
Abstract
Two types of ischemic tolerance in the brain, rapid and delayed, have been reported in terms of the interval between the conditioning and test insults. Although many reports showed that delayed-phase neuroprotection evoked by preconditioning is evident after 1 week or longer, there have been a few investigations about rapidly induced tolerance, and the reported neuroprotective effects become ambiguous 7 days after the insults. The authors examined whether this rapid ischemic tolerance exists after 7 days of reperfusion in a rat focal ischemic model, and investigated modulating effects of the adenosine A1 receptor antagonist DPCPX (8-cyclopentyl-1,3-dipropylxanthine). Preconditioning with 30 minutes of middle cerebral artery occlusion reduced infarct volume 7 days after 180 minutes of subsequent focal ischemia given after 1-hour reperfusion. The rapid preconditioning also improved neurologic outcome. These beneficial effects were attenuated by pretreatment of 0.1 mg/kg DPCPX, which did not influence the infarct volume after conditioning (30 minutes) or test (180 minutes) ischemia when given alone. The results show that preconditioning with a brief focal ischemia induces rapid tolerance to a subsequent severe ischemic insult, the effect of which is still present after 7 days of reperfusion, and that the rapid ischemic tolerance is possibly mediated through an adenosine A1 receptor–related mechanism.
Keywords
A mild ischemic insult may limit damage from a subsequent severe ischemic insult. This phenomenon was defined as ischemic tolerance and has been observed in several organs, especially in the heart (Yellon et al., 1998) and brain (Kitagawa et al., 1990; Chen and Simon, 1997). Depending on the latency between insults, two types of ischemic tolerance in the brain have been documented in the literature: rapid and delayed tolerance. The ischemic tolerance first reported in a global ischemia model of the brain by Kitagawa et al. (1990) is the delayed type, which becomes effective in a few days. Subsequent studies have shown delayed tolerance using global ischemia models (Heurteaux et al., 1995), rat middle cerebral artery (MCA) occlusion models (Chen et al., 1996; Barone et al., 1998; Chimon and Wong 1998; Puisieux et al., 2000; Mori et al., 2000; Shimizu et al., 2001), and other models (Chen and Simon, 1997; Kitagawa et al., 1997). The protective effects in delayed cerebral tolerance are evident 7 days or longer after severe second insults (Kitagawa et al., 1990, 1997; Heurteaux et al., 1995).
Only a few in vivo studies of rapid ischemic tolerance in the brain have been published (Pérez-Pinzón et al., 1997; Stagliano et al., 1999). Two minutes of conditioning global ischemia in rats produced protection against damage from a 10-minute test ischemic insult given 30 minutes after the conditioning (Pérez-Pinzón et al., 1997). However, the neuroprotective effects observed after 3 days of reperfusion disappeared when the reperfusion period was extended to 7 days. When the test ischemia duration was reduced to 7 minutes, a trend of neuroprotection by rapid preconditioning was observed even after 7-day reperfusion (Pérez-Pinzón et al., 1997).
Preconditioning with brief MCA occlusion in mice induced protection against the damage produced by a longer duration of MCA occlusion given after 30 minutes (Stagliano et al., 1999). The rapid tolerance was shown by a reduced infarct volume assessed only 24 hours later (Stagliano et al., 1999). Therefore, it remains to be determined whether the protective effects in the rapid tolerance continue for several days.
A recent human study showed that patients with repeated attacks of cerebral ischemia had a better neurologic outcome than patients without a previous ischemic attack, suggesting the existence of ischemic tolerance in humans (Moncayo et al., 2000). Investigating the mechanisms of such naturally occurring protection against ischemia may lead to development of new therapeutic strategies capable of modifying the outcome of ischemic episodes.
Although the precise mechanisms of ischemic tolerance are still unknown, adenosine or adenosine triphosphate-sensitive potassium (KATP) channels may be involved in development of this tolerance in the brain (Blondeau et al., 2000; Heurteaux et al, 1995; Pérez-Pinzón et al., 1996; Reshef et al., 2000). The first aim of the present study was to determine whether a conditioning insult by focal cerebral ischemia in rats can induce protection against subsequent severe insult given 1 hour later. We then examined whether neuroprotection, if any, that is rapidly induced by preconditioning is modified by an adenosine A1 receptor antagonist. We used 8-cyclopentyl-1,3-dipropylxanthine (DPCPX), a highly selective A1 antagonist (Lohse et al., 1987), dissolved in 50% dimethyl sulfoxide.
MATERIALS AND METHODS
Animals
This study was reviewed and approved by the Committee of the Ethics on Animal Experiment and carried out under the control of the Guideline for Animal Experiment in Yamaguchi University School of Medicine. Male Wistar rats (age, 8–9 weeks) were used. Animals were given free access to food and water, and were housed in a 12:12-hour light-dark cycle.
General preparation and middle cerebral artery occlusion
Rats were anesthetized with inhalation 3% isoflurane and 66% nitrous oxide. The tail artery was cannulated to monitor blood pressure, arterial blood gases, blood glucose, and hematocrit levels. After endotracheal intubation, rats were mechanically ventilated with 1.5% isoflurane and 66% nitrous oxide. Temperature probes were placed in the rectum and on the skull under the right temporalis muscle and temperatures were monitored (Mono-a-therm 6510; Mallinckrodt Japan, Tokyo, Japan). The cranial temperature was maintained at 37.0 ± 0.3 °C with external heating or cooling during surgical preparations and cerebral ischemia. Heparin (50 IU) was administered through the tail artery.
Middle cerebral artery occlusion was performed as described previously (Belayev et al., 1996). The right common carotid artery was exposed through a midline neck incision and gently dissected free of surrounding nerves and fascia from its bifurcation to the base of the skull. The occipital artery and superior thyroid artery branches of the external carotid artery were dissected and coagulated. The external carotid artery was dissected further distally and coagulated along with the terminal lingual and maxillary artery branches, which were then divided. The internal carotid artery was isolated and carefully separated from the adjacent vagus nerve, and the pterygopalatine artery was ligated close to its origin. A 5–0 silk suture was tied loosely around the mobilized external carotid artery stump. Every surgical wound was infiltrated with 0.5% bupivacaine.
After a stabilization period of at least 10 minutes using 1% (end-tidal) isoflurane and 66% nitrous oxide, a 3.5-cm length of 5–0 silicone-coated monofilament polypropylene suture (Prolene; Ethicon, Tokyo, Japan) was inserted through the proximal external carotid artery stump into the internal carotid artery, and was advanced to occlude the origin of the MCA 21 to 23 mm from the bifurcation of the common carotid artery until a slight resistance was felt. The silk suture around the external carotid artery was tightened around the intraluminal monofilament suture to prevent bleeding.
Experimental groups
The present study consisted of three experiments.
Experiment 1.
