
Abstract
We investigated the combined effect of increased brain topical K+ concentration and reduction of the nitric oxide(NO.) level caused by nitric oxide scavenging or nitric oxide synthase (NOS) inhibition on regional cerebral blood flow and subarachnoid direct current (DC) potential. Using thiopental-anesthetized male Wistar rats with a closed cranial window preparation, brain topical superfusion of a combination of the NO. scavenger hemoglobin(Hb; 2 mmol/L) and increased K+ concentration in the artificial cerebrospinal fluid ([K+]ACSF) at 35 mmol/L led to sudden spontaneous transient ischemic events with a decrease of CBF to 14 ± 7% (n = 4) compared with the baseline (100%). The ischemic events lasted for 53 ± 17 minutes and were associated with a negative subarachnoid DC shift of −7.3 ± 0.6 mV of 49 ± 12 minutes' duration. The combination of the NOS inhibitor N-nitro-L-arginine(L-NA, 1 mmol/L) with [K+]ACSF at 35 mmol/L caused similar spontaneous transient ischemic events in 13 rats. When cortical spreading depression was induced by KCl at a 5-mm distance, a typical cortical spreading hyperemia (CSH) and negative DC shift were measured at the closed cranial window during brain topical superfusion with either physiologic artificial CSF (n = 5), or artificial CSF containing increased [K+]ACSF at 20 mmol/L (n = 4), [K+]ACSF at 3 mmol/L combined with L-NA (n = 10), [K+]ACSF at 10 mmol/L combined with L-NA (five of six animals) or [K+]ACSF at 3 mmol/L combined with Hb (three of four animals). Cortical spreading depression induced long-lasting transient ischemia instead of CSH, when brain was superfused with either[K+]ACSF at 20 mmol/L combined with Hb (CBF decrease to 20± 20% duration 25 ± 21 minutes, n = 4), or [K+]ACSF at 20 mmol/L combined with L-NA (n = 19). Transient ischemia induced by NOS inhibition and [K+]ACSF at 20 mmol/L propagated at a speed of 3.4 ± 0.6 mm/min, indicating cortical spreading ischemia (CSI). Although CSH did not change oxygen free radical production, as measured on-line by in vivo lucigenin-enhanced chemiluminescence, CSI resulted in the typical radical production pattern of ischemia and reperfusion suggestive of brain damage (n = 4). Nimodipine (2 μg/kg body weight/min intravenously) transformed CSI back to CSH (n = 4). Vehicle had no effect on CSI(n = 4). Our data suggest that the combination of decreased NO. levels and increased subarachnoid K+ levels induces spreading depression with acute ischemic CBF response. Thus, a disturbed coupling of metabolism and CBF can cause ischemia. We speculate that CSI may be related to delayed ischemic deficits after subarachnoid hemorrhage, a clinical condition in which the release of Hb and K+ from erythrocytes creates a microenvironment similar to the one investigated here.
Keywords
Cortical spreading depression (CSD) is a depolarization wave that moves in the cerebral cortex at a speed of 2 to 5 mm/min. During CSD, a large increase of the extracellular K+ concentration ([K+]o) occurs (Kraig and Nicholson, 1978; Hansen et al., 1980). The ATP-dependent Na+-K+ pump is activated to return the extracellular K+ into the cells. To meet the increased energy demand from the activated metabolism and to clear the extracellular space from toxic products, cerebral blood flow increases during CSD (Hansen and Lauritzen, 1984). The mechanism of this CSD-induced cortical spreading hyperemia(CSH) is unknown.
Several investigators have studied the role of nitric oxide(NO.) as a possible mediator of CSH (Duckrow, 1993; Fabricius et al., 1995; Zhang et al., 1994; Wolf et al., 1996; Goadsby et al., 1992; Wahl et al., 1994; Colonna et al., 1994; 1997; Meng et al., 1995). NO. is produced by nitric oxide synthases (NOS). Increased activity of constitutively expressed nitric oxide synthase during CSD is likely because intracellular calcium is elevated. Intracellular calcium stimulates nitric oxide synthase. In the above-mentioned studies, NOS inhibitors were used to lower NO. levels. The results were controversial. In our current study, we have investigated the CBF response to CSD in the barbiturate-anesthetized rat with brain topical superfusion of two different NO.-lowering agents: the NOS inhibitor N-nitro-L-arginine (L-NA) and the NO. scavenger hemoglobin (Hb). We found that both the NOS inhibitor and the NO. scavenger reduced CSH by 34% (P < 0.05, n = 10) and 46% (P < 0.1, n = 4), respectively. When either NO.-lowering agent was combined with an increased K+ concentration in the artificial cerebrospinal fluid ([K+]ACSF), we discovered that CSH was transformed into cortical spreading ischemia (CSI). Increased [K+]ACSF without NO.-lowering agent did not alter spreading hyperemia. Cortical spreading ischemia is a newly described phenomenon in which ischemia is the consequence of neuronal/glial depolarization. We speculate that CSI is involved in the delayed ischemic deficits (DID) after subarachnoid hemorrhage (SAH) in which the release of Hb and K+ from erythrocytes produces a microenvironment similar to the one in our experiments.
METHODS
General
Male Wistar rats (n= 77; 250 to 300 g) were anesthetized with 100 mg/kg thiopental-sodium (Trapanal, BYK Pharmaceuticals, Konstanz, Germany), tracheotomized, and artificially ventilated(Effenberger Rodent Respirator, Effenberger Med-Techn. Gerätebau, Pfaffing/Attel, Germany). The left femoral artery and vein were cannulated, and a continuous intravenous saline solution (1 mL/h) was infused. Body temperature was maintained at 38°± 0.5°C using a heating pad. Systemic arterial pressure (RFT Biomonitor, Germany) and end-expiratory Pco2 (Heyer CO2 Monitor EGM I, Bad Ems, Germany) were monitored. A Compact 1 blood gas analyzer (AVL Medizintechnik GmbH, Bad Homburg, Germany) was used to serially measure PaO2, PaCO2, and pH. Anesthesia was monitored by blood pressure control and tail pinching.
A craniotomy was performed using a saline-cooled drill, the dura mater was removed, and a closed parietal cranial window was implanted (for details see Lindauer et al., 1993). The cortical surface under the window was continuously superfused with artificial cerebrospinal fluid (ACSF). The millimolar composition of the ACSF was as follows: Ca2+ 1.5; Mg2+ 1.2; HCO3− 24.5; Cl− 135; glucose 3.7; and urea 6.7. The K+ concentrations (3, 10, 20, and 35 mmol/L) determined the Na+ concentrations (152, 145, 135, and 120 mmol/L). The ACSF was equilibrated with a gas mixture containing 6.6% O2, 5.9% CO2, and 87.5% N2 leading to a Po2 of 129.1 ± 12.0 mm Hg, a Pco2 of 33.4 ± 4.6 mm Hg, and pH of 7.38 ± 0.04. The CBF was monitored continuously by a laser-Doppler flow probe (Vasamedics BPM 403 A, Troy, MI, U.S.A.) with a spatial resolution of 1 mm3 and a temporal resolution of 0.1 seconds (Dirnagl et al., 1989). The direct current (DC) potential was measured at the closed parietal window by a silver-silverchloride wire with agar bridge inserted into the space between the cortex and the coverslip. The wire was connected to an electrometer (FD 223, WPI, Sarasota, FL, U.S.A.) and an amplifier (Jens Meyer, Munich, Germany). In five animals of series II, an open cranial window was implanted frontally to monitor CBF (laser-Doppler) and DC potential (kalomel electrode). In the animals of series III through X, topical KCl (150 mmol/L) induced CSD at an open temporooccipital window at a 5-mm distance from the closed parietal window. Systemic arterial pressure, CBF, DC shift, and end-expiratory Pco2 were recorded continuously using a personal computer running the ASYST (Macmillan Software, New York, NY, U.S.A.) data acquisition software and a chart recorder (DASH IV, Astro-Med, Inc., West Warwick, RI, U.S.A.).
Hemoglobin was freshly prepared from heparinized arterial blood of Wistar rats. The blood was centrifuged (2500 g, 5 minutes, 4°C) and the plasma was discarded. The cells were washed five times with three to four volumes of cold 0.9% NaCl, and the buffy coat was removed. Cells were lysed by sonication. The suspension of lysed cells was subjected to centrifugation(15000 g, 10 minutes, 4°C) and the pellet removed. The Hb-containing supernatant was transferred by gel chromatography (Bio-Gel P-6, Bio Rad, Richmond, VA, U.S.A.) to the ACSF. Concentration and composition of Hb were measured by a radiometer(ABL system 625, Radiometer A/S, Copenhagen, Denmark) (total Hb 2.0 ± 0.3 mmol/L; oxy-Hb 95.2 ± 1.9%; CO-Hb 2.3 ± 1.3%; met-Hb 2.5 ± 2.0%; deoxy-Hb 0.0 ± 0.0%; O2-sat 100%).
Experimental design
In series I, we started with superfusion of Hb (2 mmol/L) and physiologic [K+]ACSF at 3 mmol/L for 30 minutes followed by superfusion of Hb and increased [K+]ACSF at 35 mmol/L (Fig. 1). In series II, a similar protocol was used, but Hb was replaced by L-NA (1 mmol/L, Sigma Chemicals, Deisenhofen, Germany). Equilibration time for L-NA was 120 minutes before [K+]ACSF was increased to 35 mmol/L (Fig. 2). The CBF and DC changes were measured at the parietal window.
