
Abstract
In a process called ischemic preconditioning, a brief, sublethal ischemic insult protects tissue from subsequent, more severe injury. There have been no reports of rapidly induced ischemic preconditioning. The authors sought to develop a model of cerebral ischemic preconditioning in the mouse that can be applied to transgenic and knockout animals. They found that brief middle cerebral artery (MCA) occlusion only minutes before a severe ischemic insult can induce protection from that insult. Here the investigators describe a mouse model of preconditioning using intraluminal MCA occlusion as both the conditioning and the test stimulus. One or three 5-minute episodes of ischemia given 30 minutes before MCA occlusion for 1 or 24 hours (permanent occlusion) confer significant protection as assessed by infarct volume measurements 24 hours later.
Ischemic preconditioning (IPC) occurs in several organ systems but has been classically described in the heart. Two types of IPC have been documented in the literature: early IPC, which is transient, and delayed IPC, which is sustained (Meldrum et al., 1997). Early tolerance, beginning in minutes to hours, is well documented in the myocardium (Murray et al., 1986) and recently has been shown in brain (Pérez-Pinzón et al., 1996, 1997, 1998). Delayed ischemic adaptation, requiring days to become effective, was first demonstrated in the rodent brain using a global ischemia model (Kitagawa et al., 1990; Kirino et al., 1991).
Precise mechanisms of IPC still are unknown. It has been hypothesized that the early and late windows of preconditioning may reflect changes in cellular metabolism and gene expression, respectively (Chen and Simon, 1997; Pérez-Pinzón et al., 1997). Possible mechanisms of IPC in the brain include ion channel inactivation or decreased neurotransmitter release resulting in reduced excitotoxicity, increased activity of antioxidant or anti-apoptotic enzymes, new neuroprotective gene expression, or changes in cerebral perfusion or metabolism (Chen and Simon, 1997). Adenosine, KATP channels and protein kinase C are involved in cardiac preconditioning and also have been implicated in cerebral preconditioning (Gidday et al., 1994; Yao and Gross, 1994; Heurteaux et al., 1995; Riepe et al., 1997; Pérez-Pinzón and Born, 1998).
In the brain, ischemic tolerant states have been induced by chemical (Kasischke et al., 1997; Nawashiro et al., 1997; Wiegand et al., 1997), pharmacologic (Heurteaux et al., 1995), electrical (Kobayashi et al., 1995; Matsushima et al., 1996; Taga et al., 1997), and anoxic/ischemic (Simon et al., 1993; Matsushima et al., 1995; Chen et al., 1996; Lee et al., 1997) means. In most of these states, the protection requires days to develop. Studies that used brief ischemia as the preconditioning stimulus generally observe protection with a 2- to 4-day delay, although there have been descriptions of tolerance that requires 12 to 15 days to be effective (Yanamoto et al., 1998).
In this study, we describe a rapid preconditioning model in the mouse that uses focal middle cerebral artery (MCA) occlusion as the conditioning and test stimulus. Our results are consistent with a previous report (Pérez-Pinzón et al., 1997) of an early window of preconditioning that requires only minutes to take effect in the intact brain. A mouse model of IPC provides an opportunity for the study of candidate preconditioning molecules by using homozygote and heterozygote animals with gene alterations to the preconditioned setting. Similarly, the characterization of a mouse model of IPC would allow for the discovery of novel protective factors by differential or subtractive gene analyses of normal and preconditioned brains.
MATERIALS AND METHODS
Animals
Wild-type mice (adult male C57black/6; Charles River Laboratories, Cambridge, MA, U.S.A.) weighing 18 to 25 g were given free access to food and water and housed in a climate-controlled environment.
