
Abstract
Brief occlusion of the middle cerebral artery (i.e., ischemic preconditioning; PC) induces significant brain protection to subsequent severe ischemic events. In an effort to discover genes responsible for ischemic tolerance, we have applied a new technique, suppression subtractive hybridization (SSH), to identify genes that are upregulated by PC. Using this SSH approach, a cDNA that encodes tissue inhibitor of matrix metalloproteinase-1 (TIMP-1) was identified. Time course studies using Northern analysis revealed that TIMP-1 mRNA was significantly elevated at 24 hours (3.3-fold over controls, P < 0.05, n = 5) and 2 days (4.3-fold increase, P < 0.01) after PC, corresponding to the onset of significant ischemic tolerance. Our data not only demonstrate the utility of this new polymerase chain reaction—based SSH strategy for discovery of genes differentially expressed in PC, but also suggest a potential role of TIMP-1 in PC-induced ischemic tolerance.
Keywords
A short duration of ischemia (i.e., ischemic preconditioning; PC) results in a subsequent resistance to severe ischemic tissue injury (i.e., ischemic tolerance). This phenomenon has been described in heart and brain (Kitagawa et al., 1990; Kato et al., 1991; Yellon and Baxter, 1995). Very recently, we developed a focal brain PC model using 10 minutes' occlusion of the middle cerebral artery (MCAO) followed by reperfusion in hypertensive rats (Barone et al., 1998). Pathophysiologic analysis revealed a significant reduction (more than 50%) of hemispheric infarct and neurologic deficits when permanent MCAO was performed at 24 hours to 7 days after PC (Barone et al., 1998). The molecular mechanisms responsible for this ischemic tolerance are poorly understood, although candidate genes such as heat-shock protein-70 (Kitagawa et al., 1991; Liu et al., 1993; Nishi et al., 1993) and interleukin-1 receptor antagonist (Barone et al., 1998) have been implicated in PC.
In an effort to characterize additional genes specifically regulated in PC, we applied a recently developed polymerase chain reaction—based cDNA subtractive strategy termed suppression subtractive hybridization (SSH) (Diatchenko et al., 1996) and examined the gene expression using cortical samples at 24 hours after PC, a time point when significant ischemic tolerance is established (Barone et al., 1998). As illustrated in the present work, a cDNA clone encoding tissue inhibitor of matrix metalloproteinase-1 (TIMP-1) was identified. Because TIMP-1 is known to be involved in remodeling of extracellular matrix by preferential inhibition of matrix metalloproteinases (MMP) in diverse conditions such as wound healing/scar formation, angiogenesis, and cancer metastasis, the induced expression of TIMP-1 mRNA in the cortex capable of ischemic tolerance suggests a role of TIMP-1 in ischemic injury resistance.
MATERIALS AND METHODS
Animals
Rats were housed and cared for in accordance with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1985). Procedures using laboratory animals were approved by the Institutional Animal Care and Use Committee of SmithKline Beecham Pharmaceuticals.
Focal ischemic preconditioning
Transient MCAO or sham surgery was carried out in spontaneously hypertensive rats, 300 to 350 g, under sodium pentobarbital anesthesia as described previously (Barone et al., 1992). All animals were allowed free access to food and water before and after surgery. Body temperature was maintained at 37°C using a heating pad throughout the surgical procedure and during postoperative recovery. Briefly, after craniotomy using stereotaxic procedures, the dura was opened over the right middle cerebral artery. The bent tip of a platinum-iridium wire (0.1143 mm diameter) mounted on a micromanipulator was placed under the middle cerebral artery (at the level of the inferior cerebral vein) and used to lift the artery away from the brain surface to temporarily occlude blood flow, as verified previously by monitoring cortical microvascular perfusion (Barone et al., 1995). A period of 10 minutes of temporary MCAO was used for focal ischemic PC because this short ischemic period results in no discernible injury (Barone et al., 1998).