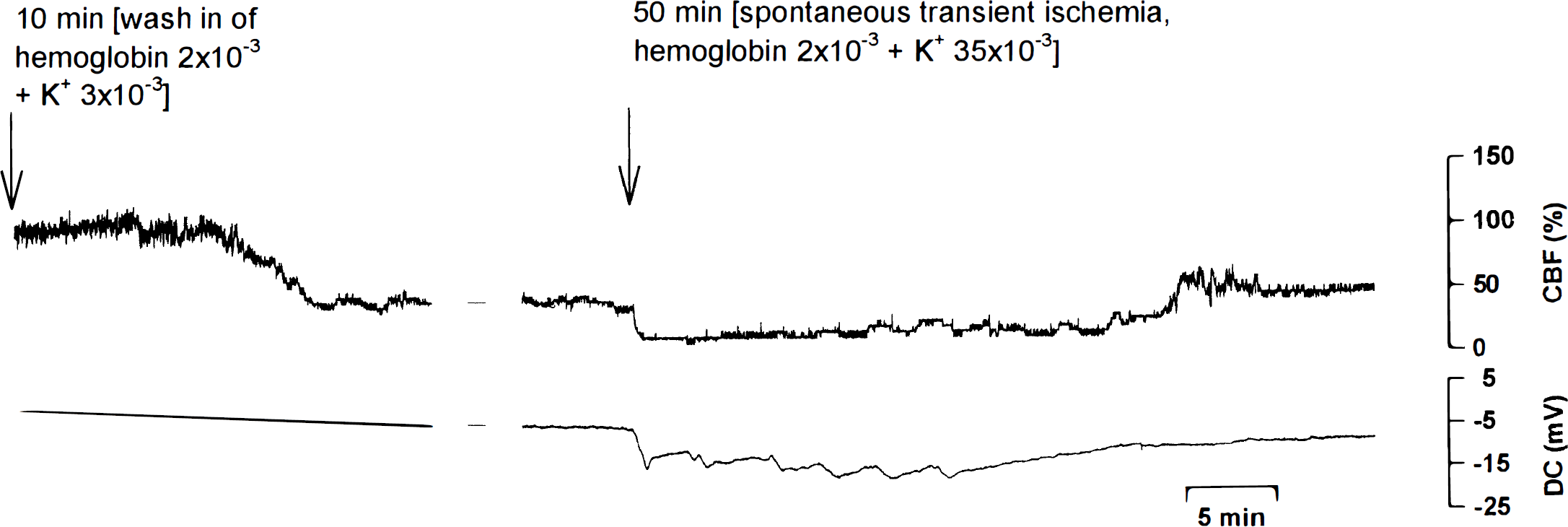
A typical experiment of series I is shown. Hemoglobin (2 mmol/L) and extracellular potassium concentration([K+]ACSF; 3 mmol/L) decreased CBF. When [K+]ACSF was increased to 35 mmol/L during continued superfusion with hemoglobin, a spontaneous transient ischemia occurred.
A typical experiment of series II is shown. Combined superfusion of the nitric oxide synthase(NOS) inhibitor N-nitro-L-arginine (L-NA; 1 mmol/L) and [K+]ACSF(20 mmol/L) induced spontaneous transient ischemic events at the closed parietal window. Simultaneous recordings of CBF and direct current (DC) potential revealed that the transient ischemic events induced one or more cortical spreading hyperemic events at an open frontal window under physiologic conditions.
In series III, KCl elicited CSD initially (control conditions), at 50 minutes (during brain topical Hb and [K+]ACSF at 3 mmol/L), and also at 80 minutes(during Hb and [K+]ACSF at 20 mmol/L). Finally, cardiac arrest by intravenous KCl was recorded (Fig. 3).
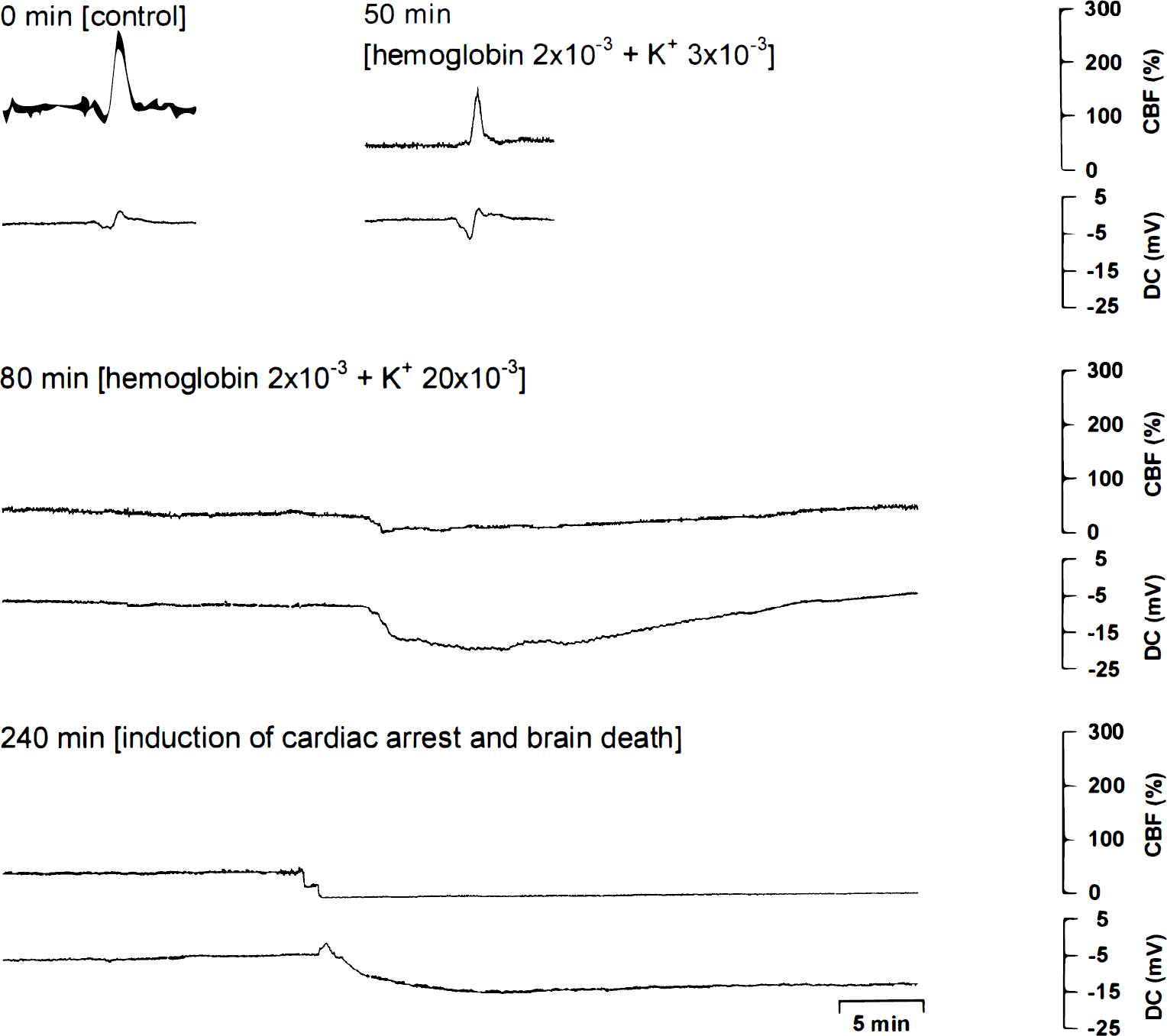
A typical experiment of series III is shown. 0 minutes: Cortical spreading depression(CSD) under physiologic conditions induced a cortical spreading hyperemia. 50 minutes: CSD during superfusion of hemoglobin (2 mmol/L) with [K+]ACSF at 3 mmol/L induced a smaller cortical spreading hyperemia and an increased negative DC shift. 80 minutes: CSD during superfusion of hemoglobin (2 mmol/L) with [K+]ACSF at 20 mmol/L induced a long-lasting transient ischemia. The CBF decrease and negative DC shift occurred almost simultaneously. An initial positive DC shift is missing. 240 minutes: Induction of cardiac arrest and brain death reveals a typical sequence of initial CBF decrease followed by a positive DC shift, with a delay by a negative DC shift. The temporal relation between CBF decrease and negative DC shift of cardiac arrest (240 minutes) is strikingly different from that of the transient ischemia(80 minutes).
In series IV through VIII, CSD was elicited at the beginning of the experiment and every 60 minutes afterward. The fourth CSD after 180 minutes was used for statistical comparison between CBF and DC response during superfusion with either physiologic ACSF (series V), or ACSF containing [K+]ACSF at 20 mmol/L (series VI), L-NA and [K+]ACSF at 3 mmol/L (series VII), L-NA and [K+]ACSF at 10 mmol/L (series VIII), or L-NA and [K+]ACSF at 20 mmol/L (series IV). Equilibration times for [K+]ACSF and L-NA were 60 minutes and 120 minutes, respectively.