Physiology
Animals were rapidly anesthetized with 2% halothane in a mixture of 70% nitrous oxide and 30% oxygen and then maintained on 1% halothane in the same mixture using a face mask (Fluotec-3 Vaporizer, Colonial Medical, Amherst, NH, U.S.A.) Regional CBF was measured over the MCA territory by laser Doppler flowmetry (Perimed, Stockholm, Sweden). A flexible 0.5-mm fiberoptic probe was affixed to the exposed skull over the ischemic cortex at 2 mm posterior and 6 mm lateral to bregma. Steady-state, baseline CBF was measured before MCA occlusion and was defined to be 100% flow. The CBF measurements were performed in all mice subjected to 1 hour of ischemia to monitor postischemic reperfusion. In the permanent ischemia group, CBF was measured for preliminary studies only. In a subset of animals (n = 3 in each group), femoral arterial catheters made of PE-10 tubing were placed to monitor mean arterial pressure and to sample arterial blood gases (P
Middle cerebral artery occlusion
The left MCA was occluded either permanently or for 1 hour using an intraluminal filament model as previously described in C57black/6 mice (Hara et al., 1996). Briefly, a silicon-coated 11-mm 8–0 nylon monofilament was inserted into the internal carotid artery and advanced to the origin of the anterior cerebral artery to occlude the MCA and the posterior communicating artery. In the transient ischemia groups, the filament was removed after 1 hour, and the tissue was reperfused. At the end of the experiments, the animals were returned to their cages.
Ischemic preconditioning
Mice were surgically prepared for MCA occlusion and laser Doppler flow measurements as described earlier. In both the preconditioned and nonpreconditioned groups (n = 10 each), the intraluminal filament was inserted into the internal carotid artery in a nonocclusive position. In the preconditioned groups, the filament was advanced for either 5 minutes or three cycles of 5 minutes separated by 10 minutes' reperfusion. Thirty minutes separated the preconditioning from the severe ischemia. The nonpreconditioned mice remained anesthetized for a similar duration while the filament remained in the nonocclusive position.
Histopathology
Mice were killed by decapitation 24 hours after ischemic onset, and brains were rapidly removed for staining with 2,3,5-triphenyltetrazolium chloride (TTC). Brains were sliced into five 2-mm thick coronal sections using a mouse brain matrix (RBM-200C, Activational Systems, Ann Arbor, MI, U.S.A.). These sections were immersed in 2% TTC dissolved in physiologic saline and were incubated in the dark for 25 minutes. The sections were transferred to buffered 10% formalin for fixation overnight. The fixed sections were scanned into an image analysis system (M4, St. Catherine, Ontario, Canada), and infarcts were traced at each level. Infarct volumes were computed using both a direct method and an indirect method that corrects for edema (contralateral hemisphere minus undamaged ipsilateral hemisphere).
Statistical analysis
Data are presented as mean ± SD. Statistical comparisons were made by one-way analysis of variance (infarct volumes, CBF measurements) followed by Bonferroni/Dunn post hoc test. StatView (Abacus Concepts, Inc., Berkley, CA, U.S.A.) was used for computer-assisted analysis. P < 0.05 was considered significant.