RNA preparation
The ipsilateral or contralateral cortex was dissected and immediately frozen in liquid nitrogen and stored at −80°C. Total RNA was prepared by homogenizing the cortical tissues in an acid guanidinium thiocyanate solution and extracting with phenol and chloroform as previously described (Chomczynski and Sacchi, 1987). Poly(A)+ mRNA was extracted with an oligo-dT cellulose column from total cellular RNA pooled from 25 animals at 24 hours after 10 minutes' MCAO or from normal cortex.
Suppression subtractive hybridization
The SSH was carried out according to the Clontech PCR-Select cDNA Subtraction Kit (Clontech, Palo Alto, CA, U.S.A.) with the following specification/modification. Three micrograms poly(A)+ mRNA from ipsilateral cortex at 24 hours after PC and from normal brain cortex was used for tester and driver cDNA synthesis, respectively. The double-stranded cDNA was extracted with phenol-chloroform and precipitated with ethanol. The pellet was resuspended in water and digested with RsaI restriction enzyme, then extracted with phenol-chloroform and resuspended in 5.5 µL H2O. Adaptor 1 and adaptor 2R were each separately ligated to 2 µL of 1:6 dilution of Rsa I—digested tester DNA at 16°C overnight. Samples were then heated at 70°C for 5 minutes to inactivate the ligase. Ligation efficiency was analyzed before the subtractive hybridization.
In the first hybridization 1.5 µL RsaI-digested driver cDNA was mixed with 1.5 µL of diluted adaptor 1–ligated tester or adaptor 2R—ligated tester (i.e., about 18 copies of driver DNA versus 1 copy of tester) in the presence of hybridization buffer, covered with mineral oil. The samples were denatured at 98°C for 1.5 minutes and immediately incubated in a thermal cycler at 68°C for 12 hours. In the second hybridization the two samples from the first hybridization were mixed in the presence of a freshly denatured (1 µL of 1:2 dilution) driver. The sample was incubated at 68°C for 18 hours. After adding 200 µL of dilution buffer the sample was incubated for an additional 7 minutes.
The primary polymerase chain reaction (PCR) was conducted in 25 µL, containing 1 µL of the diluted subtraction mixture, 1 µL PCR primer 1 (10 µmol/L), 10× PCR reaction buffer, 0.5 µL deoxyribonucleoside triphosphates mix (10 mmol/L), and 50× Advantage cDNA Polymerase Mix (Clontech), according to the manufacturer's specification. The reaction mixture was incubated at 75°C for 5 minutes to extend the adaptors, followed by 94°C for 25 seconds in a Perkin-Elmer GeneAmp PCR System 9700, then followed by 27 cycles at 94°C for 10 seconds, 66°C for 30 seconds, and 72°C for 1.5 minutes.
The primary PCR mixture was diluted 10-fold, and 1 µL was used in a secondary PCR with nested primers for the primer 1 and primer 2R. The conditions of the reaction were 94°C for 10 seconds, 68°C for 30 seconds, and 72°C for 1.5 minutes for 10 cycles.
Evaluation of subtraction efficiency
Analysis of the subtraction efficiency was carried out using the following two methods. Polymerase chain reaction amplification of glyceraldehyde-3-phosphate dehydrogenase (G3PDH) for the diluted subtracted cDNA pool versus unsubtracted control cDNA was performed according to the manufacturer's specification. The diluted unsubtracted cDNA is the mix of the ligations of the tester to adaptor 1 and adaptor R2 (before the overnight incubation). The dilution is 1 µL in 1 mL H2O, and PCR was performed for 33 cycles at 94°C for 30 seconds, 60°C for 30 seconds, and 68°C for 2 minutes. The PCR product was monitored on a 2% agarose gel for an aliquot removed from each reaction after 18, 23, 28, and 33 cycles.
In addition, the subtraction efficiency was evaluated by Southern hybridization of the first and secondary PCR amplification products from the subtracted and unsubtracted DNA to a housekeeping gene rpL32 (Wang et al., 1997).