Rats were randomly assigned to sham-operated and preconditioned groups. In the preconditioned group, the MCA was occluded for 30 minutes as a conditioning insult, and the intraluminal filament was then carefully removed so as not to strip silicone coating from the filament. The common carotid artery and internal carotid artery were inspected to ensure the return of good pulsations. Physiologic variables were measured before conditioning ischemia, 15 minutes after the onset of ischemia, and 15 and 45 minutes after the onset of reperfusion.
After 60 minutes of reperfusion, a new 5–0 silicon-coated monofilament suture was inserted through the external carotid artery into the internal carotid artery and advanced to occlude MCA. After the intraluminal suture was placed, the neck incision was closed. The catheter in the tail artery was removed and the wound was closed. The animals were then allowed to recover from anesthesia and were returned to a plastic box where the oxygen concentration was maintained at 85% to 90% with high-flow oxygen.
After 180 minutes of MCA occlusion, rats were reanesthetized, the wound was reopened, and the intraluminal suture was removed. Good pulsation of the common and internal carotid arteries was ensured, and the neck incision was closed. The thermister placed on the skull was removed and the rats were returned to the cage. For 30 minutes of postischemic reperfusion, a high concentration of oxygen was supplied and the rats were then moved to another cage in a temperature controlled (25°C) environment.
Rats in the sham-operated group underwent identical surgery but did not have conditioning ischemia. They remained anesthetized for 90 minutes after the surgical preparation and were subjected to 180 minutes of MCA occlusion.
Experiment 2.
Rats were randomly assigned to a DMSO or DPCPX-sham group, or a DPCPX-precondition group. Rats underwent the same surgical preparation described in the sham-operated group or preconditioned group of experiment 1, except for the following preischemic medications. Rats in the DPCPX-sham group received 2 mL/kg 0.1 mg/kg intraperitoneal DPCPX (a selective A1 antagonist) dissolved in 50% dimethyl sulfoxide (DMSO) for 2 minutes. In the DMSO-sham group, the same volume of 50% DMSO was administered. Ninety-five minutes after the injection, rats in both sham-operated groups were subjected to 180 minutes of MCA occlusion. In the DPCPX-precondition group, rats received 0.1 mg/kg DPCPX 5 minutes before the conditioning ischemia (30-minute MCA occlusion). After 60 minutes of reperfusion, they were subjected to 180 minutes of MCA occlusion.
In experiments 1 and 2, rats that died within 7 days after ischemic insults were excluded from further analysis and replaced in a blinded and randomized manner each group comprised 13 subjects.
Experiment 3.
This series of experiments was performed to examine the damage, if any, produced by the conditioning 30-minute ischemia and to determine whether DPCPX and DMSO influence the injury. Rats were randomly assigned to a saline-treated, dimethyl sulfoxide-treated, or DPCPX-treated groups (n = 7 in each group). In each group, rats received 0.9% saline, 50% dimethyl sulfoxide, or DPCPX 0.1 mg/kg intraperitoneally. Five minutes later, all rats received 30-minute MCA occlusion and remained anesthetized for 90 minutes of reperfusion. Animals were then allowed to recover from anesthesia and were returned to their cages.
Preliminarily, 0.5 mg/kg DPCPX was administered to five rats, and we observed a tendency toward exacerbated damage produced by 30-minute MCA occlusion. Therefore we selected the dose 0.1 mg/kg DPCPX.
Neurologic evaluation
Neurologic evaluation was performed 2, 4, and 7 days after ischemia by an investigator who was blinded to the experimental group design. The neurologic findings were scored using a four-point scale (Bederson et al., 1986): a score of 0 indicated no observable neurologic deficit, a score of 1 indicated a failure to extend left forepaw fully, a score of 2 indicated decreased resistance to lateral push without circling, and a score of 3 indicated decreased resistance to lateral push with circling.
Infarct volume measurement
Animals were anesthetized with isoflurane and killed 7 days after MCA occlusion by an intracardiac injection of thiopental. Brains were rapidly removed and sectioned coronally at 2-mm intervals, immersed in 2% 2,3,5-triphenyltetrazolim hydrochloride (TTC), dissolved in 0.9% saline, and incubated at 37 °C for 25 minutes. The sections were transferred to a 4% formaldehyde buffer for 72 to 96 hours before measurements of infarct volume were made.
The sections were analyzed for infarct size by an observer who was blinded to the experimental group design. The hemispheric infarct area in each section was calculated by subtracting the area of normal, TTC-staining brain in the ipsilateral ischemic hemisphere from the contralateral nonischemic area. This technique minimizes the effect of edema on measurement of infarct size (Swanson et al., 1990). Infarct volume was then calculated by summing the infarct area over all sections and multiplying by the slice thickness.
Histologic sections were prepared from the same surface of the slice and stained with hematoxylin and eosin for confirmation of the TTC determinations of infarct volume.
Statistical analysis
Nonparametric analyses (Mann-Whitney test and Kruskal-Wallis test) were used to determine significant differences in infarct volume and neurologic deficit scores between groups. The correlation between neurologic scores and infarct volume was tested using Spearman ordinal rank analysis. The Fisher exact test was used to compare the survival rates between groups. Differences in physiologic parameters were analyzed using repeated-measures analysis of variance. Bonferroni correction was applied for post hoc testing. P < 0.05 was considered significant. All values are expressed as mean ± SD.
RESULTS
Survival rates and physiologic variables
In experiments 1 and 2, 19 rats died after neurologic deterioration within 96 hours after ischemia (4 in the sham-operated group, 5 in the preconditioned group, 2 in the DMSO-sham group, 4 in the DPCPX-sham group, and 4 in the DPCPX-precondition group). No animals died 4 to 7 days after ischemia. There were no significant differences in mortality rate among groups. All rats in experiment 3 survived for 7 days after ischemia
There were no significant differences in body weights at the start of experiments or on the seventh day after insults among groups (mean preischemic and postischemic weights, 309 ± 25 and 245 ± 45 g, respectively).
Physiologic variables in each experiment are presented in Tables 1 to 3.