In series IX, the protocol of series IV initially was used. After the CSD at 180 minutes, superfusion with L-NA and [K+]ACSF at 20 mmol/L continued, and nimodipine (2 μg/kg body weight/min, BAYER, Leverkusen, Germany) or vehicle(23.7 volume-percent alcohol) was infused intravenously. Equilibration time for nimodipine was 60 minutes.
In series X, brain cortex oxygen free radical production was measured by enhanced chemiluminescence (CL). We compared CSH under physiologic conditions with CSI during L-NA and [K+]ACSF at 20 mmol/L (equilibration times for L-NA and [K+]ACSF at 20 mmol/L, as in series IV). The DC potential was measured at an open frontal window by a calomel electrode.
Nitric oxide synthase assay
Immediately after the experiment, tissue specimens were removed from the cortex under the parietal window and from the contralateral cortex in situ by conization to a depth of 1 mm. They then were immediately frozen in liquid N2 for later analysis.
The NOS assay was based on assessing the conversion of L-[3H]-arginine to L-[3H]-citrulline. Tissue samples of 10 mg/150 μL buffer were homogenized in a Potter-Elvehjem homogenizer with a Teflon pestle, four to five strokes, in ice-cold 0.05 mol/L TRIS buffer (pH 7.4) containing 1.15% (weight/volume) KCl, 1 mmol/L ethylenediamine tetraacetic acid, 5 mmol/L glucose, 0.1 mmol/L D,L-dithiothreitol, 200 U/mL superoxide dismutase, 2 mg/L pepstatin A, 10 mg/L trypsin inhibitor, and 44 mg/L PMSF. The homogenate was centrifuged for 10 minutes in a refrigerated bench microcentrifuge at 2000 g(4°C). The protein concentration was measured at 224 and 236.5 nm using serum albumin as a standard. The detection limit was 5 μg/mL.
Determination of NOS activity was performed in a reaction buffer: 50 mmol/L HEPES buffer (pH 7.4), which contained 1 mmol/L ethylenediamine tetraacetic acid, 1.25 mmol/L CaCl2, 1 mmol/L D,L-dithiothreitol, 1 μmol/L flavin adenine dinucleotide, 1 μmol/L calmodulin, 15 μmol/L 6R-tetrahydrobiopterin, and 1 μmol/L L-[3H]arginine(1 μCi final concentration). The supernatant was added to obtain a final protein concentration of 15 to 30 μg/mL in a reaction volume of 150 μL. The reaction was initiated by adding the cofactor NADPH to a final concentration of 1 mmol/L. Samples were incubated for 30 minutes at 37°C. The reaction was terminated by adding 1 mL of ice-cold 100 mmol/L HEPES buffer (pH 5.5) containing 10 mmol/L EGTA(stop-buffer). The total volume (1 mL) was applied to a 0.5 mL Dowex AG50 WX-8 column(Na+-form), which had been equilibrated with the stop-buffer (minus EGTA). L-[2,3-3H]-citrulline was eluted twice with 0.5 mL stop-buffer (minus EGTA), and the radioactivity was determined by liquid scintillation counting.
The enzyme percent inhibition was calculated by subtraction of enzyme activity in the contralateral cortex (100%) minus enzyme activity in the cortex of the window which was superfused with L-NA.
Enhanced chemiluminescence
Chemiluminescence was recorded using a cooled (−20°C) photomultiplier (Hamamatsu R943-02) with a dark count of 3 cps at −20°C. The counts of the photomultiplier were amplified and counted by a Hewlett-Packard Universal counter (HP 5316 B) connected to a personal computer for data recording and storage. To exclude photons from other sources than brain CL, the animal in the stereotactic frame was housed in a dark box and was covered with aluminum foil. Only the cranial window was left unshielded and was positioned under a reflector, reflecting the photons from the exposed brain onto the photonsensitive area of the photocathode (for details, see Dirnagl et al., 1995).
Lucigenin (N,N′-dimethylacridinium, Sigma Chemicals) is a standard CL enhancer that produces low levels of light (450 to 500 nm) on reaction with intracellular oxygen free radicals, being particularly sensitive to superoxide radicals. Compared with other methods of radical detection, the advantage of the CL principle is that it is sensitive and has high temporal resolution, therefore allowing the on-line registration of rapid changes of oxygen free radical production.
Data analysis
Data were analyzed by comparing relative changes of CBF and oxygen free radical production and absolute changes of DC. The CBF changes were calculated in relation to baseline at the onset of the experiment (100%). Mean value comparisons between two sets of data were made using Student's t test. The data were compared for multiplicative effects using a logarithmic transformation. All data in the text, table, and figures are given as mean value ± SD.
RESULTS
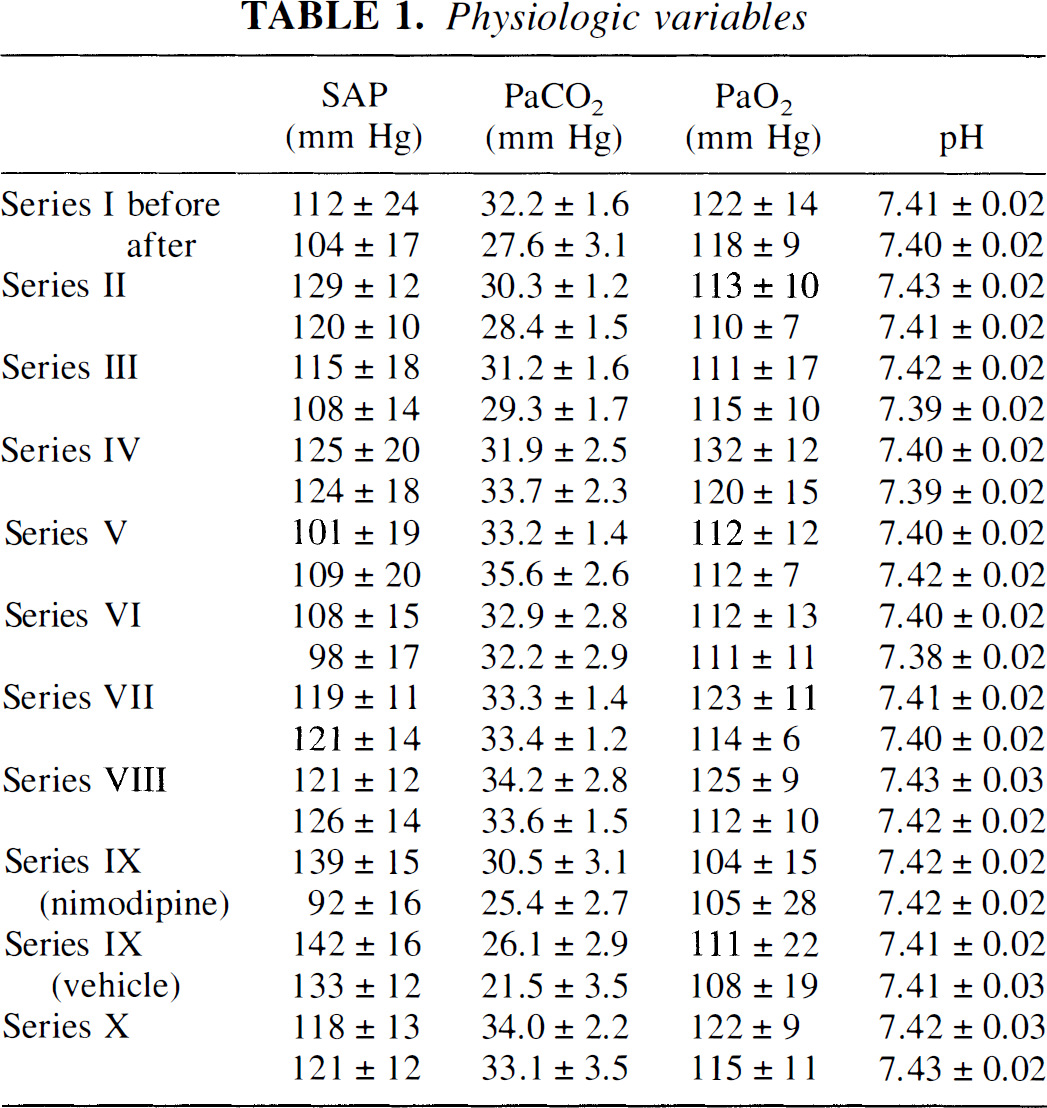
Physiologic variables before and at the end of the experiments are given in Table 1. Neither brain topical application of L-NA, Hb nor that of increased [K+]ACSF had any significant influence on the systemic variables. Intravenous nimodipine decreased blood pressure significantly (P < 0.01). In the long experiments of series IX, the animals became mildly hypocapnic (P < 0.05). The Pco2 was not significantly different between nimodipine- and vehicle-treated animals.