Products of the selective secondary PCR were cloned into a pCR 2.1 vector using T/A cloning kit (Invitrogen, Carlsbad, CA, U.S.A.). The ligation was transformed into an Escherichia coli, IVNαF'.
Screening of the subtracted cDNA
Screening of the subtracted cDNA sample was carried out partially using the PCR-Select Differential Screening Kit (Clontech) against randomly selected bacterial colonies cultured overnight in a 96-well plate and blotted onto the 96-well bio-dot apparatus (Bio Rad, Hercules, CA, U.S.A.). The membranes were treated with 0.5 mol/L NaOH, 1.5 mol/L NaCl for 4 minutes and with 0.5 mol/L Tris-HCl, also for 4 minutes. Then the membranes were washed for 30 minutes in 0.2× SSC and 0.2% sodium dodecyl sulfate (SDS) at 63°C. Prehybridization was carried out at 42°C in a buffer containing 5× SSPE (750 mmol/L NaCl, 50 mmol/L NaH2P
Northern analysis
For Northern analysis, 30 µg/lane total cellular RNA was electrophoresed through formaldehyde agarose gel and transferred to a GeneScreen Plus membrane (NEN Life Science Products, Boston, MA, U.S.A.). cDNA fragments for TIMP-1 were released from the plasmid and gel purified. rpL32 cDNA was generated by reverse transcription—PCR as described previously (Wang et al., 1997). The cDNA probes were uniformly labeled with [α-32P]dATP (3,000 Ci/mmol, Amersham Corp., Erlington Heights, IL, U.S.A.) using a random-priming DNA labeling kit (Boehringer Mannheim, Indianapolis, IN, U.S.A.). Hybridization and washing were performed as described in detail previously (Wang et al., 1997).
Statistical analysis
Statistical evaluation was performed on five complete sets of cortical samples from each time point using one-way analysis of variance followed by a Fisher's protected t test. The results are expressed as mean ± standard deviation. Significance was accepted for P < 0.05 by comparing the relative mRNA levels in the ipsilateral cortex after PC with the sham-operated cortex.
RESULTS
The PCR-based subtraction was used to subtract cortical samples 24 hours after PC from normal cortex. The subtraction efficiency was evaluated using two housekeeping genes, G3PDH and rpL32, of which the rpL32 has been consistently demonstrated for its expression in normal, ischemic, and PC cortex (Wang et al., 1995; Barone et al., 1998). After we ensured that the housekeeping genes had been extensively removed in the subtracted pools (data not shown), the subtracted cDNA was subcloned into a pCR 2.1 vector and transformed into bacteria, and the clones were analyzed by comparative Southern dot-blot hybridization. The dot-blot hybridization once again confirmed the efficiency of the subtraction by showing that a large number of genes (in addition to the two housekeeping genes as described above) had been removed by SSH (compare Fig. 1A, hybridized to the subtracted probe, with B, hybridized to the normal cortical probe) and some low abundantly expressed genes are significantly enriched (data not shown). Noticeably, one clone (indicated with an arrow in Fig. 1) was markedly induced in the PC samples over the normal controls. The increased expression of this clone was confirmed by Northern analysis, and DNA sequencing identified it as TIMP-1.
