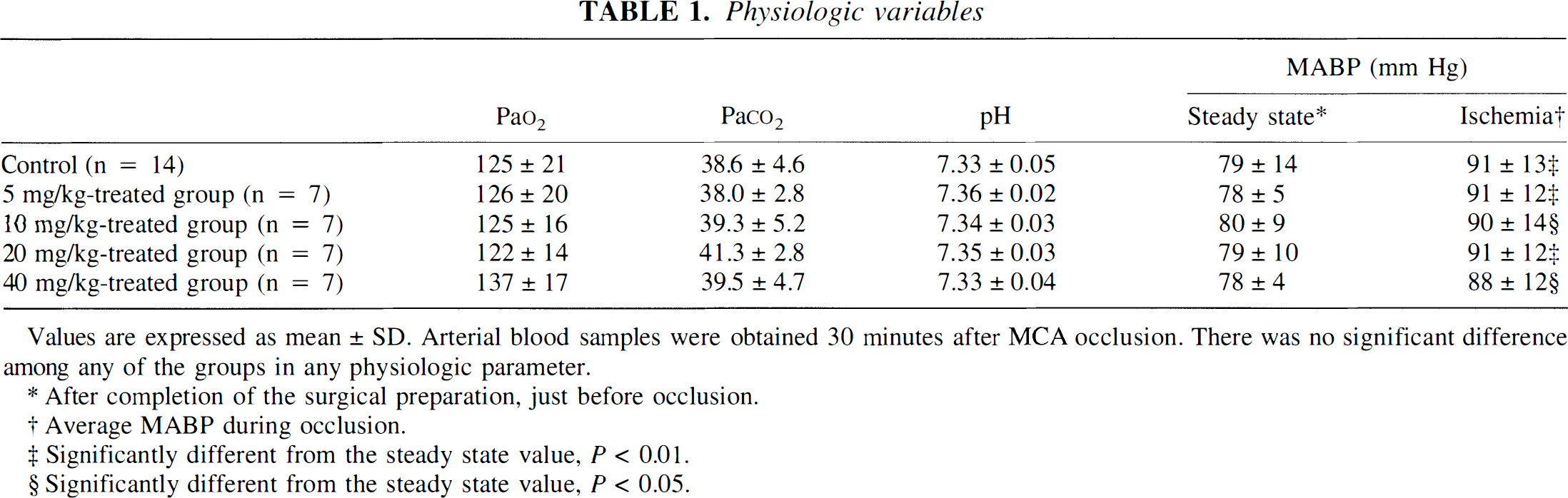
Values are expressed as mean ± SD. Arterial blood samples were obtained 30 minutes after MCA occlusion. There was no significant difference among any of the groups in any physiologic parameter.
After completion of the surgical preparation, just before occlusion.
Average MABP during occlusion.
Significantly different from the steady state value, P < 0.01.
Significantly different from the steady state value, P < 0.05.
Effects of the right middle cerebral artery and bilateral common carotid artery occlusion on cerebral blood flow
Since the blood flow values obtained from the laser Doppler are in arbitrary units, only percent changes from the baseline (before occlusion) are reported. Right MCA and bilateral CCA occlusion produced a significant decrease in relative blood flow in the right parietal cortex to 20.8 ± 7.7% of the baseline in the control group (n = 5), 18.7 ± 7.4% in the 5 mg/kg–treated group (n = 7), 21.4 ± 7.7% in the 10 mg/kg–treated group (n = 7), and 19.3 ± 11.2% in the 40 mg/kg–treated group (n = 7). There were no significant differences in the blood flow response to occlusion among the four groups. In addition, blood flow showed no significant changes throughout the entire occlusion period in any group.
Effects of PARS inhibition on focal cerebral ischemia
The cauterization of the distal portion of the right MCA with bilateral temporary CCA occlusion consistently produced a well-recognized cortical infarct in the right MCA territory of each animal. There was an apparent uniformity in the distribution of the damaged area as measured by 2,3,5-triphenyltetrazolium chloride staining in each group (Fig. 1). Inhibition of PARS leads to a significant decrease in the damaged volume in the 5 mg/kg–treated group (106.7 ± 23.2 mm3, P < 0.002), the 10 mg/kg–treated group (76.4 ± 16.8 mm3, P < 0.001), and the 20 mg/kg–treated group (110.2 ± 42.0 mm3, P < 0.02) compared with the control group (165.2 ± 34.0 mm3). However, there was no significant difference between the control and the 40 mg/kg–treated group (135.6 ± 44.8 mm3) (Fig. 2).
