
Abstract
In rats, spreading depolarization induces vasodilation/hyperemia in naïve tissue but the inverse response when artificial cerebrospinal fluid is topically applied to the brain containing (a) a nitric oxide–lowering agent and (b) elevated K+. The inverse response is characterized by severe vasoconstriction/ischemia. The perfusion deficit runs together with the depolarization in the tissue (=spreading ischemia). Here, we found in male Wistar rats that pre-treatment with artificial cerebrospinal fluid containing elevated K+ in vivo led to a selective decline in α2/α3 Na+/K+-ATPase activity, determined spectrophotometrically ex vivo. Moreover, spreading ischemia, recorded with laser-Doppler flowmetry and electrocorticography, resulted from artificial cerebrospinal fluid containing a nitric oxide–lowering agent in combination with the Na+/K+-ATPase inhibitor ouabain at a concentration selectively inhibiting α2/α3 activity. Decline in α2/α3 activity results in increased Ca2+ uptake by internal stores of astrocytes, vascular myocytes, and pericytes since Ca2+ outflux via plasmalemmal Na+/Ca2+-exchanger declines. Augmented Ca2+ mobilization from internal stores during spreading depolarization might enhance vasoconstriction, thus, contributing to spreading ischemia. Accordingly, spreading ischemia was significantly shortened when intracellular Ca2+ stores were emptied by pre-treatment with thapsigargin, an inhibitor of the sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA). These findings might have relevance for clinical conditions, in which spreading ischemia occurs such as delayed cerebral ischemia after subarachnoid hemorrhage.
Keywords
Introduction
Spreading depolarizations (SD) occur abundantly in numerous diseases such as traumatic brain injury (TBI), intracerebral spontaneous hematoma (ICH), aneurysmal subarachnoid hemorrhage (aSAH), and malignant hemispheric stroke (MHS).1,2
Under physiological conditions, a marked rise of regional cerebral blood flow (rCBF) (=spreading hyperemia) is coupled to SD in rats and higher mammals, which is followed by oligemia.3,4 By contrast, the inverse hemodynamic response is often observed in patients with aSAH, MHS, or TBI.5–7 The inverse response is characterized by severe vasoconstriction and hypoperfusion up to ischemia instead of vasodilation and hyperemia.8,9 The SD-triggered perfusion deficit runs together with the depolarization wave in the tissue (=spreading ischemia (SI)) where it causes a delay in the energy-dependent recovery from SD. If SI is sufficiently prolonged, it leads to widespread cortical necrosis. 10
In animal experiments, SI resulted from topical application of artificial cerebrospinal fluid (ACSF) containing an elevated K+ concentration ([K+]ACSF) in combination with either the nitric oxide synthase (NOS) inhibitor NG-nitro-L-arginine (L-NNA) or the nitric oxide (NO)-scavenger hemoglobin (Hb) to the brain cortex. 9 Similar conditions are likely responsible for SI after arterial occlusion11–13 because focal ischemia leads to a baseline elevation of the extracellular K+ concentration ([K+]o) before it induces SD 14 and molecular oxygen is required for NO synthesis. 15
[K+]o increases to a peak level of ∼50 mmol/L during SD. At concentrations above ∼20 mmol/L, [K+]o produces marked vasoconstriction in a dose-dependent fashion, which is strongly augmented in absence of NO. 16 Hence, it would provide a plausible explanation how elevated [K+]ACSF contributes to SI if [K+]o reached a higher peak level during SD because the SD-induced rise of [K+]o starts from a higher baseline level of [K+]o. However, this was previously ruled out both in in vivo and neocortical slice experiments,17,18 suggesting that elevated [K+]ACSF contributes to SI by another mechanism.
Baseline [K+]o is controlled by spatial buffering of astrocytes and Na+/K+-ATP-ase (NaKA) activity. NaKA is activated by a short-lasting rise in [K+]o, whereas a prolonged rise caused a decline in NaKA activity in astroglial culture. 19 Here, we investigated whether decline in NaKA activity is a plausible mechanism by which elevated [K+]ACSF contributes to SI.
We observed that prolonged elevation of baseline [K+]o in vivo led to selective decline in α2/α3 NaKA activity. This effect can be mimicked by the NaKA inhibitor ouabain. Under simultaneous NO depletion, a low concentration of this drug was sufficient to trigger the switch from spreading hyperemia to SI similar to elevated baseline [K+]o. Ouabain did not significantly increase baseline [K+]o at this concentration, suggesting that elevated baseline [K+]o is not a primary mechanism. Notably, concentrations of ouabain, or respectively, baseline [K+]o, triggering the switch from spreading hyperemia to SI under NO depletion, were lower than concentrations for evoking SD by their depolarizing effect on neurons. Based on previous observations that ouabain augments internal Ca2+ stores, we used thapsigargin to deplete these stores and observed attenuation of SI. Overall, our findings support a role for internal Ca2+ stores in mediating the effects of ouabain to induce SI. This could designate store-operated Ca2+ signaling as a new therapeutic target to prevent SI in patients.