RESULTS
Physiology
Physiologic parameters post-IPC (after IPC or sham IPC) and post-ischemia (30 minutes after induction of ischemia)
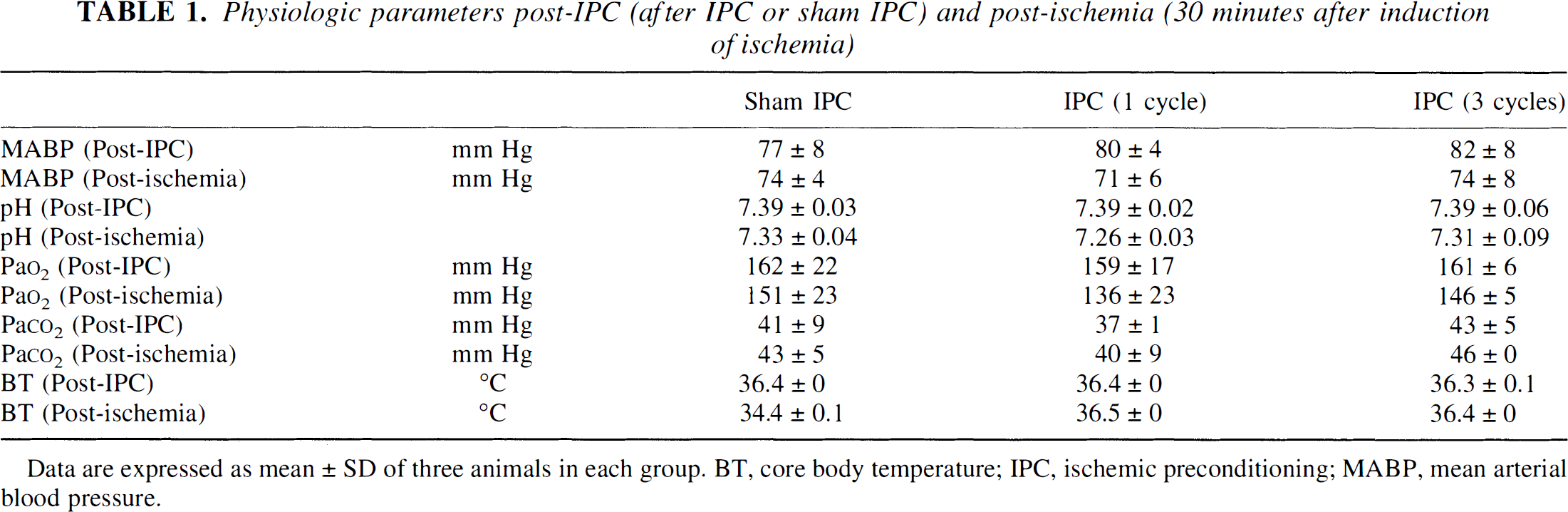
Data are expressed as mean ± SD of three animals in each group. BT, core body temperature; IPC, ischemic preconditioning; MABP, mean arterial blood pressure.
Representative regional CBF recordings over the lateral parietal cortex are provided in Fig. 1. Baseline CBF is arbitrarily defined as 100%. Severe ischemia was achieved when flow was reduced to less than 20% of baseline. A nonpreconditioned animal with 1 hour of transient ischemia is shown in Fig. 1A. During the sham preconditioning, the filament was placed in a nonocclusive position with no effect on baseline flow. In 75% of the preconditioned mice, CBF returned to normal levels after each preconditioning cycle (Fig. 1B). These mice were included in the histopathologic analyses. In the remaining 20% of animals, CBF became progressively reduced with each cycle of IPC (to less than 70% of baseline during reperfusion) as seen in Fig. 1C. These mice were excluded from the study. The flowmetry data show that with or without preconditioning, severe flow reductions (less than 20%) were achieved during the test ischemia.