Physiologic variables
In series I, we investigated the effect of combined superfusion with Hb (2 mmol/L) and [K+]ACSF at 3 and 35 mmol/L on CBF and DC potential (n = 4). A typical experiment is shown in Fig. 1. The Hb and physiologic [K+]ACSF decreased CBF to 58 ± 6% compared with baseline (100%; P < 0.05). When [K+]ACSF was increased from 3 to 35 mmol/L during continued superfusion with Hb, CBF decreased to 49 ± 3%. Starting from this level of CBF, sudden spontaneous transient ischemic events developed (amplitude of negative DC shift −7.3 ± 0.6 mV; duration of negative DC shift 49 ± 12 minutes; lowest CBF level 14 ± 7%; duration of hypoperfusion 53 ± 17 minutes). The CBF level after the transient ischemia increased to 88 ± 14%.
Vasoconstriction by free Hb results from its ability to scavenge NO: Therefore, in series II, we tested whether spontaneous transient ischemic events also may occur when Hb is replaced by the NOS inhibitor L-NA (1 mmol/L) (n = 13). The L-NA and [K+]ACSF at 35 mmol/L decreased CBF to 94 ± 13%. Starting from this level of CBF, spontaneous transient ischemic events developed as in series I (lowest CBF level 22 ± 12%; duration of hypoperfusion 8.1 ± 1.7 minutes). When transient ischemia developed at the closed parietal window, CBF and DC potential measurements at a second open frontal window under physiologic conditions demonstrated with a delay, the typical pattern of a spreading depression (hyperemia 329 ± 141%; amplitude of negative DC shift −10.8 ± 3.0 mV [kalomel electrode; n = 5; Fig. 2).
In the normal cascade of ischemia, the decrease of CBF induces first a neuronal hyperpolarization and then a depolarization with a delay of several tens of seconds. The characteristic sequence of the initial CBF decrease, followed by a positive DC shift and a delayed negative DC shift, is shown in Fig. 3 (240 minutes, CBF, and DC potential after cardiac arrest). The transient ischemic events in series I and II do not fit into the normal picture of ischemia. Their pattern is characterized by an almost simultaneous onset of CBF decrease and negative DC shift. An initial positive DC shift is missing (compare Fig. 3 [80 versus 240 minutes]). The probability of CSD occurrence was increased by our experimental design since [K+]ACSF was increased to 35 mmol/L. Therefore, we hypothesized that the negative DC shift was caused by a spreading depression mechanism, and the transient ischemia was a paradoxical CBF response to CSD.
To prove this hypothesis, in series III, we tested whether CSD induces transient ischemia under the condition of brain topical Hb (2 mmol/L) and increased [K+]ACSF (20 mmol/L; n = 4). The CSD was elicited by KCl. When CSD arrived at the closed parietal window, CBF and DC changes were measured. A typical experiment is shown in Fig. 3. At physiologic ACSF, CSD induced CSH (289 ± 53%) and a negative DC shift of −2.8 ± 1.5 mV (Fig. 3: 0 minutes). Superfusion of Hb and physiologic [K+]ACSF decreased CBF to 61 ± 20% (P < 0.1), reduced CSH to 154 ± 67% (P < 0.1), and increased the DC potential significantly to −6.0 ± 1.8 mV(P < 0.05) (Fig. 3: 50 minutes). A transient ischemia was not observed with one exception. When [K+]ACSF was increased from 3 to 20 mmol/L during continued superfusion with Hb, CSD induced transient ischemic events in all animals (amplitude of negative DC shift −9.0 ± 1.8 mV; duration of negative DC shift 28 ± 19 minutes; lowest CBF level 20 ± 20%; duration of hypoperfusion 25 ± 21 minutes) (Fig. 3: 80 minutes). The CBF level after the transient ischemia increased to 108 ± 54%. In Fig. 4, the statistical analysis is summarized.
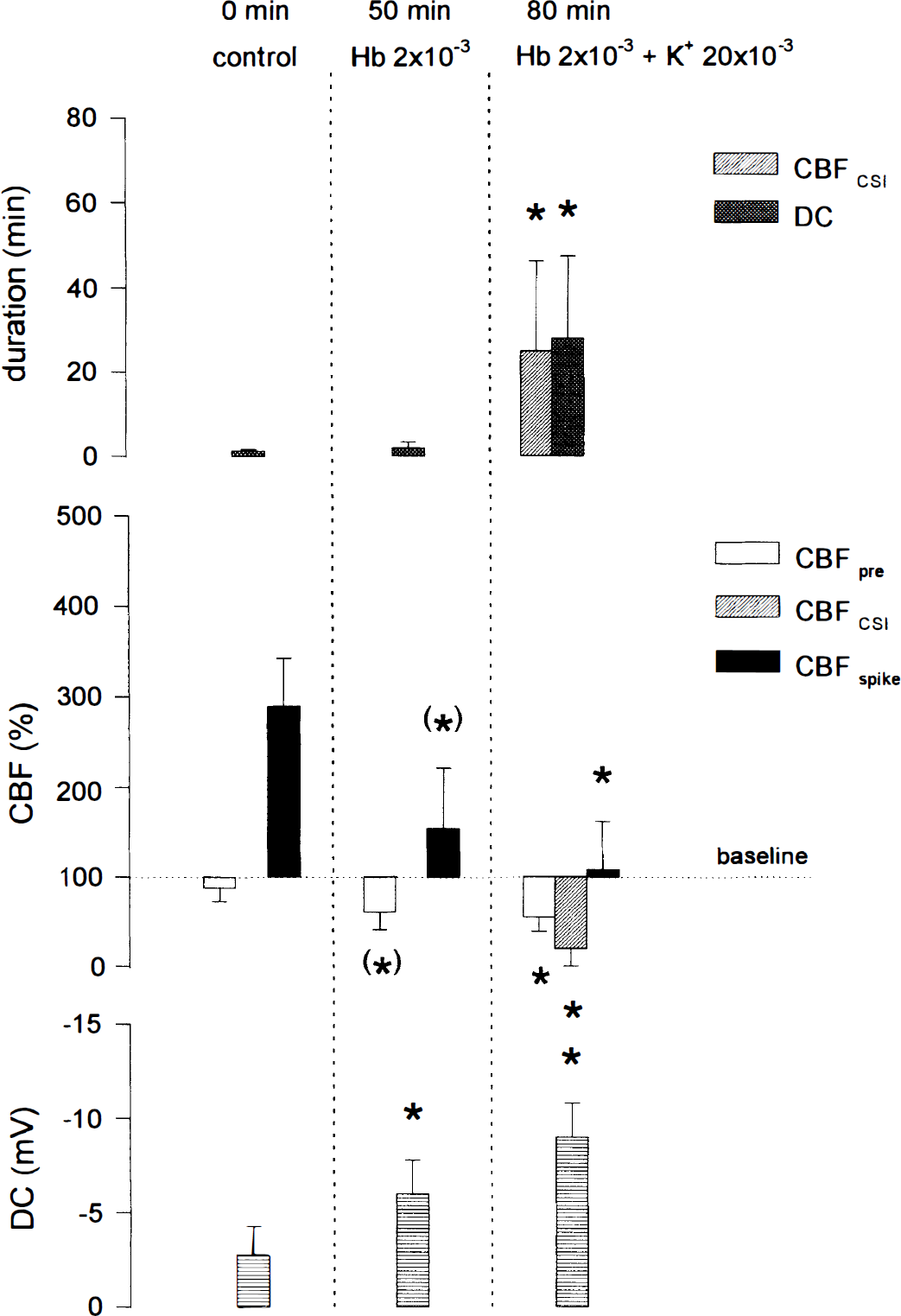
Statistical analysis of series III. The cortical spreading ischemia during superfusion of hemoglobin (Hb, 2 mmol/L) and [K+]ACSF at 20 mmol/L is completely different from the normal CBF response to CSD. CBFCSI, lowest CBF level during transient cortical spreading ischemia; CBFpre, resting CBF level immediately before CSD; CBFspike, highest CBF level during cortical spreading hyperemia or highest reperfusion level after transient ischemia; DC, negative DC shift; *, level of significance of at least P < 0.05; (*), statistical trend (P < 0.1). Bars indicate mean value ± SD. Concentrations in mol/L.
In series IV, the transformation of CSH into transient ischemia was confirmed by combined superfusion of the NOS inhibitor L-NA and increased [K+]ACSF at 20 mmol/L (n = 19). A typical experiment is shown in Fig. 5A. Both L-NA and [K+]ACSF at 20 mmol/L decreased CBF to 95 ± 14%. As in series III, CSD induced transient ischemia (amplitude of negative DC shift−4.8 ± 3.0 mV; duration of negative DC shift 34 ± 44 minutes; lowest CBF level 33 ± 21%; duration of hypoperfusion 18 ± 26 minutes). The CBF level after the transient ischemia increased to 177 ± 77%. To complement our studies, we measured the spreading of the CBF response in five animals with two laser-Doppler flow probes. As can be seen in Fig. 5B, CSH under physiologic conditions travels at similar speed (3.6 ± 0.7 mm/min) as CSD-induced ischemia under the condition of NOS inhibition and increased [K+]ACSF(3.4 ± 0.6 mm/min). This finding clearly demonstrates the transformation of CSH into CSI.