Methods
Animal preparation, rCBF measurement, and electrophysiology in vivo
The reporting of animal experiments complies with the ARRIVE Guidelines. All animal experiments were approved by the Landesamt für Arbeitsschutz, Gesundheitsschutz und technische Sicherheit Berlin [LAGetSi] (G0346/98 and G0156/05). Naïve, male, wild-type Wistar rats (n = 146; 250–400 g, ∼10–16 weeks old, supplied by Charles River, Germany) were anesthetized with 100 mg/kg body weight thiopental-sodium intraperitoneally (Trapanal, BYK Pharmaceuticals, Konstanz, Germany), tracheotomized, and artificially ventilated (Effenberger Rodent Respirator, Effenberger Med.-Techn. Gerätebau, Pfaffing/Attel, Germany). The left femoral artery was cannulated, and continuous saline solution (0.5 ml/h) was infused. Body temperature was maintained at 38.0 ± 0.5℃ using a heating pad. Systemic arterial pressure (RFT Biomonitor, Zwönitz, Germany) and endexpiratory partial pressure of carbon dioxide (pCO2; Heyer CO2 Monitor EGM I, Bad Ems, Germany) were monitored. Arterial pO2, pCO2, and pH were serially measured using a Compact 1 Blood Gas Analyzer (AVL Medizintechnik GmbH, Bad Homburg, Germany). Adequacy of the level of anesthesia was assessed with testing motor and blood pressure responses to tail pinch. Animals were killed with intravenous KCl infusion after the experiment.
Parietally, a craniotomy was performed using a saline-cooled drill as previously reported. 9 The dura mater was removed. The craniotomy site was either covered with a cover slip (closed window) (groups 4–15, 23, 24) or left open (groups 1–3, 21, 22). In groups 16–18, two separated open windows were implanted. The composition of control ACSF in mmol/L was NaCl 127.5, KCl 3.0, CaCl2 1.5, MgSO4 1.2, NaHCO3− 24.5, glucose 3.7, and urea 6.7. 20 Increasing [K+]ACSF from 3 to 20, 35, or 130 mmol/L determined the decrease in [Na+]ACSF to maintain isoosmolarity from 152 to 135, 120, and 25 mmol/L, respectively. The ACSF was equilibrated with a gas mixture containing 6.6% O2, 5.9% CO2, and 87.5% N2. Regional CBF was continuously monitored with laser-Doppler flow probes (Periflux 4001, Perimed AB, Järfälla, Sweden). The subarachnoid steady (direct current (DC)) potential was measured with an Ag/AgCl wire inserted into the space between cortex and coverslip. Ion-selective microelectrodes were used to measure changes of the intracortical DC potential and [K+]o in the open window. They were prepared from double-barrelled theta-glass capillaries as previously described (Fa. H. Kugelstätter, Garching, Germany). 16 Potassium Ionophore I-Cocktail A (Fluka/Sigma-Aldrich, Deisenhofen, Germany) was used as ion-exchanger. The measuring depth in the cerebral cortex was 300 µm. Electrodes were connected to a differential amplifier (Jens Meyer, Munich, Germany). Regional CBF, subarachnoid DC shift, and the voltage signals from the microelectrodes were continuously recorded using a personal computer and a chart recorder (DASH IV, Astro-Med, Inc, West Warwick, RI, USA).
A stimulus isolator in constant voltage mode (ISO Flex; AMPI Instruments), controlled by a Master-8 stimulator (AMPI Instruments, Jerusalem, Israel), delivered an electrical stimulus of 20 Hz for 10 s (200 pulses; width: 100 µs; 1.5–5 V) via a bipolar platinum stimulation electrode in groups 21 and 22.
Ouabain and L-NNA were solved in ACSF using sonication. Thapsigargin was first solved in 97% ethanol and then added to the ACSF. The resulting ethanol concentration in the ACSF was 0.1%. The closed window preparations allowed imaging of the pial arterioles with a CCD camera (Leica DFC300 FX Digital Color Camera, Leica Microsystems AG, CH-9435, Heerbrugg, Switzerland) connected to the microscope (Leica MZ16 FA, Leica Microsystems) using Leica Application Multifocus module software.
Induction of SD in vivo
In groups 4–17, a burr hole (ø: ∼2 mm) was drilled lateral to the temporal ridge and the dura mater was incised. There, a droplet containing KCl at 1 mmol/L was applied and rinsed shortly thereafter to trigger SD unless it had already occurred spontaneously (cf. results section). In groups 18, 23, and 24, SDs always developed spontaneously. In groups 21 and 22, SD was triggered in presence of ouabain (50 µmol/L) by electrical stimulation using the protocol above.