Regional CBF recordings over the middle cerebral artery (MCA) core territory using laser Doppler flowmetry. The baseline blood flow before ischemia is defined as 100%. Representative recordings are shown for animals without ischemic preconditioning (IPC) 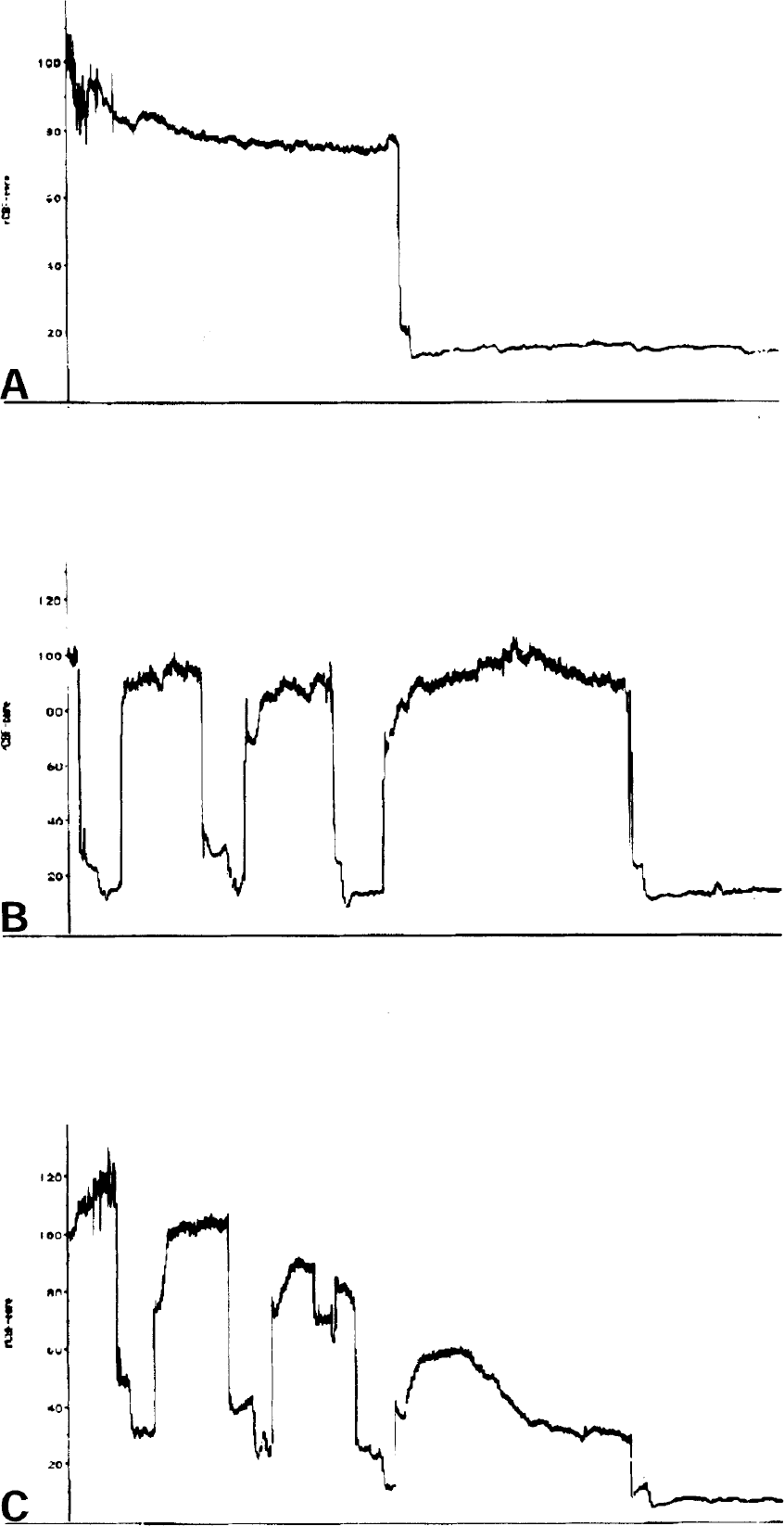
Figure 2 indicates the mean of relative CBF values at the indicated time points for the transient ischemia groups with and without preconditioning. Laser Doppler CBF was measured in all mice in these groups. There were no significant differences in regional CBF at any of the times measured.