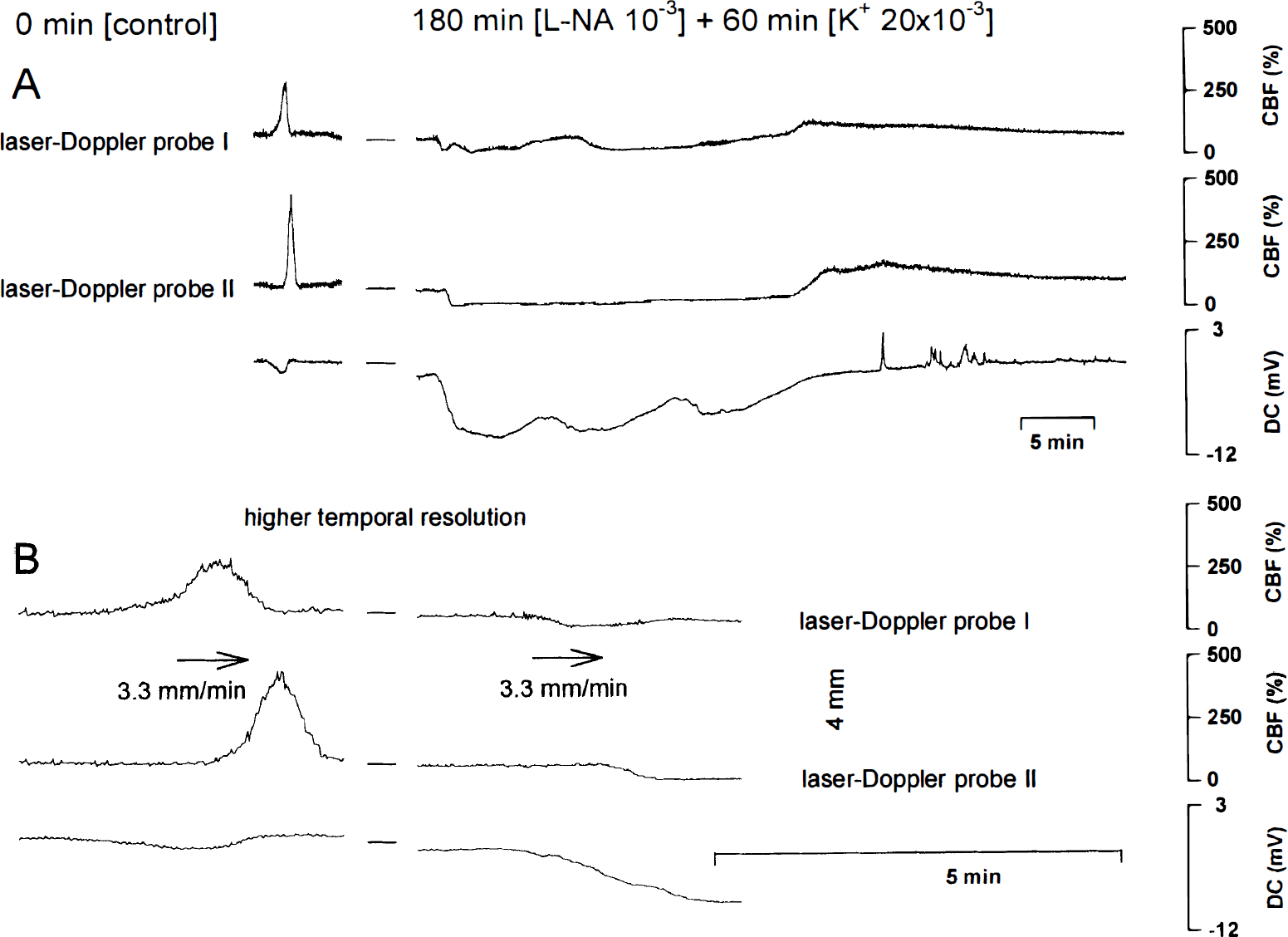
A typical experiment of series IV is shown.
In series IV through VIII, CBF and DC responses to CSD were compared during superfusion with either physiologic ACSF (series V, n = 5), or ACSF containing [K+]ACSF at 20 mmol/L (series VI, n = 4), L-NA and [K+]ACSF at 3 mmol/L(series VII, n = 10), L-NA and [K+]ACSF at 10 mmol/L) (series VIII, n = 6), or L-NA and [K+]ACSF at 20 mmol/L (series IV, n = 19). The statistical analysis is summarized in Fig. 6.

The statistical analysis of series IV through VIII is shown. CBFCSI ▨, lowest CBF level during cortical spreading ischemia; CBFCSI duration, duration of CSI; CBFpre □, resting CBF level immediately before CSD; CBFspike ▪, highest CBF level during cortical spreading hyperemia or highest reperfusion level after cortical spreading ischemia; DC , negative DC shift; *, level of significance of at least P < 0.05. Bars indicate mean value ± SD. Concentrations in mol/L.
We found that both the NO.-lowering agent and an increase of [K+]ACSF to at least 20 mmol/L are necessary for the induction of CSI (with one exception in series VIII). Thus, both the NO.-lowering agent and [K+]ACSF are involved in the modulation of the vascular reactivity, which predisposes to CSI. In series VI, the CBF response to K+ after CSD was decreased compared with our reference values without preceding CSD (P < 0.05) (Dreier et al., 1995). This observation is consistent with decreased vascular reactivity after CSD (Lauritzen, 1984; Wahl et al., 1987). Similarly to Hb, [K+]ACSF at 20 mmol/L without L-NA increased the negative DC shift significantly (P < 0.05, series VI), whereas L-NA and [K+]ACSF at 3 mmol/L did not (series VII). A small initial hypoperfusion, as described by Duckrow (1993) and Fabricius and colleagues(1995), was found in 5 of 10 animals in series VII (L-NA and [K+]ACSF at 3 mmol/L) and in 2 of 6 animals in series VIII (L-NA and [K+]ACSF at 10 mmol/L). Both L-NA and [K+]ACSF at 3 mmol/L reduced CSH by 34% (P < 0.05, series VII). In seven animals of series VII, we compared Ca2+-dependent NOS activity in the cortex superfused with L-NA with the contralateral hemisphere. The NOS activity in the contralateral cortex was 10.2 ± 4.4 pmol/L/min/mg protein; NOS activity in the L-NA-treated cortex was 0.9 ± 0.6 pmol/L/min/mg protein. Thus, inhibition of NOS activity with L-NA was 91%.
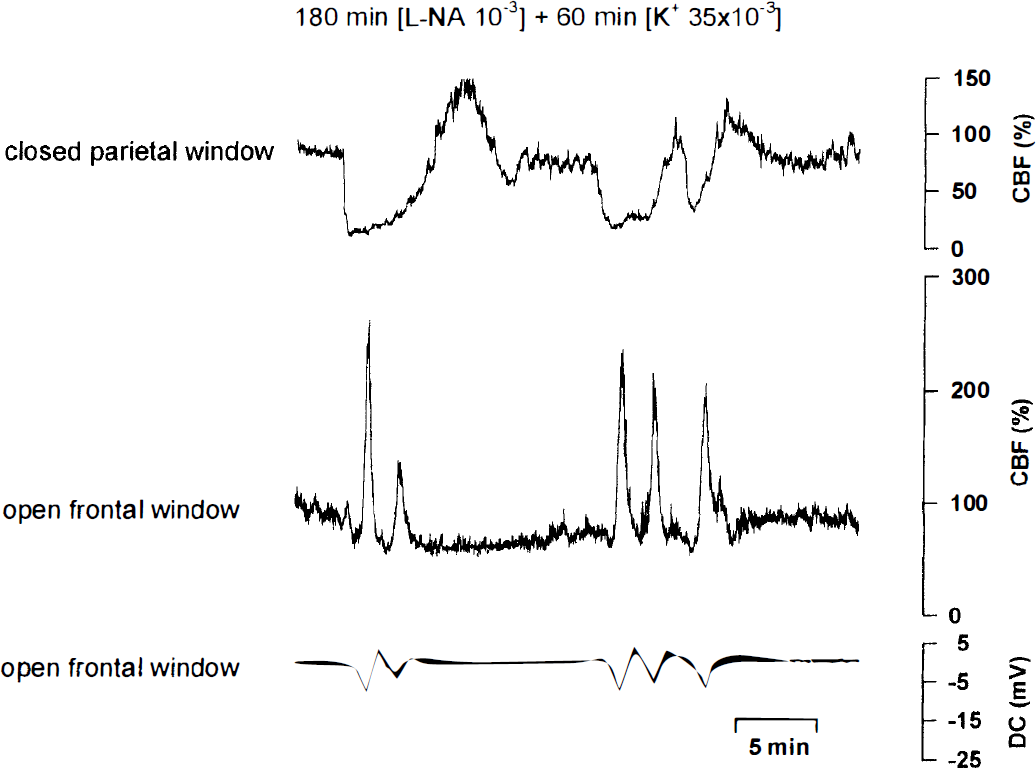
In series IX, we investigated the effect of nimodipine (2 μg/kg body weight/min intravenously) on CSI (n = 4). A typical experiment is shown in Fig. 7. Under physiologic conditions, CSD induced CSH (Fig. 7: 0 minutes). Later, during superfusion with combined NOS inhibition and [K+]ACSF at 20 mmol/L, CSD induced CSI (Fig. 7: 180 minutes). Administration of nimodipine reestablished an almost normal CBF response to CSD despite continued NOS inhibition and [K+]ACSF at 20 mmol/L (Fig. 7: 300 minutes). In contrast, the vehicle control with alcohol (23.7 volume percent) showed an increase of CSI, which was not statistically significant (n = 4). The statistical analysis is shown in Fig. 8.