Physiological variables in sham-operated and preconditioned groups
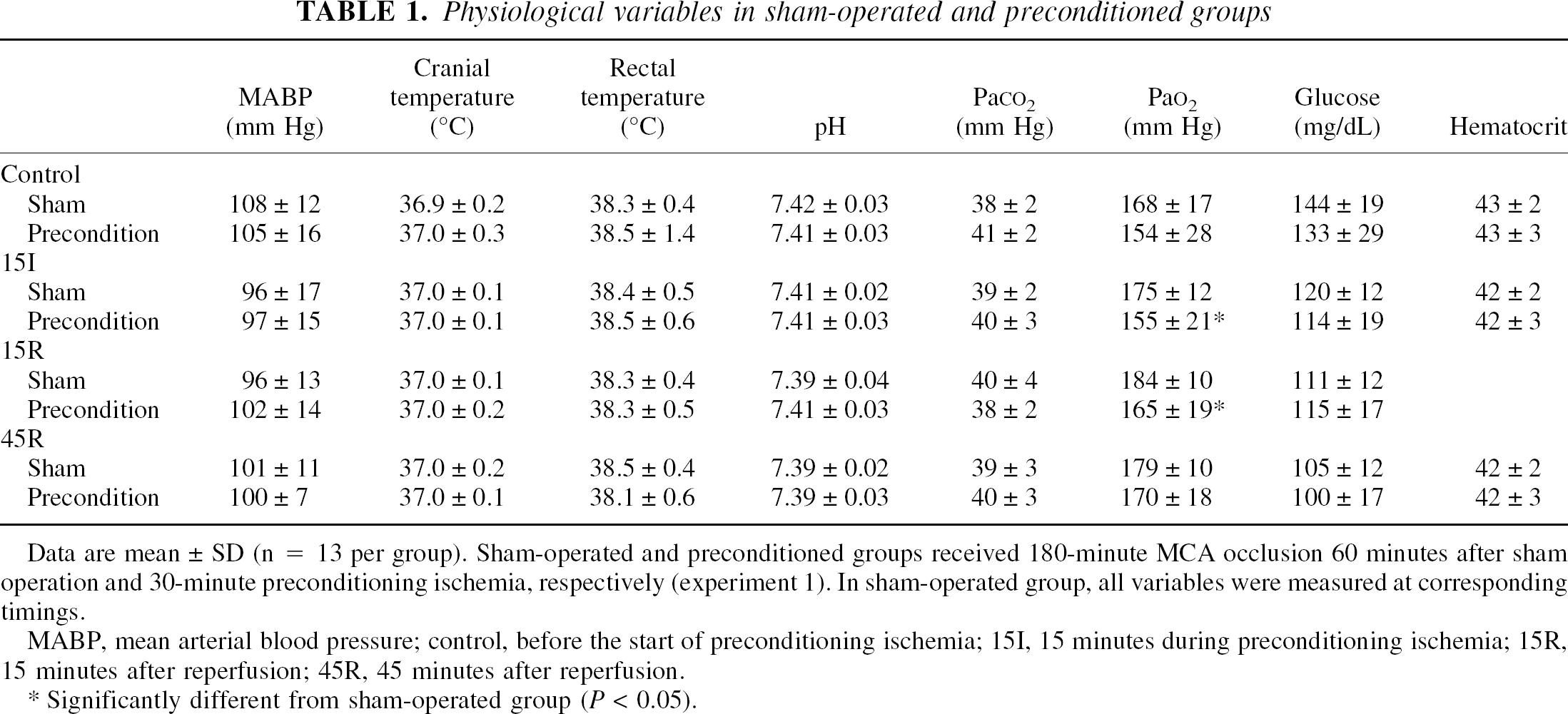
Data are mean ± SD (n = 13 per group). Sham-operated and preconditioned groups received 180-minute MCA occlusion 60 minutes after sham operation and 30-minute preconditioning ischemia, respectively (experiment 1). In sham-operated group, all variables were measured at corresponding timings.
MABP, mean arterial blood pressure; control, before the start of preconditioning ischemia; 15I, 15 minutes during preconditioning ischemia; 15R, 15 minutes after reperfusion; 45R, 45 minutes after reperfusion.
Significantly different from sham-operated group (P < 0.05).
Physiological variables in A1 antagonist- and solvent-treated groups
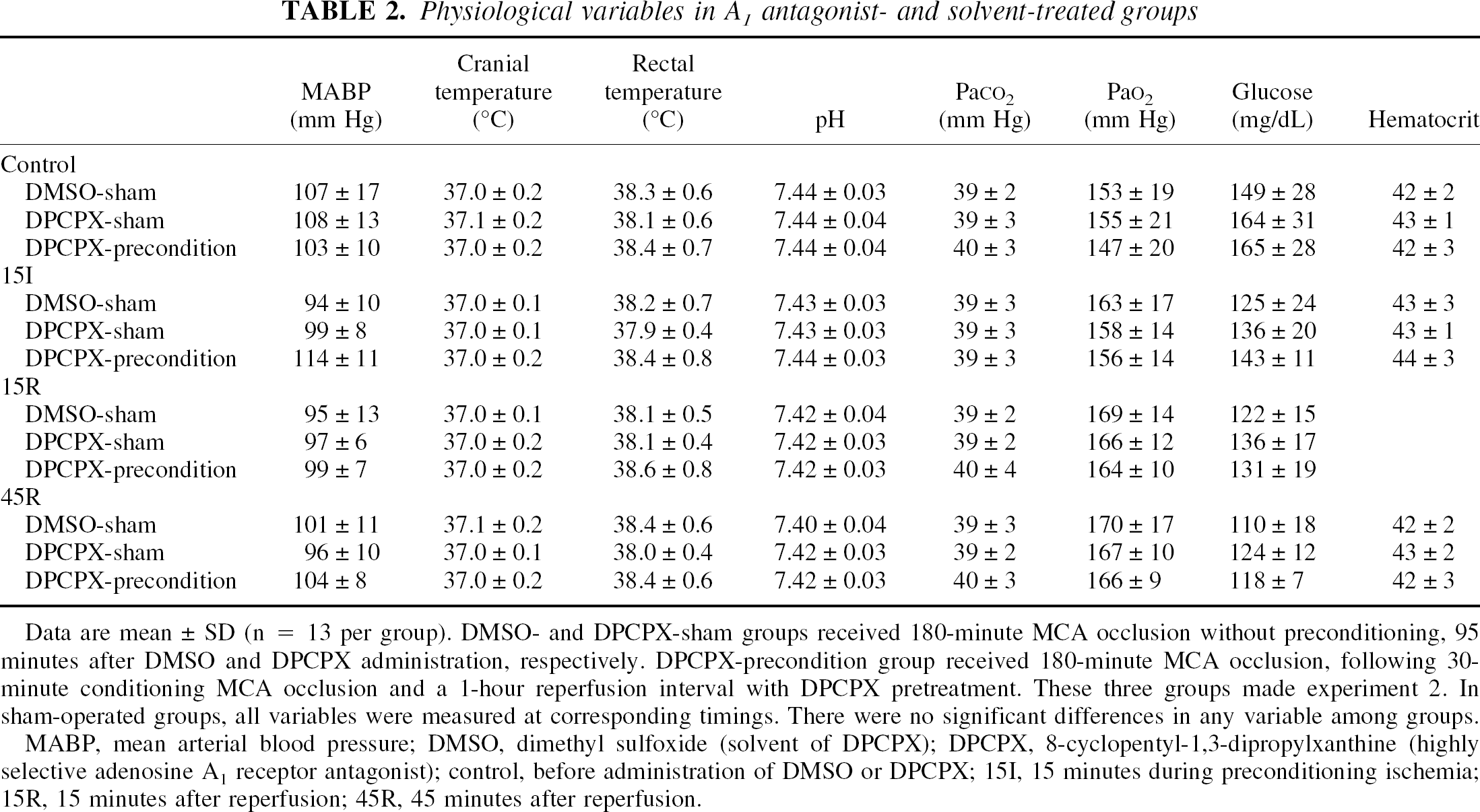
Data are mean ± SD (n = 13 per group). DMSO- and DPCPX-sham groups received 180-minute MCA occlusion without preconditioning, 95 minutes after DMSO and DPCPX administration, respectively. DPCPX-precondition group received 180-minute MCA occlusion, following 30-minute conditioning MCA occlusion and a 1-hour reperfusion interval with DPCPX pretreatment. These three groups made experiment 2. In sham-operated groups, all variables were measured at corresponding timings. There were no significant differences in any variable among groups.