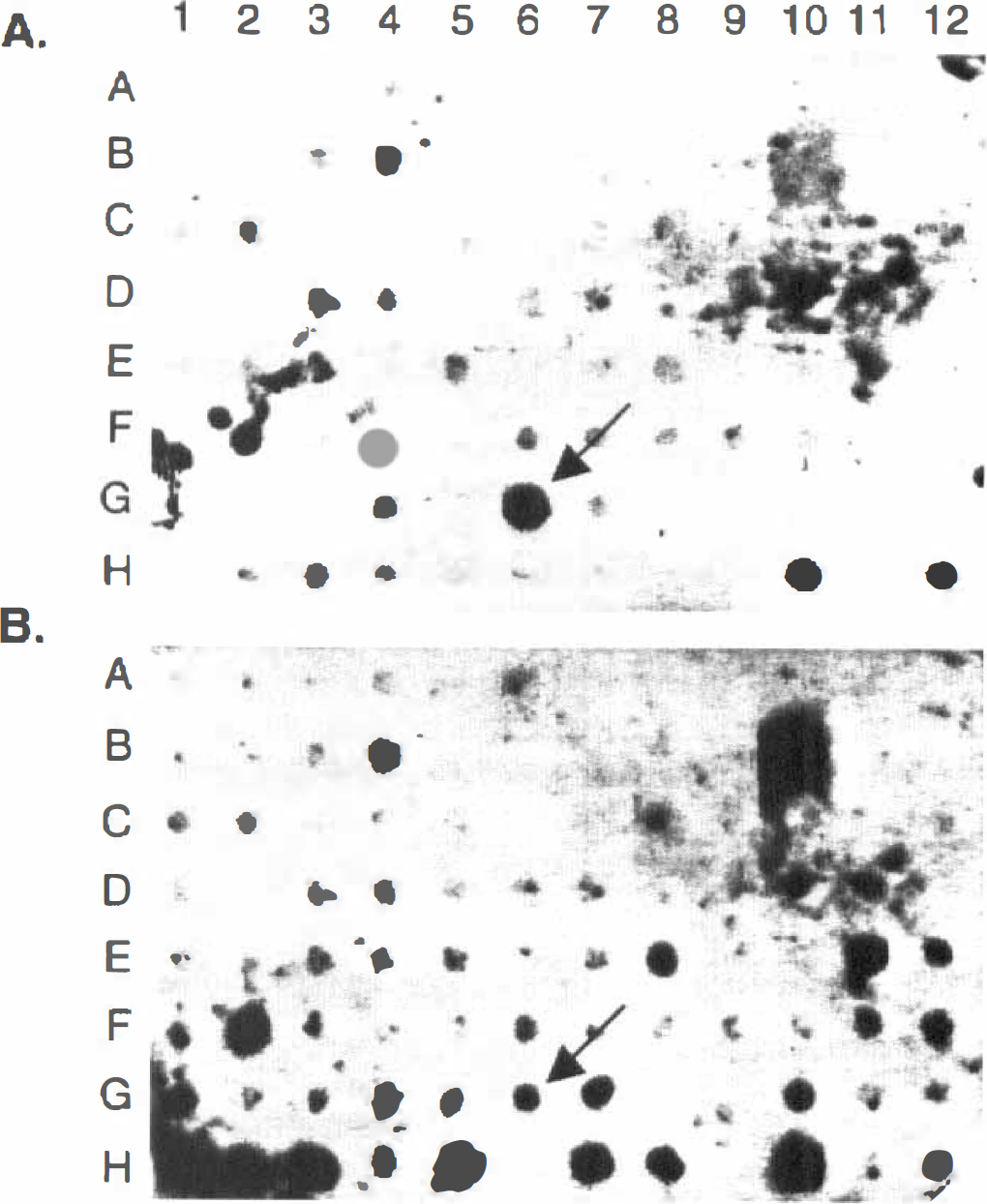
Comparative Southern analysis of cDNA clones identified by the suppression subtractive hybridization. cDNA clones were generated by suppression subtractive approach and cultured in a 96-well dish. The bacterial cultures were transferred onto a nylon membrane of a dot-blot apparatus and then analyzed by Southern hybridization.
The temporal expression of TIMP-1 mRNA after PC was examined. A representative autoradiograph of Northern blot for TIMP-1 mRNA expression in the cortex at various times after PC and in sham-operated cortical samples is illustrated in Fig. 2A. The quantitative data for TIMP-1 mRNA (n = 5), after normalizing to a housekeeping gene rpL32, are summarized graphically in Fig. 2B. Sham-operated samples were taken from animals 24 hours after surgery. As shown in Fig. 2, a very low level of TIMP-1 mRNA was detected in the contralateral cortex, and it was slightly elevated in sham-operated samples. A significant induction of TIMP-1 mRNA was observed at 24 hours (3.3-fold increase over control, P < 0.05) and 2 days (4.3-fold increase, P < 0.01) in the ipsilateral cortex after PC (Fig. 2).