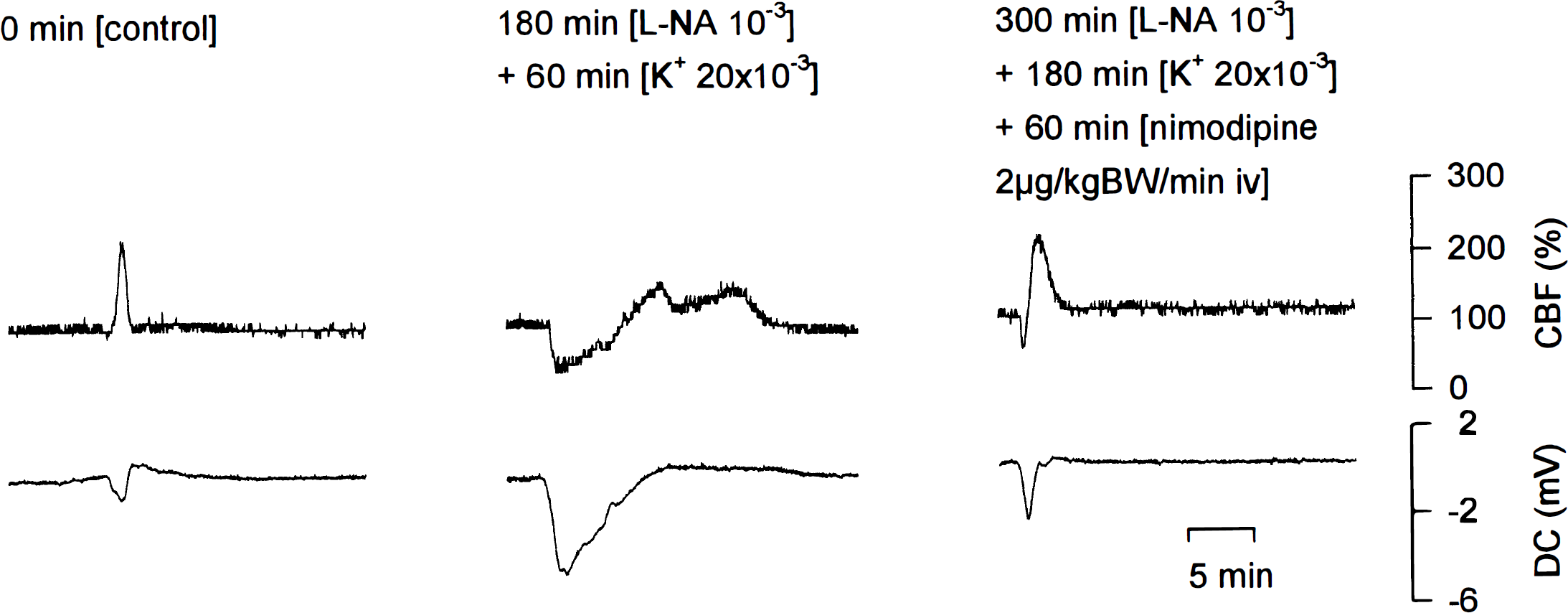
A typical experiment of series IX is shown. 0 minutes: CSD under physiologic conditions induced cortical spreading hyperemia. 180 minutes: CSD during superfusion of L-NA(1 mmol/L) with [K+]ACSF at 20 mmol/L induced cortical spreading ischemia. 300 minutes: Nimodipine intravenously (2 μg/kg body weight/min) almost completely abolished cortical spreading ischemia despite continued brain topical L-NA (1 mmol/L) and [K+]ACSF at 20 mmol/L.
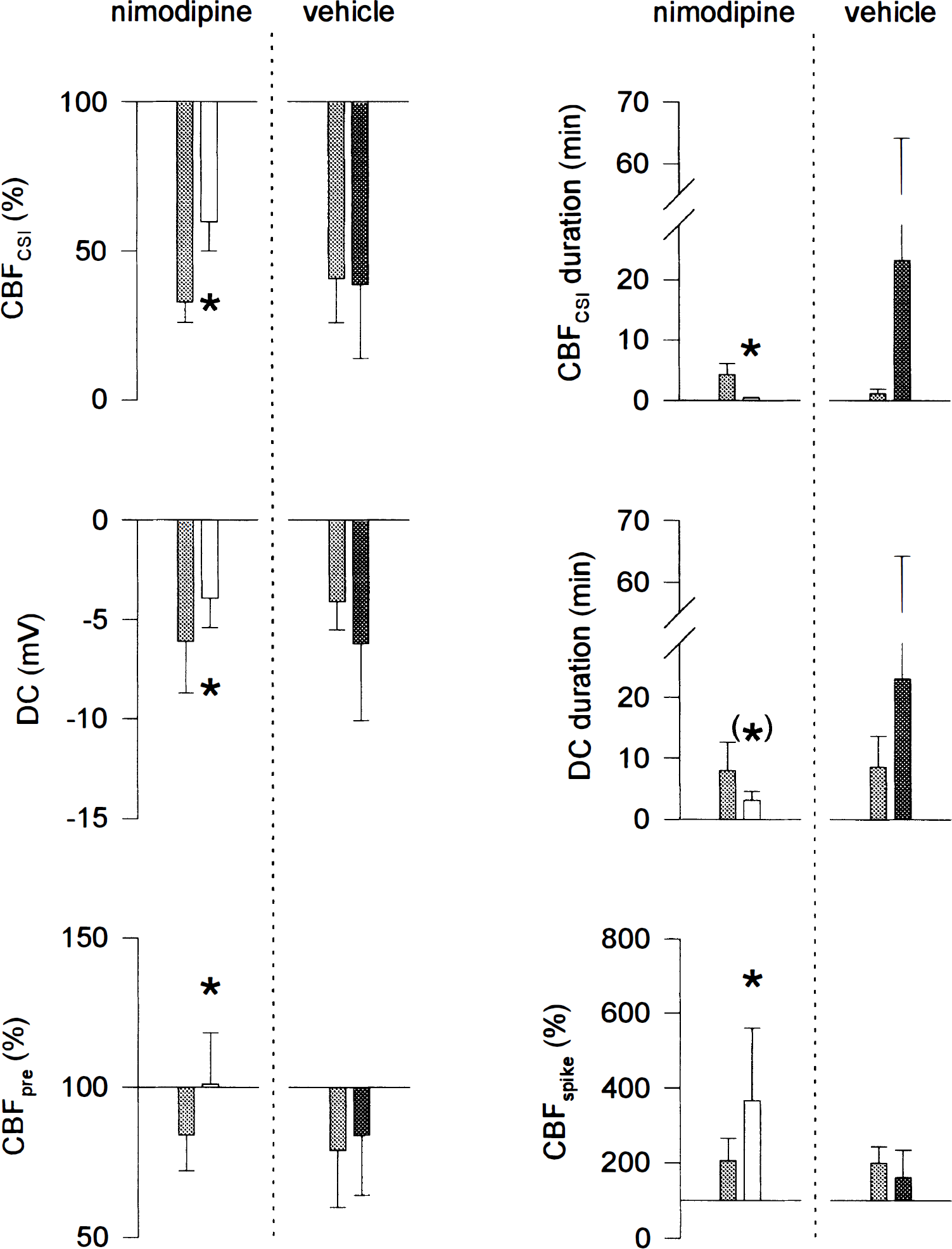
The statistical analysis of series IX is shown. In four animals we tested the effect of nimodipine (2 μg/kg body weight/min intravenously) during superfusion of L-NA (1 mmol/L) and [K+]ACSF at 20 mmol/L on cortical spreading ischemia. In four other animals the effect of the vehicle (23.7 volume-percent alcohol) was tested. Whereas nimodipine almost completely abolished cortical spreading ischemia, the vehicle showed a tendency to aggravate CSI, which was not statistically significant. CBFCSI, lowest CBF level during CSI; CBFCSI duration, duration of cortical spreading ischemia; CBFpre, resting CBF level immediately before CSD; CBFspike, highest CBF level in response to CSD, DC, negative DC shift; *, level of significance of at least P < 0.05;(*), statistical trend of P < 0.1. Bars indicate mean value ± SD. Concentrations in mol/L. (▓) control (180 min [L-NA 10−3]+ 60 min [K+ 20 × 10−3]). (□) 60 min [nimodipine 2μg/kgBW/min iv] + 300 min [L-NA 10−3] + 180 min [K+ 20 × 10−3]. (▪) 60 min [vehicle iv] + 300 min [L-NA 10−3] + 180 min [K+ 20 × 10−3].
To investigate whether CSI may have potential to cause tissue damage, in series X, we compared the pattern of oxygen free radical production during CSH with that of CSI. The CSH did not change radical production, whereas CSI produced a pattern typical of ischemia and reoxygenation (initial dip by −56 ± 22% followed by increase above baseline to +284 ± 58%, Fig. 9, n = 4).