Laser Doppler CBF values. Regional CBF values were not significantly different across groups of animals subjected to 1 hour of transient ischemia (○) No IPC, 1 cycle IPC (□), or 3 cycles IPC (⚲). Periods of ischemic preconditioning (IPC), reperfusion, (R), and ischemia (ISCH) are labeled here. Baseline before ischemia is 100%. Data are presented as mean ± SD of eight animals in each group.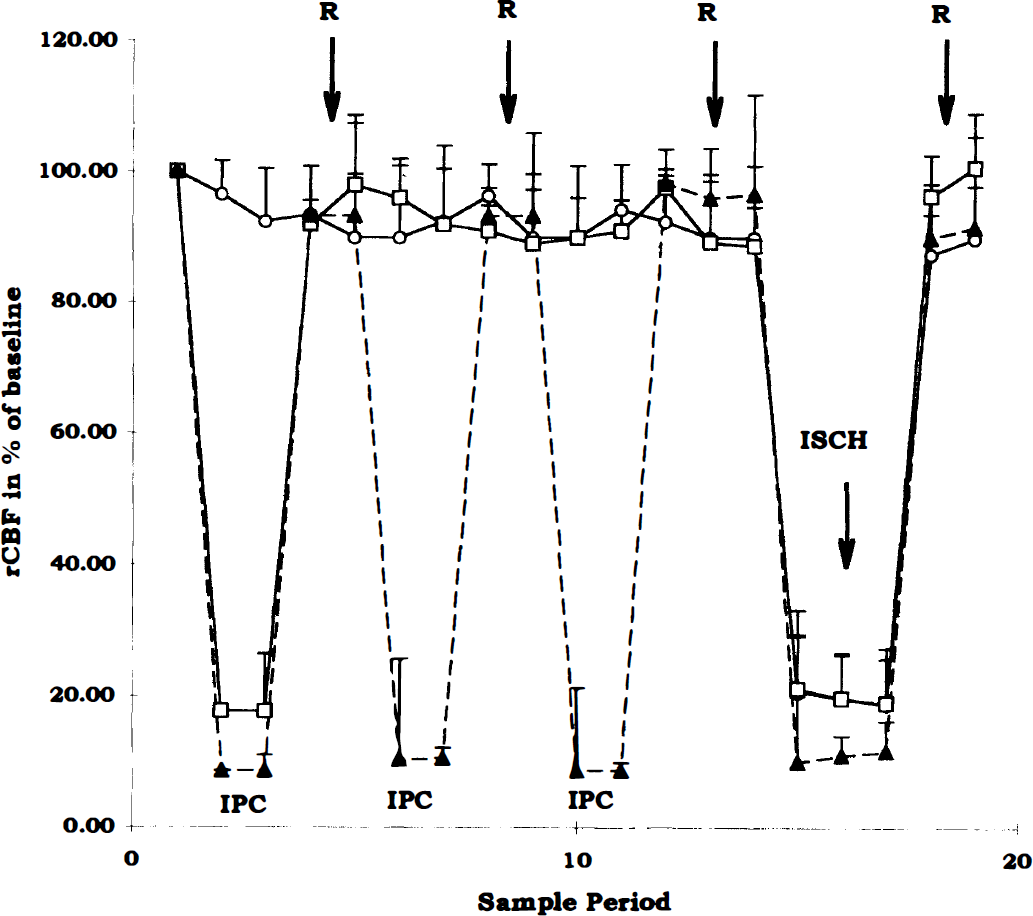
Histopathology
One or three 5-minute cycles of focal ischemia does not produce observable histopathologic damage as assessed by TTC staining at 24 hours. In a separate series of animals, 20% of mice that undergo three cycles of IPC have scattered pyknotic and TUNEL-positive neurons 3 days in hematoxylin and eosin-stained sections of the striatum (data not shown).
One and three cycles of 5-minute focal ischemia protected the brain from subsequent transient and permanent MCA occlusion. Statistically significant differences were observed in the mice preconditioned with three cycles of ischemia, and a trend existed for moderate protection in those preconditioned with one cycle (Fig. 3). Indirect infarct volume data are presented as the mean of eight animals in each group. Also, infarct volumes determined directly were significantly smaller in the mice with three cycles of IPC compared with no IPC (for permanent ischemia, 142.1 ± 38 mm3 versus 87.8 ± 19 mm3, P < 0.01; for transient ischemia, 141.5 ± 20 mm3 versus 95.3 ± 32 mm3, P < 0.01). The infarct volumes are comparable between the permanent and transient ischemia models. This may result from exacerbation of injury in the 1-hour group by the surgical time required (1.5 to 2 hours) for the monitoring of CBF in all of these animals.