MABP, mean arterial blood pressure; DMSO, dimethyl sulfoxide (solvent of DPCPX); DPCPX, 8-cyclopentyl-1,3-dipropylxanthine (highly selective adenosine A1 receptor antagonist); control, before administration of DMSO or DPCPX; 15I, 15 minutes during preconditioning ischemia; 15R, 15 minutes after reperfusion; 45R, 45 minutes after reperfusion.
Physiological variables in 30-minute ischemia groups
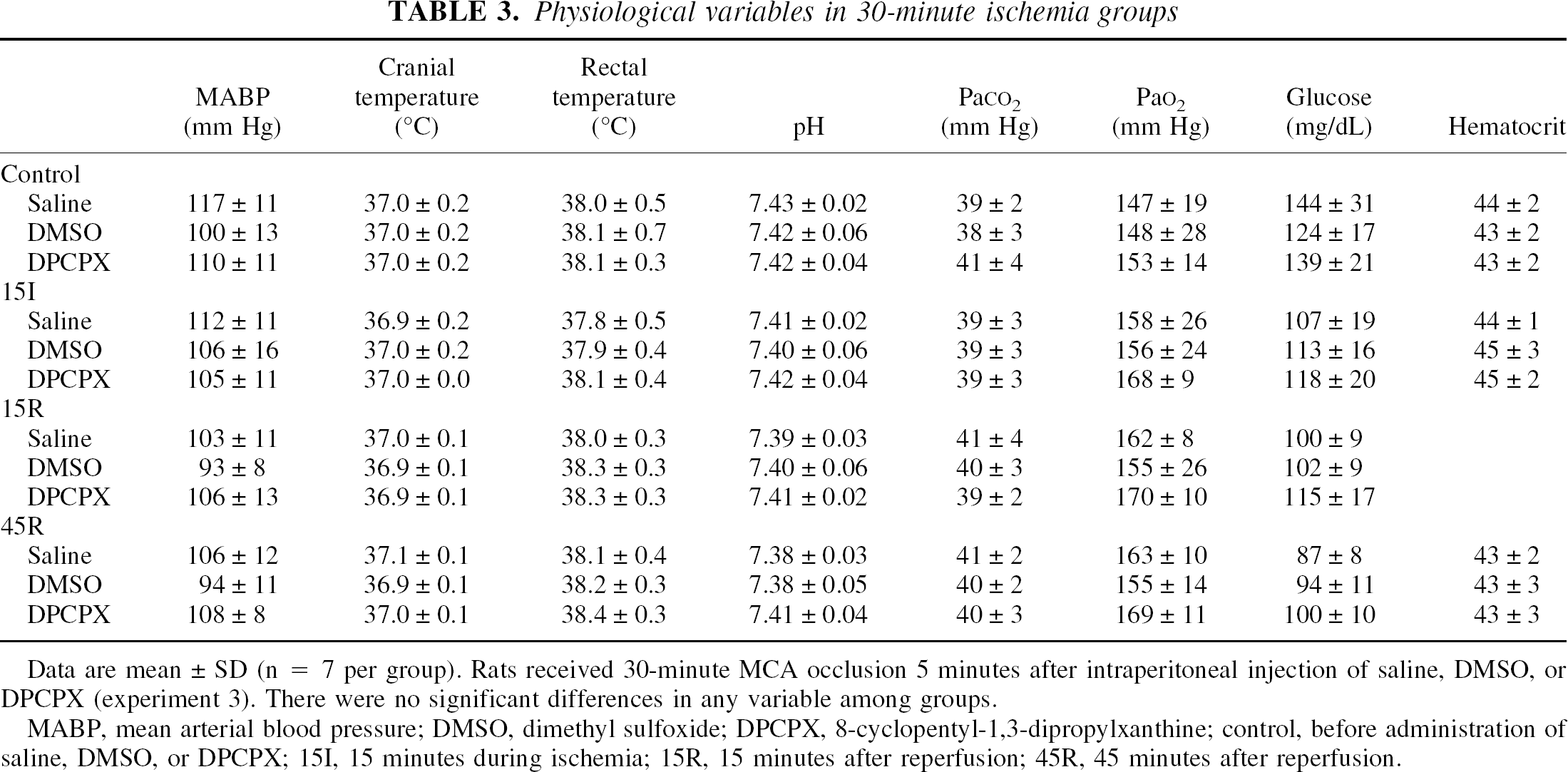
Data are mean ± SD (n = 7 per group). Rats received 30-minute MCA occlusion 5 minutes after intraperitoneal injection of saline, DMSO, or DPCPX (experiment 3). There were no significant differences in any variable among groups.
MABP, mean arterial blood pressure; DMSO, dimethyl sulfoxide; DPCPX, 8-cyclopentyl-1,3-dipropylxanthine; control, before administration of saline, DMSO, or DPCPX; 15I, 15 minutes during ischemia; 15R, 15 minutes after reperfusion; 45R, 45 minutes after reperfusion.
There were no significant differences between groups in each experiment in mean arterial blood pressure, arterial pH, arterial blood gases, hematocrit level, blood glucose level, rectal temperature, or cranial temperature. Between-group differences in arterial oxygen tension in experiment 1 were noted, though values in all rats ranged between 110 to 200 mm Hg.