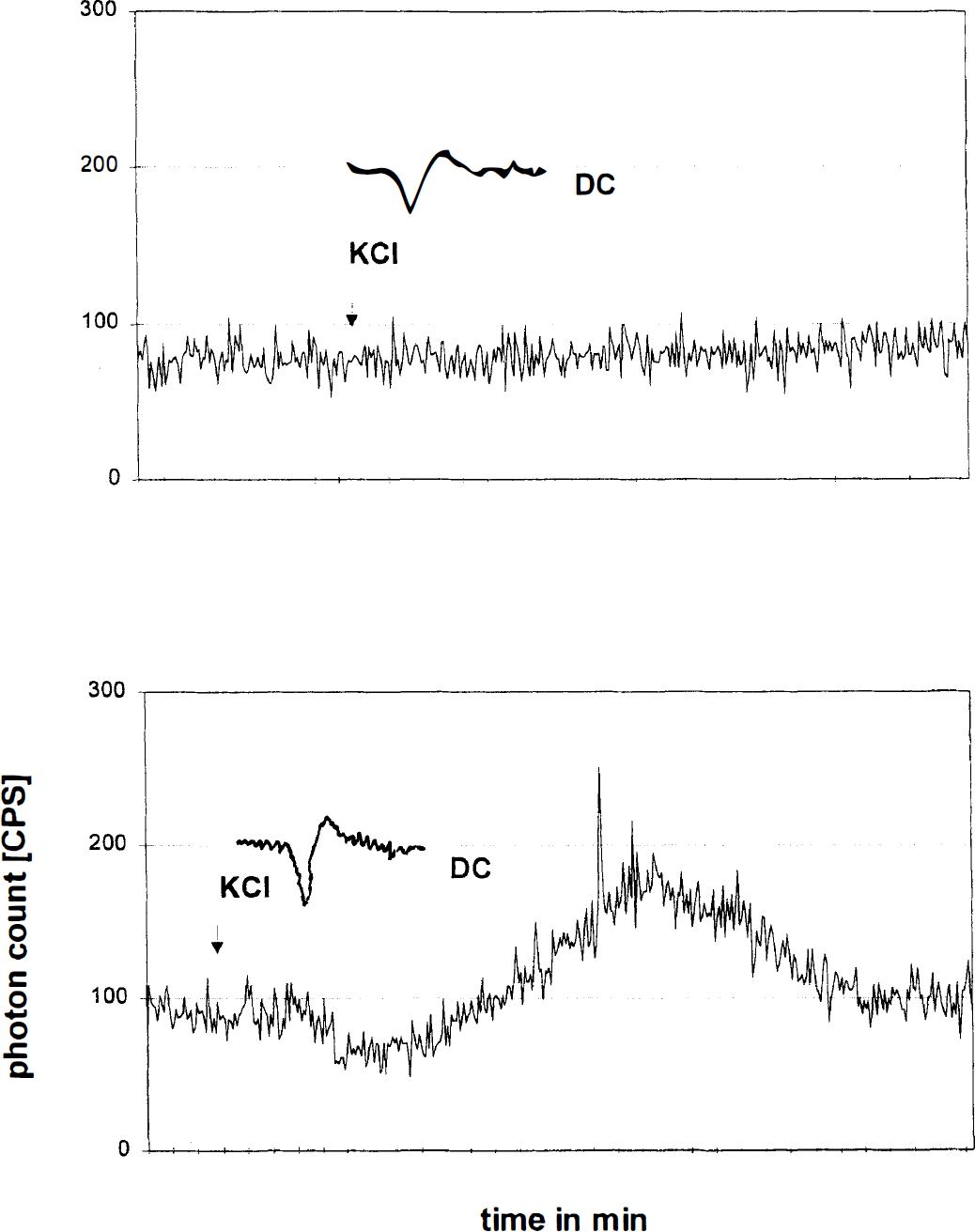
A typical experiment of series X is shown. In the upper plot, the oxygen free radical production is measured by enhanced chemiluminescence during normal CSD. No change of the radical production is observed. In the lower plot, the oxygen free radical production is measured during cortical spreading ischemia. In the lower plot, during CSI, the typical radical production pattern of transient ischemia and reoxygenation is characterized by an initial dip during ischemia followed by free radical burst during reperfusion. The DC potential was simultaneously measured by a kalomelelectrode positioned frontally, at a distance from the closed window. Therefore the DC potentials of the two plots are similar.
DISCUSSION
The effect of NOS inhibition on the CBF response to CSD is controversial. Duckrow(1993) demonstrated a modulation of the CBF response with a short initial hypoperfusion and an insignificant reduction of CSH in the awake rat by NOS inhibition. The difference of the mean values of CSH between the untreated group and the group treated with N-nitro-L-arginine-methyl-ester was more pronounced than in our study, but the statistical methods differ and his sample size was smaller. This may explain the statistical difference between our study and the study by Duckrow. Fabricius and others (1995) confirmed the short initial hypoperfusion in the anesthetized rat but did not find a reduction of CSH. Zhang and coworkers (1994) and Wolf and associates (1996) did not observe any effect of NOS inhibition on CSH in the anesthetized rat. Goadsby and associates(1992) reported a complete block of CSH in the anesthetized cat but without preceding hypoperfusion. Colonna and others (1997) reported a reduction of CSH in the anesthetized rabbit. Wahl and colleagues (1994) described a significant reduction of CSD-induced pial arteriolar dilation in the anesthetized cat, and Colonna and associates (1994) in the rabbit. In our study, both NO. scavenging and NOS inhibition reduced CSH by approximately 40% and provoked a short initial hypoperfusion in some animals. The reason for the controversial results concerning NOS inhibition on CSH are not clear. Differences in anesthesia, species, preparation, or the route and time of drug application may be responsible (Zhang et al., 1994). We emphasize that within the same laboratory using the same anesthesia and similar animals, systemic NOS inhibition with L-NA did not affect CSH (Wolf et al., 1996), but in this study, brain topical L-NA reduced CSH. However, Duckrow (1993) also used systemic NOS inhibition and observed effects similar to this study. Therefore, a definite explanation cannot be given. Hypothetically, NO. may have a low threshold at which NO. deprivation becomes effective. In this study, NOS inhibition was 91%, which was higher than the effect reported by Zhang and colleagues (1994). This hypothesis could be tested by combined topical application of Hb and NOS inhibitor or an increased concentration of NOS inhibitor when no effect is observed in a certain model. The cranial window must be large to avoid interfering effects by neighboring naive cortical areas. A threshold effect would point to a permissive action of NO. during CSD rather than agonist-induced NOS activation(see later). In summary, although NO. is not the only mediator of the vascular response to CSD in the rat, under certain conditions it may be involved. When [K+]ACSF is increased, lowering of NO. transforms CSH into CSI.
Brain topical superfusion with increased [K+]ACSF at 35 mmol/L combined with a NO.-lowering agent induced spontaneous transient ischemic events (series I and II). Their negaive DC-shift occurred almost simultaneously with the CBF decrease and was not preceded by a positive DC-shift in contrast to the classic sequence of ischemia. We hypothesize that the depolarization is not the result of ischemia in this case, but of a spreading depression mechanism induced by increased [K+]ACSF. Consequently, the transient ischemia is interpreted as a paradoxical CBF response to CSD. We support this hypothesis in the experiments conducted in series III through VIII. In these experiments, [K+]ACSF was maximally increased to 20 mmol/L to avoid spontaneous CSD at the parietal window. Instead, at a 5-mm distance from the parietal window, CSD was elicited at will by KCl application. When CSD arrived at the parietal window, CBF and DC response were measured. Although CSD induced a CSH under physiologic conditions, cortical spreading ischemia (CSI) was induced by the combination of the NO.-lowering agent and [K+]ACSF at 20 mmol/L. Spreading of CSI was measured with two laser-Doppler flow probes and was identical to spreading of CSH.
The generation of CSI depends on a decrease of the NO. level. This was shown indirectly by two different NO.-lowering agents. N-Nitro-L-arginine is an N-substituted L-arginine analogue that inhibits the NOS (Iadecola et al., 1994a). Hemoglobin binds NO. with an affinity 1500 times higher than its affinity for oxygen (Gibson and Roughton, 1957) at its heme iron and reactive sulfhydril groups at cysteineβ93. Deoxygenation causes transition of S-nitroso-hemoglobin from the R(oxygenated) to the T(deoxygenated) structure. The S-nitroso-hemoglobin contracts blood vessels in the R structure while it relaxes them in the T structure. The vasodilation in the T structure is dependent on glutathione and results from release of NO. from the binding at cysteineβ93 (Stamler et al., 1997; Jia et al., 1996). Thus, erythrocytes may exert a delicate action on vascular tone caused by NO. binding and release by Hb, dependent on oxygen tension. When Hb is released into the extracellular space by hemolysis, the fine-tuned regulation is lost, and Hb acts essentially as a NO. scavenger (Perutz, 1996). The evolutionary advantage likely is a protracted control of bleeding by strong vasoconstriction. Thus, free Hb injected into the subarachnoid space decreases cGMP in the vascular walls to levels similar to that produced by the NOS inhibitor N-nitro-L-arginine-methyl-ester (Edwards et al., 1992), causing acute vasoconstriction (Petruk et al., 1972) and reduction in CBF (Watkins et al., 1994). The decrease of CBF by free Hb in our study is consistent with these findings.