Infarct volumes in mice measured by the indirect method. Infarct volumes are shown for permanent 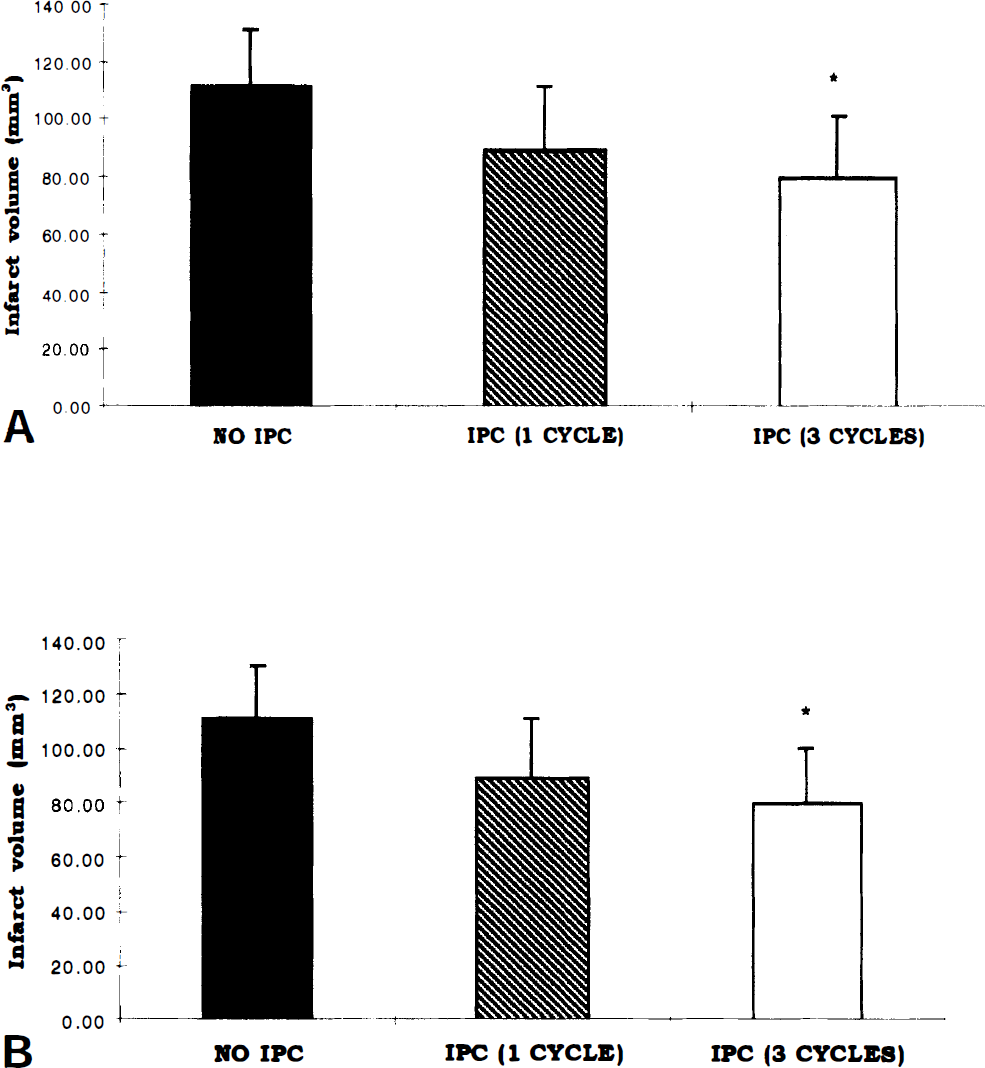
DISCUSSION
Despite recent advances in the understanding of the pathophysiologic mechanisms of experimental ischemic stroke, the available treatment options for patients are limited. The careful study of ischemic tolerance, a naturally occurring phenomenon in the brain, may provide new approaches to the treatment of cerebral ischemia. This report describes a novel model of rapid IPC in mice. To our knowledge, this is the first demonstration of rapid tolerance in mice and the first to use focal IPC in this setting. The development of a mouse model of IPC should facilitate the study of endogenous neuroprotective mechanisms in the brain using transgenic and knockout mice.