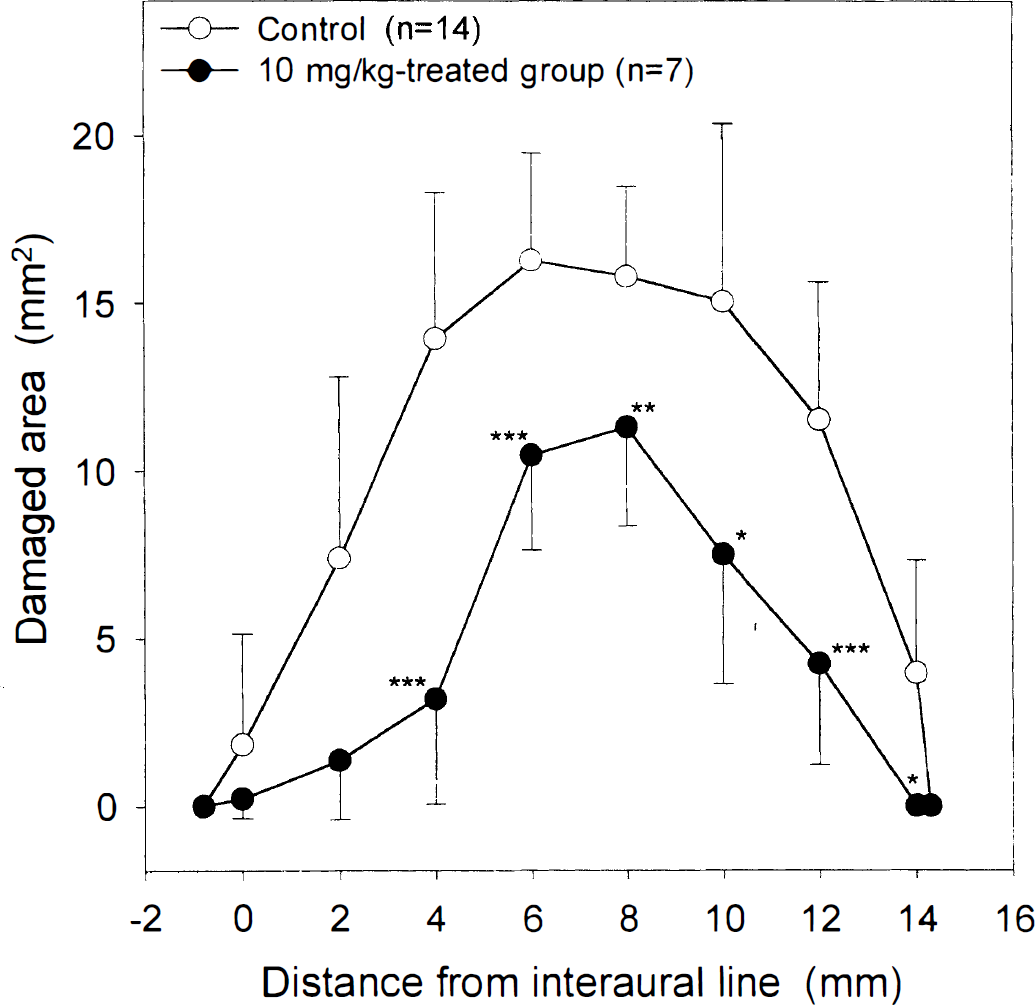
The distribution of the cross-sectional infarct area at representative levels along the rostrocaudal axis as measured from the interaural line in nontreated animals and in animals treated with 10 mg/kg of 3,4-dihydro 5-[4-(1-piperidinyl) butoxy]-1(2H)-isoquinolinone. The area of damage is expressed as mean ± SD. Significant differences between the 10 mg–treated group and the control group are indicated (*P < 0.05, **P < 0.02, ***P < 0.005). The 5 mg/kg and 20 mg/kg curves fall approximately halfway between the control and 10 mg/kg curves, whereas the 40 mg/kg curve is close to the control. These curves are omitted for clarity.