During CSD, stimulation of NOS by increased intracellular calcium may occur. Thus, NOS inhibition and NO. scavenging may interfere with the CBF response to CSD by inhibition of a NO. concentration peak during CSD. On the other hand, decrease of the basal NO. level may abolish the permissive function of NO. for other vasodilators. A permissive function was reported, for example, for CO2, K+, and alpha 2-adrenoceptor-mediated vasodilation (Iadecola et al., 1994b; Iadecola and Zhang, 1996; Bryan et al., 1995; Dreier et al., 1995). Suggestion of a permissive effect of NO. during CSD was provided by Colonna et al. (1997). They suggested neuronal NOS as the source of NO. for this effect, since 7-nitroindazole reduced CSH similar to L-NA. Whether endothelial NOS is not inhibited by 7-nitroindazole is controversial (Fabricius et al., 1996).
The dependence of CSI on the artificial increase of [K+]ACSF to 20 mmol/L is surprising, since [K+]o increases in the cortex to 60 mmol/L during normal CSD (Kraig and Nicholson, 1978; Hansen et al., 1980). By an unknown mechanism, the increase of [K+]ACSF may potentiate the small initial hypoperfusion that is induced by NO.-lowering agents (Duckrow, 1993; Fabricius et al., 1995). The increase of the subarachnoid DC potential by increased [K+]ACSF indicates a decrease of the electrical resistance between cortex and subarachnoid space. Electrical coupling of the cortical and pial extracellular space may have implications for vascular ion channels during CSD. The direct effect of K+ on cerebral circulation is complex. In concentrations below 20 mmol/L, it acts as a vasodilator through inward rectifier K+ channels, and above 20 mmol/L, as a vasoconstrictor (Edwards et al., 1988; McCulloch et al., 1982; Kuschinsky et al., 1972). A synergistic vasodilating effect of mildly increased [K+]o and sodium nitroprusside was reported for the rat aorta (Rapoport and Murad, 1983). Consistently, vasoconstriction by highly increased [K+]o was attenuated by sodium nitroprusside (Rapoport et al., 1985). An attenuation of K+ induced vasoconstriction by NO. also was shown in the canine basilar artery (Minato et al., 1995). In contrast, the effect of increased [K+]o on NO.-mediated cerebral vasodilation is only marginal. Although NO.-induced smooth muscle hyperpolarizations in the rabbit basilar artery (Plane and Garland, 1994) and middle cerebral artery (Parsons et al., 1991) are antagonized by high levels of [K+]o, smooth muscle relaxation is not. The hyperpolarization is caused by activation of potassium channels and therefore dependent on [K+]o. However, the independence of the NO.-mediated smooth muscle relaxation from[K+]o supports that NO. is an efficient antagonist against K+-induced vasoconstriction. Therefore, a decrease of the NO. level may promote acute vasoconstriction by the CSD-induced extracellular K+-peak, thus producing hypoperfusion and CSI. Further vasoconstrictors besides K+ that are involved in the vascular response to CSD are prostaglandins (Shibata et al., 1991; Meng et al., 1995). The NO. antagonizes different potent vasoconstrictors (e.g., endothelins and serotonin). In addition, interference of decreased NO. levels or increased subarachnoid K+ levels with vasodilation (e.g., by CGRP from the trigeminovascular system) (Colonna et al., 1994; Wahl et al., 1994) is conceivable.
Both the depolarization and the CBF response of CSI take much longer than normal CSD. This finding may be explained by the depolarization-induced ischemia. Under these conditions, the Na+-K+ ATPase fails to return the increased extracellular K+ ions into the cells (Rosenthal and Somjen, 1973; Mayevsky et al., 1982). Thus, repolarization does not occur and a vicious circle with vasoconstriction and ischemia is established. In summary, depolarization, in the case of CSI, is not the result but the cause of ischemia in contrast to anoxic depolarization or periinfarction depolarization (Nedergaard and Astrup, 1986; Back et al., 1994). Failure of repolarization is similar in CAI and classic ischemia but it does not occur in normal CSD. If the primary event of CSD occurs in astrocytes (Nedergaard, 1994), CSI is an astrocyte-induced ischemia.
In series X, we confirm the data by Meng and Busija (1996) that the oxygen free radical production during normal CSD is not changed. The pattern of the oxygen free radical production during CSI is consistent with the typical pattern of transient cerebral ischemia in other in vivo models (Dirnagl et al., 1995) and hypoxia in brain slices (Schreiber et al., 1995). This suggests that CSI may cause brain damage.
Clinical relevance of CSD is most frequently discussed in relation to classic migraine (Leão and Morison, 1945; Lauritzen and Olesen, 1984; Lauritzen, 1994). Migraine is associated in rare cases with strokes beginning with a migrainous aura that leads to a persistent neurologic deficit (Broderick, 1997). Such a sequence may be consistent with a CSD-induced ischemia. Complicated migraine and predominantly cortical brain infarcts are particularly impressive in patients with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS syndrome) (Rosen et al., 1990; Matthews et al., 1991). Association of some of the stroke-like episodes with seizures is interesting, since seizure-like events and CSD often occur in the same pathophysiologic models, such as in the low Mg2+ model in rat (Mody et al., 1987) and human brain slices (Avoli et al., 1991). A stroke principle based on disturbed coupling between metabolism and cerebral blood flow may offer a pharmacologic treatment, such as nimodipine or blockade of CSD.
One of our experimental protocols suggests another connection with a clinical condition. In the erythrocyte, Hb (21.03± 0.75 mmol/L; Stobbe, 1991) and K+ (102.4 ± 3.9 mmol/L; Beutler, 1995) are the protein and ion, respectively, with the highest concentration. It is tempting to link the induction of CSI by Hb and K+ in the subarachnoid space with DID after SAH. Closely associated with the evolution of hemolysis after SAH, vasospasm and DID develop (MacDonald and Weir, 1991). The severity of this complication correlates with the amount of subarachnoid blood observed in fissures and cisterns in the computer tomogram (Fisher et al., 1980; Brouwers et al., 1993). Of the patients without therapy, 30% develop DID after SAH, which cause death in 15% (Kassell et al., 1990). The pathomorphologic substrate of DID are cortical laminar and bell-shaped infarcts in over 90% (Stoltenburg-Didinger and Schwarz, 1987; Stoltenburg-Didinger, 1997; Neil-Dwyer et al., 1994). The infarcts develop preferentially in areas covered with blood (Stoltenburg-Didinger and Schwarz, 1987; Stoltenburg-Didinger, 1997).
The mechanism of these infarcts is unknown. There is controversy whether structural changes of the vessels, vasoconstriction, or thrombosis may be responsible (Mohr et al., 1992). Vasoconstriction by free Hb as a single factor is not sufficient to induce ischemia (White et al., 1987). A more complex mechanism is likely. In intracerebral hematomas, [K+]o can increase with a delay from 3 to 50 mmol/L (Ohta et al. 1983). Thus, the necessary elements of CSI, Hb, and K+ may be present in the subarachnoid blood clot when hemolysis occurs after SAH.
Metaanalysis of seven randomized, double-blind, placebo-controlled trials shows that the L-type calcium channel blocker nimodipine decreases the risk of poor outcome in patients with aneurysmal SAH by 42% (Tettenborn and Dycka, 1990). This effect results from the decrease of DID but not from angiographically visible vasospasm (Allen et al., 1983; Espinosa et al., 1984; Petruk et al., 1988). The transformation of CSI back to an almost normal CSH by nimodipine may further strengthen a link to the cortical infarcts after SAH. Interestingly, nimodipine blocks K+- and serotonin-induced vasoconstriction efficiently, but vasoconstriction by Hb or prostaglandins is antagonized poorly (Towart, 1981; Nosko et al., 1986). This may indicate a role of CSD-induced extracellular K+ rise or serotonin release for the acute CBF decrease of CSI.
The first combined recording of DC potential, [K+]o, and CBF changes in a human, typical of CSD, were reported by Mayevsky and colleagues (1996) in 1 of 14 patients with hemorrhagic brain trauma. During their observation period, the recorded spreading depressions showed a transition from a normal CBF response to a CBF response with “initial hypoperfusions.” The first CBF responses with initial hypoperfusion of the patient were similar to the CBF responses during NOS inhibition and normal subarachnoid K+ in the rat reported by Duckrow (1993). With time, the initial hypoperfusions in the patient became deeper and longer, and the following hyperperfusion decreased in amplitude and increased in duration. Although the initial hypoperfusions of the patient remained less pronounced than during CSI, a relation of both may be possible. Mayevsky and colleagues (1996) recorded at the right frontal cortex, and the computed tomography scan revealed a contusional hemorrhage at the right parietal cortex.
In summary, we have shown that during CSD there exists a complex relation between extracellular K+ and NO: Under normal conditions, CSD produces hyperemia. When levels of NO. are decreased and subarachnoid K+ is increased, CSD produces long-lasting CSI by induction of acute microcirculatory vasospasm caused by disturbed coupling between metabolism and CBF. We speculate the DID after subarachnoid hemorrhage, a condition in which K+ and the NO. scavenger Hb are released into the subarachnoid space, may be related to CSI.
Footnotes
Acknowledgements
The authors thank Nuran Akgören for reading and discussing the manuscript.