Our results indicate that brief, mild, focal MCA occlusion can produce tolerance to a subsequent, severe ischemic insult in the same vascular territory. Relative CBF measurements in the core ischemic territory do not show changes in the primary hemodynamic insult, although they do not rule out changes in detailed flow patterns in other areas such as the ischemic penumbra or the posterior circulation. A model of late preconditioning against MCA occlusion also failed to show differences in regional CBF, which could be correlated with neuroprotection (Matsushima et al., 1995). Taken together with our data, preconditioning does not appear to significantly affect the degree of vascular compromise posed by the test insult.
Protection at 24 hours is present in both permanent and transient MCA occlusion, indicating that a pathophysiologic mechanism common to both injuries is potentially targeted by IPC. Although it was unanticipated, the infarct volumes reported in these studies are similar for both ischemic settings. The transient ischemia animals were subjected to long durations of anesthesia (1.5 to 2 hours) and laser doppler flow measurements, which may have exacerbated the injury. The infarct area may be overestimated in this group because of the inclusion of both pan-necrotic tissue and areas of scattered cellular injury using TTC-stained sections. Despite these effects, preconditioning confers greater protection in the permanent ischemia setting, which may result from the greater degree of excitotoxic (rapid necrotic) injury in this model. In agreement with these observations, a series of preliminary experiments with focal IPC before intrastriatal N-methyl-
It is not known whether the protection conferred will extend to later time points. Protection was transient after rapid (Pérez-Pinzón et al., 1997) and delayed (Corbett and Crooks, 1997) global IPC. An additional consideration comes from the observation that 30 minutes of continuous ischemia (Du et al., 1996) produces late apoptotic death that is not present at 1 day. Although we have determined that IPC alone results in TUNEL-positive cells only when reperfusion between conditioning cycles is incomplete, the possibility of delayed apoptotic neuronal death resulting from the combination of mild and severe ischemia remains. These areas require future work.
The protection in this model is lost when the delay between conditioning and test stimulus is extended to 2 hours (data not shown). The precise maximum delay that retains protection is not known, but a window of less than 2 hours implies that the dominant mechanism in rapid preconditioning may not involve changes in gene expression. In a model of delayed preconditioning, with 24 hours between the conditioning and the test stimulus, our preliminary data indicate that one cycle of focal IPC protects against permanent ischemia (N.E.S, unpublished observations, 1999). It seems that this focal–focal model in mice allows for manipulation of the time delay between conditioning and test stimuli to include intervals of days. Late protective mechanisms against cerebral ischemia that require new gene expression and protein synthesis can be investigated using this time window in mice.
Several potential limitations of this model deserve mention. The intraluminal filament approach to preconditioning can be technically difficult in some mice, as demonstrated in the CBF recordings in Fig. 1C. Animals in which multiple preconditioning episodes led to a significant reduction in cerebral perfusion were discarded (two to three per group). The ischemia in these animals may result from local vasospasm, endothelial denudation, interanimal vascular variability, or a combination of these. This type of injury rarely occurs with a single episode of IPC, an advantage of the single-episode model. Although others have shown that brief, repetitive global ischemia causes edema and cumulative injury in the gerbil (Tomida et al., 1987), we did not observe tissue damage in our model when reperfusion was adequate (as assessed by laser Doppler flowmetry). Clearly, if hypoperfusion persists after even a brief ischemic episode, the effects can be deleterious.
By investigating the mechanisms of naturally occurring protection against ischemia, we may be able to identify pathways that are amenable to therapeutic intervention after stroke or that can be mimicked to prevent at-risk patients from experiencing ischemia in surgical or clinical settings.