Infarct volume and neurologic outcome
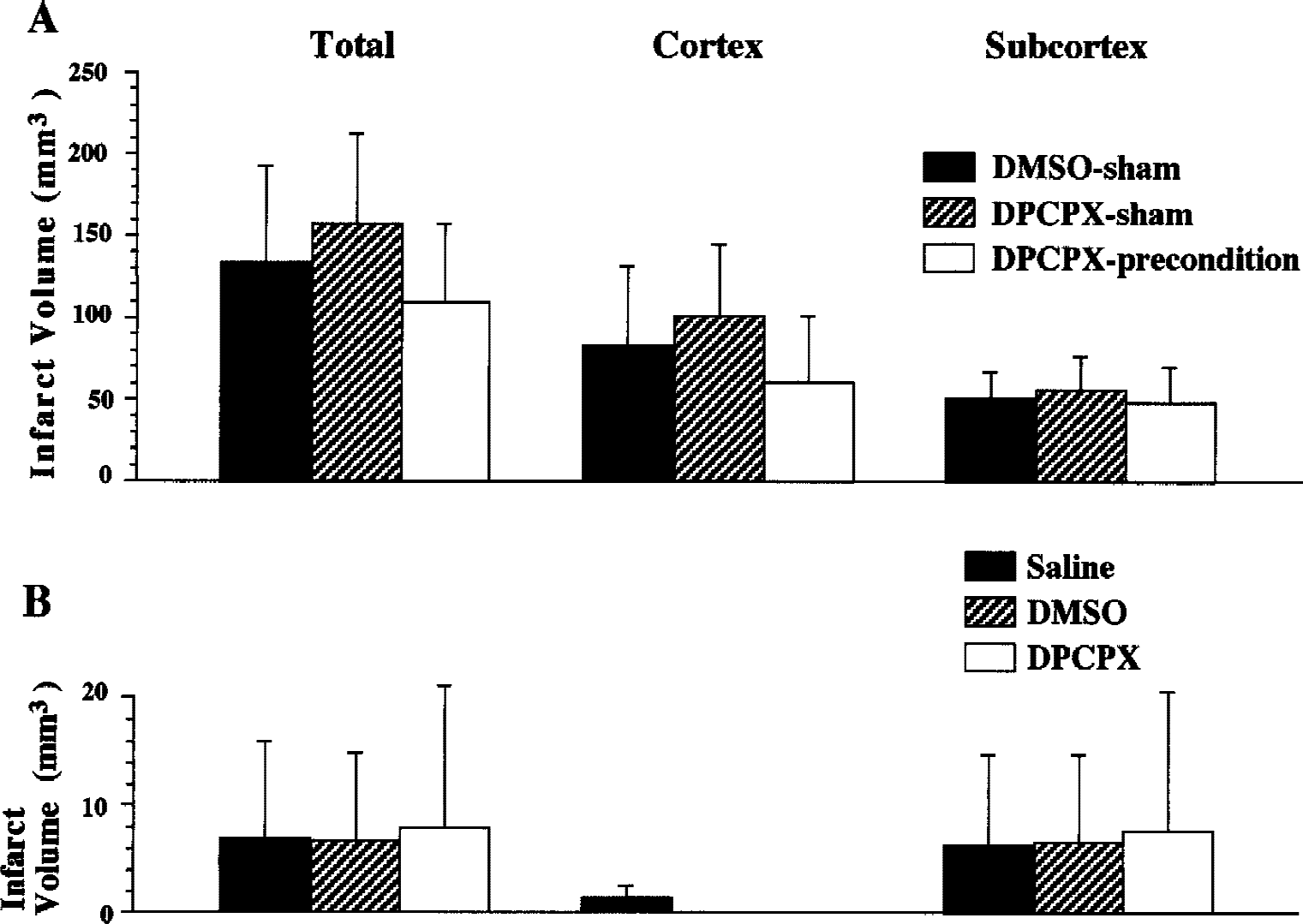
Figures 1 to 4 show infarct size, microphotographs, and neurologic outcome in each experiment.
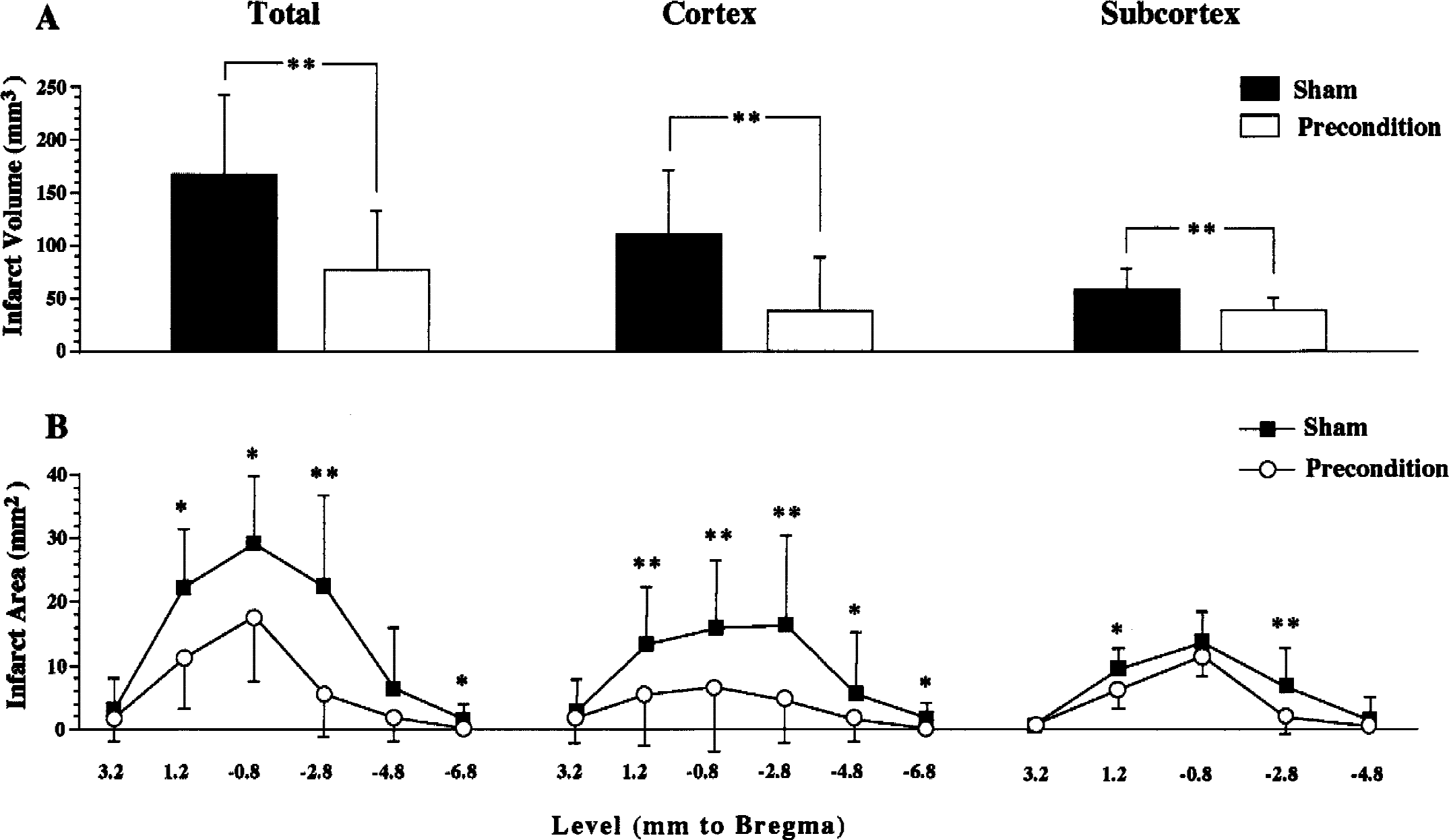
Effects of ischemic preconditioning (30-minute middle cerebral artery [MCA] occlusion) on infarct size after 180-minute MCA occlusion given at a 1-hour reperfusion interval (experiment 1).