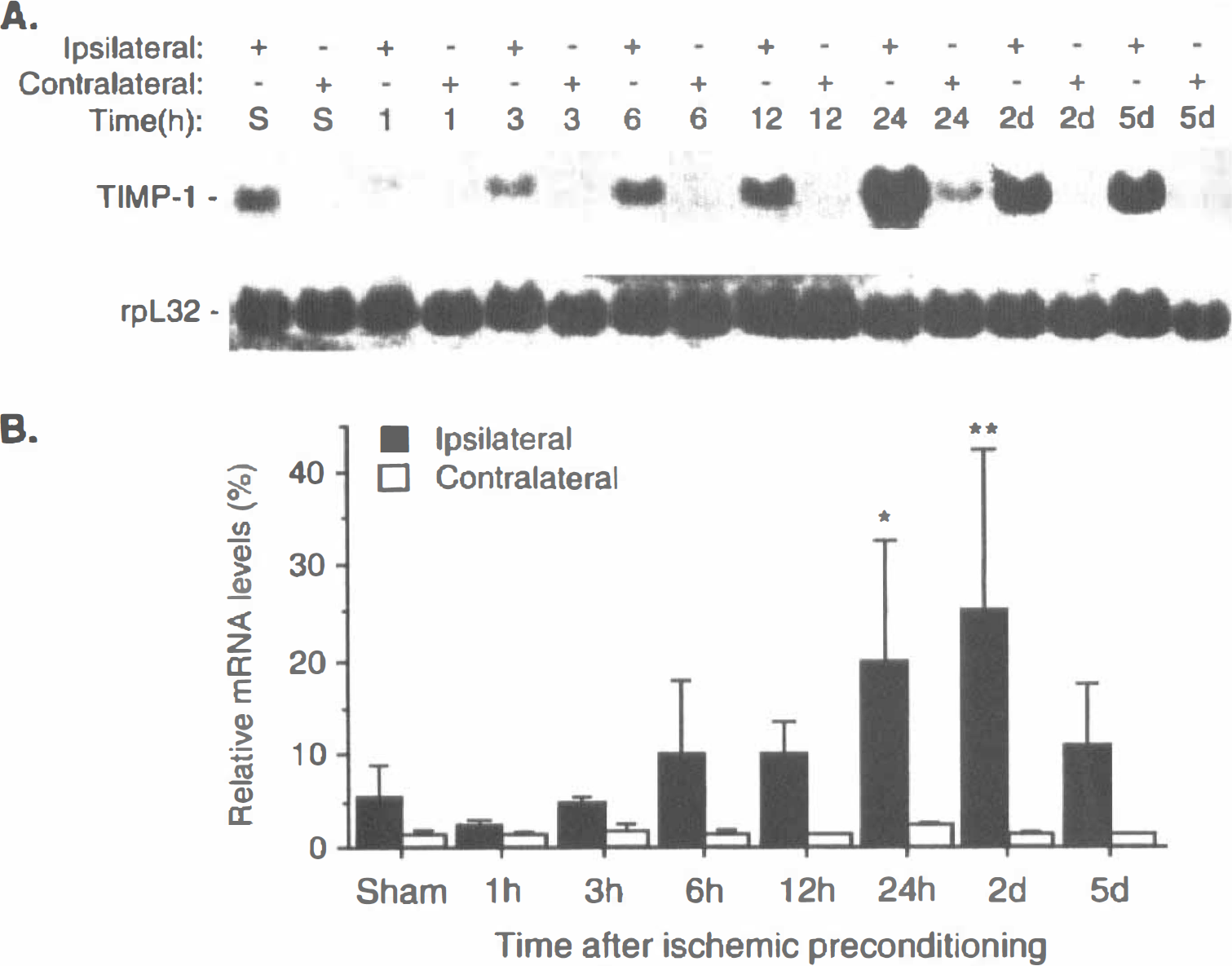
Temporal expression of TIMP-1 mRNA in cortical samples after 10 minutes' middle cerebral artery occlusion (MCAO) in rats.