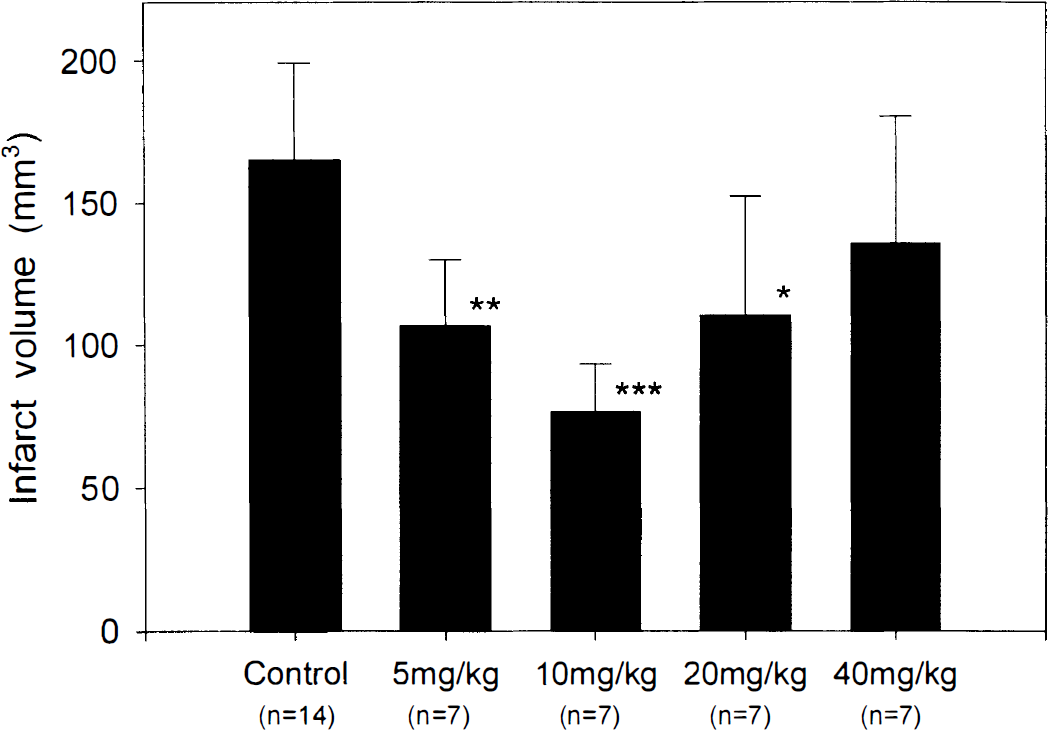
The effect of intraperitoneal administration of 3,4-dihydro 5-[4-(1-piperidinyl) butoxy]-1(2H)-isoquinolinone on the infarct volume. The volumes of infarct are expressed as mean ± SD. Significant differences between the treated groups and the control group are indicated (*P < 0.02, **P < 0.002, ***P < 0.001).
DISCUSSION
In the current study, we demonstrate for the first time that in vivo administration of a PARS inhibitor leads to a significant reduction of infarct volume in a focal cerebral ischemia model in the rat. This suggests that the activation of PARS may play an important role in the pathogenesis of brain damage in cerebral ischemia through intracellular energy depletion.
In our focal ischemia model, the ligation of the right distal MCA and bilateral temporary CCA occlusion for 90 minutes produced a large and well-recognized infarction. It has been shown that the ligation of the right distal MCA only (without CCA occlusion) does not produce a cortical infarction in Long-Evans rats (Chen et al., 1986). When both CCA also are temporarily occluded, as in the current study, the infarct volume becomes 188 ± 18 mm3 (means ± SEM) (Liu et al., 1989), and when both the MCA and the CCA are reopened after 90 minutes of occlusion, the volume of infarction is comparable (171 ± 16 mm3) (Yip et al., 1991). These values are similar to that observed in our study. After release of the carotid occlusions, we observed a good recovery of blood flow (sometimes hyperemia) in the right MCA territory of all animals (data not shown). Reperfusion of the ischemic tissue results in the formation of NO and peroxynitrite, in addition to oxygen-derived free radicals. All of these radicals have been shown to cause DNA strand breaks and activate PARS (Zhang et al., 1995; Szabó et al., 1996). It has been recently demonstrated that during stroke in mice, PARS is activated as revealed by a substantial increase of poly(ADP-ribose) polymer detected by immunostaining (Eliasson et al., 1997).
A series of PARS inhibitors have been shown to protect rat cortical cell cultures from NO-mediated neurotoxicity acting through NMDA-receptor with relative potencies paralleling their ability as PARS inhibitors (Zhang et al., 1994). Several previous in vitro studies have shown that PARS inhibitors are capable of protecting cells against glutamate-mediated toxicity (Miller et al., 1993; Cosi et al., 1994), NO-mediated toxicity (Radons et al., 1994; Inada et al., 1995), reactive oxygen injury (Thies and Autor, 1991; Aalto and Raivio, 1993), hydrogen peroxide–induced injury (Schraufstatter et al., 1986), and peroxynitrite-mediated injury (Szabó et al., 1996). These data show that PARS inhibition is neuroprotective and indicate that NAD levels may be responsible for this neuroprotection. It has been recently reported that inhibitors of poly(ADP-ribosyl)ation also provide good protection from NO-induced injury with recovery of CA1-evoked responses in rat hippocampal slices (Wallis et al., 1993). In addition, pancreatic islet cells from mice with an inactivated PARS gene do not show NAD depletion after exposure to DNA-damaging radicals, and are more resistant to the toxicity of both NO and reactive oxygen intermediates (Heller et al., 1995; Wang et al., 1995). The PARS inhibitors prevent the rapid fall in NAD and ATP pools. This preservation of the ATP pool apparently has a permissive effect on energy-dependent processes in the cell, which can salvage injured tissue from intracellular energy depletion.