Infarct volume after 180-minute middle cerebral artery (MCA) occlusion with or without preconditioning (
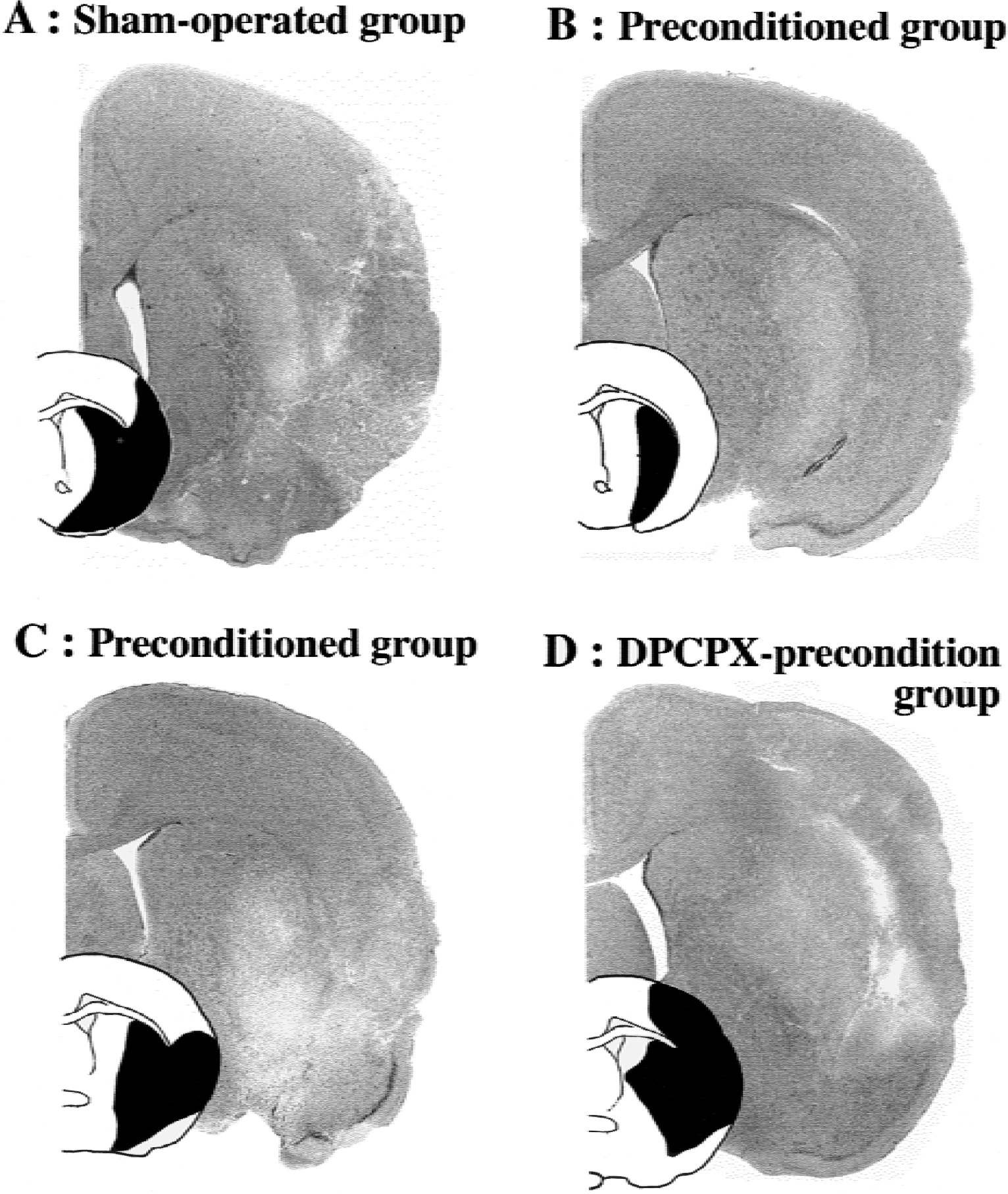
Representative microphotographs (hematoxylin and eosin staining) and schematic drawings in the corresponding section illustrating infarct area in black.
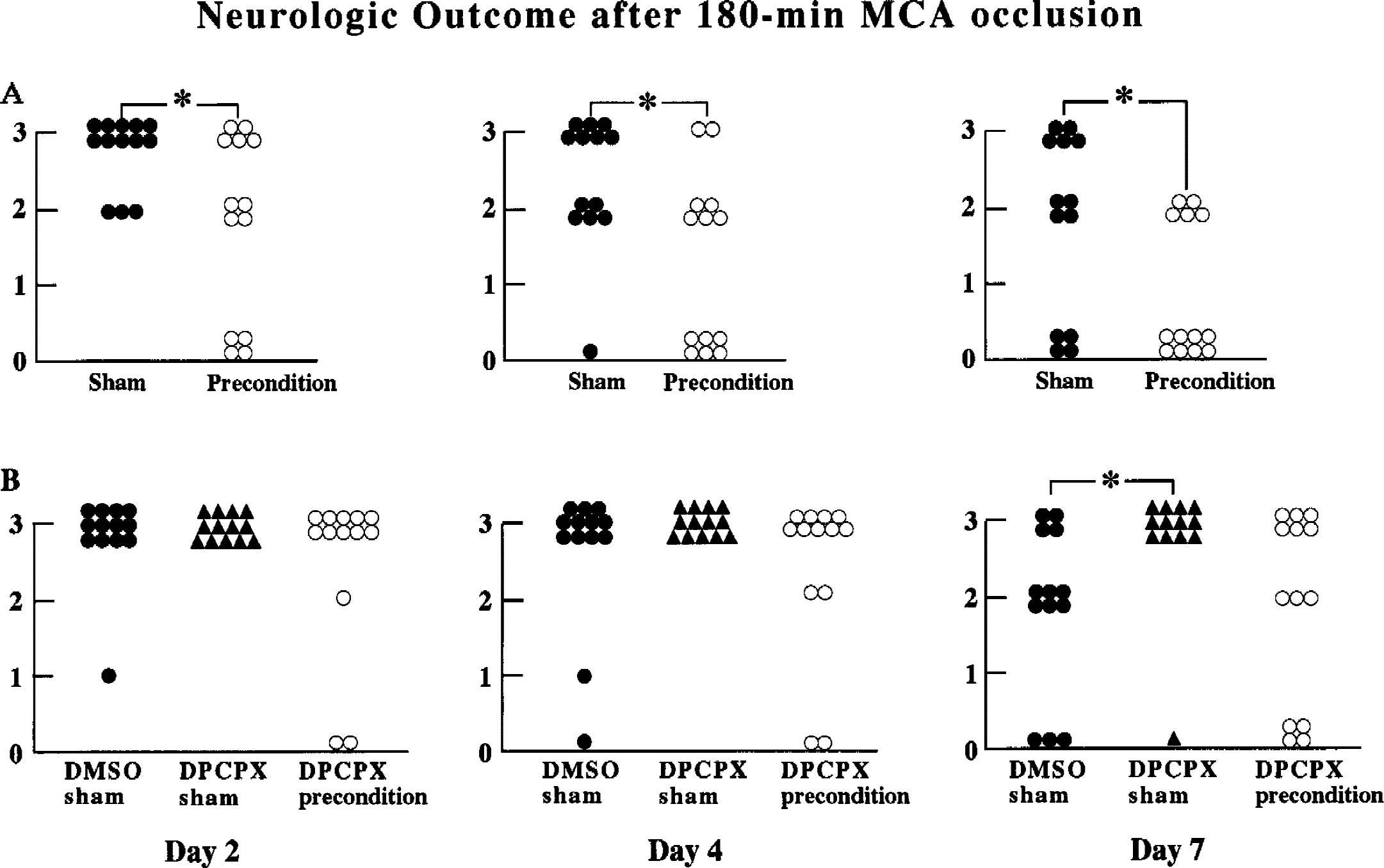
Neurologic outcome 2, 4, and 7 days after 180-minute middle cerebral artery (MCA) occlusion with or without preconditioning. A score of 0 indicates normal functioning, 1 indicates forepaw flexion, 2 indicates decreased resistance to lateral push, and 3 indicates circling.