DISCUSSION
The present work illustrates the application of SSH strategy for the cloning of altered gene expression after PC. It is one of the few successful applications for novel gene discoveries since the initial description of this technique (Diatchenko et al., 1996). Compared with other techniques that have been widely used for novel gene discovery, including mRNA differential display (Liang and Pardee, 1992; Wang et al., 1995) and conventional subtractive library screening (Hedrick et al., 1984; Wang et al., 1998), the major advantage of SSH strategy is its sensitivity and reproducibility for identifying differentially expressed genes. Comparatively, the mRNA differential display technique is sensitive but generates a high incidence of false positives. Although the traditional subtraction approach is reliable, it is biased to detect more abundant genes. It has been shown that the combination of both PCR amplification, especially the suppression PCR (Diatchenko et al., 1996), and cDNA subtraction followed by differential hybridization as described in the present work allowed us to identify both low (for the enrichment of suppression PCR) and high abundant messages that are differentially expressed. For example, in addition to TIMP-1, we have cloned the abundantly expressed HSP-70 gene and low abundantly expressed genes encoding for cell surface proteins in the PC-induced cortical samples using this SSH technique (our unpublished observations).
The identification of TIMP-1 expression in PC is a novel finding. Tissue inhibitor of matrix metalloproteinase-1 is a specific inhibitor for a group of zinc-dependent proteolytic enzymes known as MMP, including MMP-1, MMP-2, MMP-3, and MMP-9, but most preferably for MMP-9. Both MMP and TIMP have been widely implicated in the process of tissue remodeling under pathologic conditions such as wound healing/scar formation, angiogenesis, and cancer metastasis. Focal brain ischemia elicits a robust inflammatory reaction marked by significant leukocyte infiltration, along with disruption and reconstruction of the extracellular brain matrix (Hallenbeck et al., 1986; Clark et al., 1993; Garcia et al., 1994). The stroke-induced expression of MMP-2 (gelatinase A) and MMP-9 (gelatinase B) and their increased proteolytic activities have been demonstrated previously (Rosenberg et al., 1996; Romanic et al., 1998), and MMP-9 contributes significantly to brain injury produced by focal stroke (Romanic et al., 1998). A remarkable parallel induction of TIMP-1 mRNA with MMP-9 after focal stroke has also been demonstrated (Wang et al., 1998). The induced expression of TIMP-1 after focal stroke can play a role in inhibiting MMP-9 activity. In the present PC model, a short duration of MCAO does not by itself result in ischemic damage, but instead induces ischemic tolerance (Barone et al., 1998). The significant induction of TIMP-1 mRNA was observed at 24 and 48 hours after PC, a time when significant brain protection because of PC is observed (Barone et al., 1998), whereas the expression of MMP-9 mRNA was not detected in the PC cortex by Northern hybridization and PCR amplification (our unpublished observation). These data suggest that the induced expression of TIMP-1 by PC may contribute to the PC-induced brain protective effect (i.e., by preventing the ischemia-induced proteolytic cascade of MMP or by a mechanism unrelated to increased MMP activity). However, the level of TIMP-1 peptide expression and the exact role of the induced expression of TIMP-1 in PC remains to be explored.
In conclusion, the present study demonstrated a successful application of PCR-based SSH for discovery of altered gene expression in ischemic tolerance. The elevated expression of TIMP-1 may suggest its role in ischemic tolerance by inhibiting ischemia-induced MMP activity.
Footnotes
Acknowledgments
The authors thank Raymond White for excellent technical assistance on the animal model and Ganesh Sathe for DNA sequencing.