In our study, we treated animals with 3,4-dihydro 5-[4-(1-piperidinyl) butoxy]-1(2H)-isoquinolinone 2 hours before the MCA occlusion and 2 hours after the MCA occlusion (30 minutes after the reopen of the bilateral CCA occlusion). There is evidence that single injections of PARS inhibitors reduce the infarct size caused by ischemia and reperfusion of the heart or skeletal muscle in rabbits (Thiemermann et al., 1997). In these studies, a single injection of 3-aminobenzamide, another PARS inhibitor (10 mg/kg), either 1 minute before occlusion or 1 minute before reperfusion, causes similar reductions in infarct size in the heart (32% or 42%). Other PARS inhibitors, including 1,5-dihydroxyisoquinoline (1 mg/kg), which belongs to a same series of PARS inhibitors as 3,4 dihydro 5-4[(1-piperidinyl) butoxy]-1(2H)-isoquinolinone, also reduce infarct size by 38% to 48%. These findings suggest that PARS inhibitors are able to salvage previously ischemic tissue, even when administered with the onset of reperfusion.
The PARS inhibition has been shown to sensitize cancer cells for radiation and chemotherapy, as well as to prevent damage to normal cells under ischemia–reperfusion injury (Oleinick and Evans, 1985; Griffin et al., 1995; Thiemermann et al., 1997). The exact mechanism underlining these two paradoxical effects of PARS inhibition is not clear. Accumulating evidence seems to suggest that the effect of PARS inhibition depends on the status of the cell in its replication cycle and the nature and extent of DNA damage that causes PARS activation. It has been shown that 3-aminobenzamide has dramatically different effects on x-ray–induced cytogenetic damage in human lymphocytes, depending on the stage of the cell cycle in which cells are irradiated (Wiencke and Morgan, 1987). In rapidly dividing cells such as cancer cells, PARS inhibition may delay the repair of extensive DNA damage caused by radiation and may lead to unchecked DNA replication causing cell death. In quiescent cells, such as nondividing neurons, PARS inhibition may prevent energy depletion as a result of free-radical damage to DNA that activates PARS, and thus render cells more resistant to subsequent insults (Gaal et al., 1987). Secondly, PARS appears to be part of the necessary repair mechanisms in cells irradiated with sublethal to lethal doses. Excessive turnover of PARS, which can deplete cells of their NAD pools, may be a response to supralethal doses of ionizing radiation or ischemic injury, and the loss of NAD may contribute to cell death (Oleinick and Evans, 1985).
It is not clear why a high dose (40 mg/kg) of 3,4 dihydro 5-4[(1-piperidinyl) butoxy]-1(2H)-isoquinolinone is less neuroprotective. The U-shaped dose–response curve (Fig. 2) suggests dual effects of the compound. In a study using mice whose PARS genes were inactivated, it was found that the infarct volume resulted from MCA occlusion was substantially reduced (Eliasson et al., 1997). This suggests a secondary effect of this compound at higher doses, which, instead of inhibiting PARS, may exacerbate the neuronal damage.
Although this study demonstrates that the PARS inhibitors 3,4-dihydro 5-[4-(1-piperidinyl) butoxy]-1(2H)-isoquinolinone provides neuroprotection after cerebral ischemia, the mechanism by which this compound provides this protection requires further investigation. Demonstration that in vivo administration of PARS inhibitors can attenuate the decrease of ATP/NAD levels during ischemia, although complicated by the significant changes in these substances that normally accompanies hypoxia/ischemia, will provide additional support that this compound can inhibit PARS in vivo.
PARS plays an important role in the repair of DNA damage. However, mice lacking PARS and poly(ADP-ribosyl)ation are healthy and fertile. Embryonic fibroblasts derived from PARS knockout mice have been shown to be able to repair DNA damage. These mice, however, are susceptible to the spontaneous development of skin disease, since ∼30% of older mice develop epidermal hyperplasia (Wang et al., 1995). Although lack of PARS may lead to side effects, acute inhibition of PARS can be of therapeutic value to prevent the ischemic tissue injury. The substantial reduction in infarct volume observed in this study indicates that PARS may play an important role in ischemic celldamage.