Experiment 1.
Infarct volume assessed 7 days after 180-minute MCA occlusion was significantly smaller in rats preconditioned with 30-minute occlusion applied 1 hour previously, compared with the volume observed in the sham-operated group. The reduction of infarct volume induced by the rapid preconditioning was marked in the cerebral cortex, though the difference between preconditioned and sham-operated groups was also significant in the subcortical regions.
The preconditioning predominantly salvaged circumferential regions around ischemic core in the cortex, but the infarction in the middle caudate was not significantly decreased (Figs. 1 and 3).
With hematoxylin and eosin staining, we observed somewhat more selectively damaged neurons around the infarcted area in the preconditioned group. However, precise evaluation of the selective neuronal damage was not performed because involvement of postmortem histologic change could not be precluded in the present study.
Postischemic neurologic outcome in the preconditioned group was significantly better than that in the sham-operated group during the 7-day observation period.
There was weak but significant correlation between neurologic deficit scores measured 7 days after ischemia and infarct volume (P = 0.028; Fig. 5).
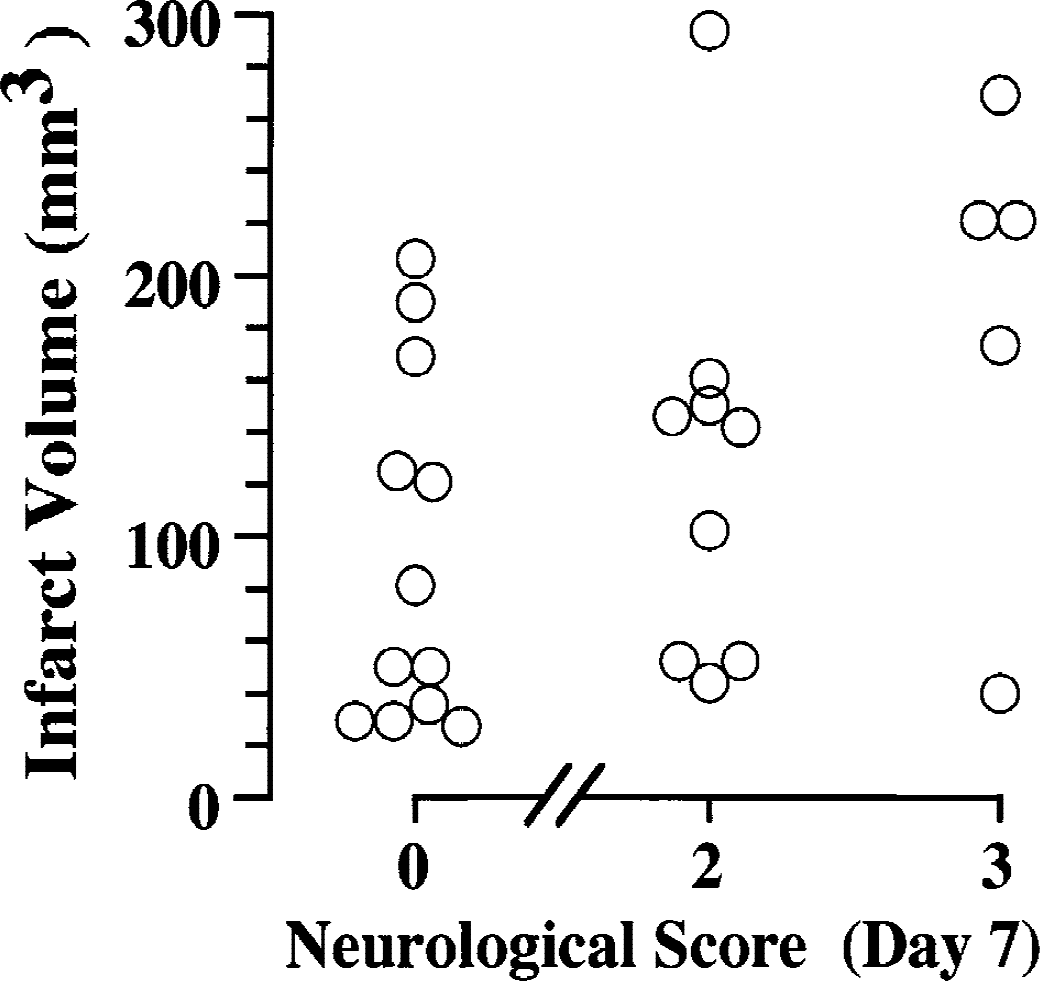
Correlation between infarct volume and neurologic outcome 7 days after 180-minute middle cerebral artery occlusion with or without preconditioning (P < 0.05, experiment 1). A neurologic score of 0 indicates normal functioning, 1 indicates forepaw flexion, 2 indicates decreased resistance to lateral push, and 3 indicates circling. No rat had a score of 1 on the seventh day in this experiment.
Experiment 2.
There were no significant differences in cortical, subcortical, or total volume of infarction among groups, indicating that DPCPX attenuated beneficial effects elicited by preconditioning observed in experiment 1.
There were no differences in neurologic outcome among groups, except that the seventh-day neurologic outcome in the DPCPX-sham group was significantly poorer than that in DMSO-sham group.
Experiment 3.
No apparent infarct was observed in three or four of seven rats in each group. Seven days after the insults (30-minute MCA occlusion), no neurologic deficits were observed in four, six, and six rats in the saline-treated, DMSO-treated, and DPCPX-treated group, respectively.
There were no significant differences in infarct volume or neurologic outcome among groups.
DISCUSSION
The results of the present study show that a brief period of focal cerebral ischemia in rats produces rapid tolerance to subsequent ischemic insults in the same vascular territory, and that the beneficial effect of ischemic preconditioning is attenuated by a selective adenosine A1 receptor antagonist, DPCPX. The selective adenosine A1 receptor antagonist at the dose studied had no influence on the infarct size after the conditioning insult or the severe ischemic insult given separately. These results indicate that the rapid ischemic tolerance is mediated, at least in part, through adenosine A1 receptor–related mechanisms.
Although DMSO has been reported to influence outcome from cerebral ischemia (de la Torre, 1983), the dose of DMSO used as a solvent in this study showed no apparent influence on infarct volume after 30-minute or 180-minute MCA occlusion.
We measured infarct volume 7 days after ischemia using TTC staining and confirmed this measurement by hematoxylin and eosin staining of the same slice. Although it has been reported that macrophages infiltrated into chronically infarcted area can be stained red with TTC (Liszczak et al., 1984), only a little discoloration in the white area that was considered to be indicative of infarction did not complicate the delineation of the infarct area in the present study. Similarly to our study, other investigators applied TTC staining to evaluate injury volume 7 days after insults and reported the validity (Sager et al., 2000; Schäbitz et al., 1996) and correlation with damages (Assaf et al., 1997).
Neuroprotection induced by preconditioning is dependent on the intensity of the conditioning stimulus and the interval between insults. It has been reported in rats that the duration of MCA occlusion as a precondition that best induces tolerance in a delayed phase is 30 minutes, which can be delivered as a single insult or during three separate 10-minutes periods (Chen et al., 1996). Concerning intervals, we preliminarily observed a tendency toward exacerbation of damage at an interval of 30 minutes between insults, and variable results at a 45-minute interval. From these previous reports and from our observations, we selected the 30-minute MCA occlusion duration of conditioning ischemia and a reperfusion interval of 60 minutes. We used a single insult as a conditioning ischemia because multiple ischemic episodes might lead to significant reduction in cerebral blood flow during reperfusion (Stagliano et al., 1999).
In general, ischemic tolerance was evident in the brain when the interval between conditioning and test insults was prolonged to a few days (Chen and Simon, 1997). Using MCA occlusion models in rats, several studies (Chen et al., 1996; Barone et al., 1998; Chimon and Wong 1998; Puisieux et al., 2000; Mori et al., 2000; Shimizu et al., 2001) reported that 24-hour to 72-hour latency between insults was necessary to develop tolerance, whereas a shorter interval (2–24 hours) was insufficient. However, none has examined preconditioning effects when insults given at intervals shorter than 2 hours in the rat models.
In contrast to the present results, repeated sublethal global ischemia has been reported to result in extensive neuronal damage, which is most severe when such ischemia is repeated at 1-hour intervals in rats and gerbils (Nakano et al., 1989; Kato et al., 1991; Inoue et al., 1992). Repeated focal cerebral ischemia induced by unilateral carotid artery occlusion when given at 1-hour intervals also produced cumulative damage in gerbils (Kato et al., 1992). Why the same 1-hour interval between insults was adequate for the development of rapid tolerance in the current study is not readily explained.
However, a few in vivo studies have shown rapid tolerance with a short interischemic latency. Stagliano et al. (1999) reported in mice that three 5-minute cycles of MCA occlusion separated by 10-minute reperfusion produced tolerance to subsequent 1-hour or 24-hour period of MCA occlusion given after 30 minutes of reperfusion. The neuroprotective effects of preconditioning were observed at 24 hours after test ischemia. Pérez-Pinzón et al. (1997) also reported rapid induction of ischemic tolerance using a global cerebral ischemia model in rats. A 2-minute conditioning forebrain ischemia produced protection against 10-minute test ischemia given 30 minutes later. However, the neuroprotection observed after 3 days of reperfusion almost disappeared at 7 days after ischemia (Pérez-Pinzón et al., 1997). To our knowledge, the present study is the first to show that neuroprotective effects rapidly induced by focal ischemic preconditioning continue for several days, and is also the first in vivo report indicating the involvement of an adenosine A1 receptor in the rapid cerebral tolerance, though the role of adenosine has been suggested in in vitro studies and in delayed brain tolerance.
Consistent with our results, short-intervals (30-minute to 2-hour) preconditioning-induced neuronal protection has been reported in in vitro studies using brain slices, (Pérez-Pinzón et al., 1996; Schurr et al., 1986), and the involvement of adenosine is indicated (Pérez-Pinzón et al., 1996). Preconditioning with short anoxic insult improved evoked potential recovery from subsequent severe anoxic insult given after 30 minutes of reoxygenation in rat hippocampal slices (Pérez-Pinzón et al., 1996). Transient superfusion with DPCPX during the conditioning anoxia lessened the improvement of evoked potential recovery (Pérez-Pinzón et al., 1996).
The role of adenosine A1 receptors was also supported by the finding that DPCPX pretreatment abolished a delayed ischemic tolerance in the rat model of global ischemia (Heurteaux et al., 1995). The delayed tolerance induced by ischemic preconditioning was also blocked by glibenclamide, a specific blocker of KATP channels (Heurteaux et al., 1995). In addition, glibenclamide inhibited neuroprotection produced by an adenosine A1 receptor agonist and an opener of KATP channels (Heurteaux et al., 1995). Furthermore, the opening of KATP channels is suggested to be mandatory for the acquisition of rapid and delayed ischemic tolerance by adenosine in neuronal cultures (Reshef et al., 2000).
Adenosine is massively produced during cerebral ischemia and presumably activates KATP channels through adenosine A1 receptor activation (Bischofberger et al., 1997; von Lubitz, 1999; Wan et al., 1999). Activation of KATP channels has been reported to block neurotransmitter release and glutamate excitotoxicity in hippocampal neurons (Abele and Miller, 1990; Lauritzen et al., 1997; Schmidt-Antomarchi et al., 1990), and to provoke neuroprotection against ischemia (Blondeau et al., 2000; Heurteaux et al., 1993, 1995; Reshef et al., 2000). Therefore, the rapid tolerance observed in the present study is likely to be attributed to the liberation of adenosine during conditioning ischemia and to the activation of KATP channels by adenosine A1 receptors, but the involvement of other mechanisms can not be excluded.
Recently, it was reported that rapid preconditioning prevented microglial activation after global cerebral ischemia in rats (Pérez-Pinzón et al., 1999), and that adenosine can block harmful effects, such as the release of tumor necrosis factor from microglia (Schubert et al., 1997). Taken together, the inhibitory effects of adenosine on microglial activation might be involved in development of rapid tolerance.
Although cerebral blood flow was not measured in the current study, no differences in blood flow were found in rapid tolerance to MCA occlusion in mice (Stagliano et al., 1999). Similarly, in a delayed phase of tolerance, there were no differences in cerebral blood flow in the tolerant regions between sham-operated and preconditioned animals before and during test focal ischemia (Chen et al., 1996; Barone et al., 1998).
In summary, the present study shows that rapid tolerance to focal cerebral ischemia can be induced and is possibly mediated by the activation of adenosine A1 receptors. This finding may help to develop new effective approaches to cerebral protection against ischemic injury.