Supplemental methods
NaKA assay, Hb preparation, neocortical brain slice experiments as well as isolation and cannulation of the rat middle cerebral artery (MCA) are described in the Supplemental Methods.
Data analysis
Data were analyzed by comparing relative changes of rCBF and absolute changes of DC potential and [K+]o. Regional CBF changes were calculated in relation to baseline (=100%). A zero level was established at the end after global cerebral ischemia. All data in text and figures are given as median, first quartile, and third quartile. The statistical tests are given in the text. P < 0.05 was accepted as statistically significant. Experiments were only performed when windows were watertight and free of significant hemorrhage or air bubbles. Animals were excluded from analysis in rare cases when the animal died prematurely, e.g. due to a spontaneous blockade of the tracheal tube during the recording period, or when air bubbles accumulated in the closed cranial window interfering with recording procedures. No randomization was performed. Experimenters analyzed their data in an un-blinded fashion. In addition, data including raw data were reviewed and discussed with JD. The starting point of the study was not hypothesis-driven but the incidental finding that SI occurs in response to brain topically applied ouabain and L-NNA.
Results
The physiological variables remained within normal limits in all groups.
High baseline [K+]o selectively reduces α2/α3 NaKA activity
Elevated [K+]ACSF at 20 mmol/L selectively reduces α2/α3 NaKA activity.

Groups 2 and 3 were compared with control group 1 using one-way ANOVAs and post-hoc Bonferroni tests (***P < 0.001).
[K+]ACSF: K+ concentration of the artificial cerebrospinal fluid; NaKA: Na+/K+-ATPase; L-NNA: NG-nitro-L-arginine.
SD triggers SI when NaKA is inhibited and NO is absent
We then determined the dose-response curve of the shift from the normal to the inverse hemodynamic response to SD under increasing concentrations of ouabain (group 7: 0 mol/L; group 8: 500 nmol/L; group 9: 5 µmol/L; group 10: 50 µmol/L; each n = 7) topically co-applied with the NOS inhibitor L-NNA (1 mmol/L) to the brain (Figures 1–3). We also determined the dose-response curve to ACSF containing increasing concentrations of ouabain (group 12: 0 mol/L; group 13: 5 µmol/L; group 14: 50 µmol/L; each n = 7) in presence of the NO scavenger Hb (∼2 mmol/L) (Figure 3). Furthermore, we determined the dose-response curve of the shift from the normal to the inverse hemodynamic response to SD under increasing concentrations of Hb (group 6: 0 mol/L; group 15: 230 [190, 250] µmol/L; group 14: 2.05 [1.94, 2.17] mmol/L; each n = 7) topically co-applied with ouabain (50 µmol/L) (Figure 3). The effect of Hb (∼2 mmol/L) was approximately equivalent to the effect of L-NNA (1 mmol/L). The effect of ouabain (5 µmol/L) in presence of L-NNA (group 9; n = 7) was approximately equivalent to the effect of elevated [K+]ACSF (20 mmol/L) in presence of L-NNA (group 11, n = 8) (Figure 3). In absence of an NO-lowering agent, topical application of ouabain at either 5 µmol/L (group 5; n = 7) or 50 µmol/L (group 6; n = 7) did not shift the normal to the inverse hemodynamic response to SD (Figure 3).
Representative examples of an SD associated with SH under control conditions (a) and an SD associated with SI under L-NNA (1 mmol/L) and ouabain (50 µmol/L) (b). The dotted lines indicate the duration of ischemia and hyperemia, respectively. The down-going vertical arrow in panel (b) helps the reader to perceive the time delay, which occurs between the onsets of SI at the two spatially separated laser-Doppler flow probes. This time delay is explained by the propagating nature of SD/SI in the tissue. It may be added that the time course in (b) shows at least four different events of SD/SI, which are melting into each other. Acute SD-induced vasospasm of the microcirculation is the process underlying SI and can be observed through the microscope as reported previously.
9
However, here, SI was induced by topical application of ACSF to the brain containing L-NNA (1 mmol/L) in combination with ouabain (50 µmol/L) instead of elevated [K+]ACSF. It is seen that SI starts rostrally (right part of the photos) and spreads caudally (left part of the photos). Arrows mark a pial artery. Decreased rCBF is reflected by whitening of the cortex, which expands with the SD. Below the photos of the cortical surface, the time course of the changes in rCBF (laser-Doppler) at both the rostral and the caudal part of the window and the changes in the DC potential are displayed. The numbered dotted lines match the numbers of the photos above. Comparison of DC and rCBF changes due to inhibition of NaKA and/or NO depletion (groups 4–15). For each parameter, all groups were statistically compared with the control group using an ANOVA on ranks and post-hoc Dunnʼs tests. Significant differences are marked with an asterisk. Notably, in all groups characterized by pharmacological inhibition of NaKA activity in combination with NO depletion, the durations of hypoperfusion and SD were significantly prolonged.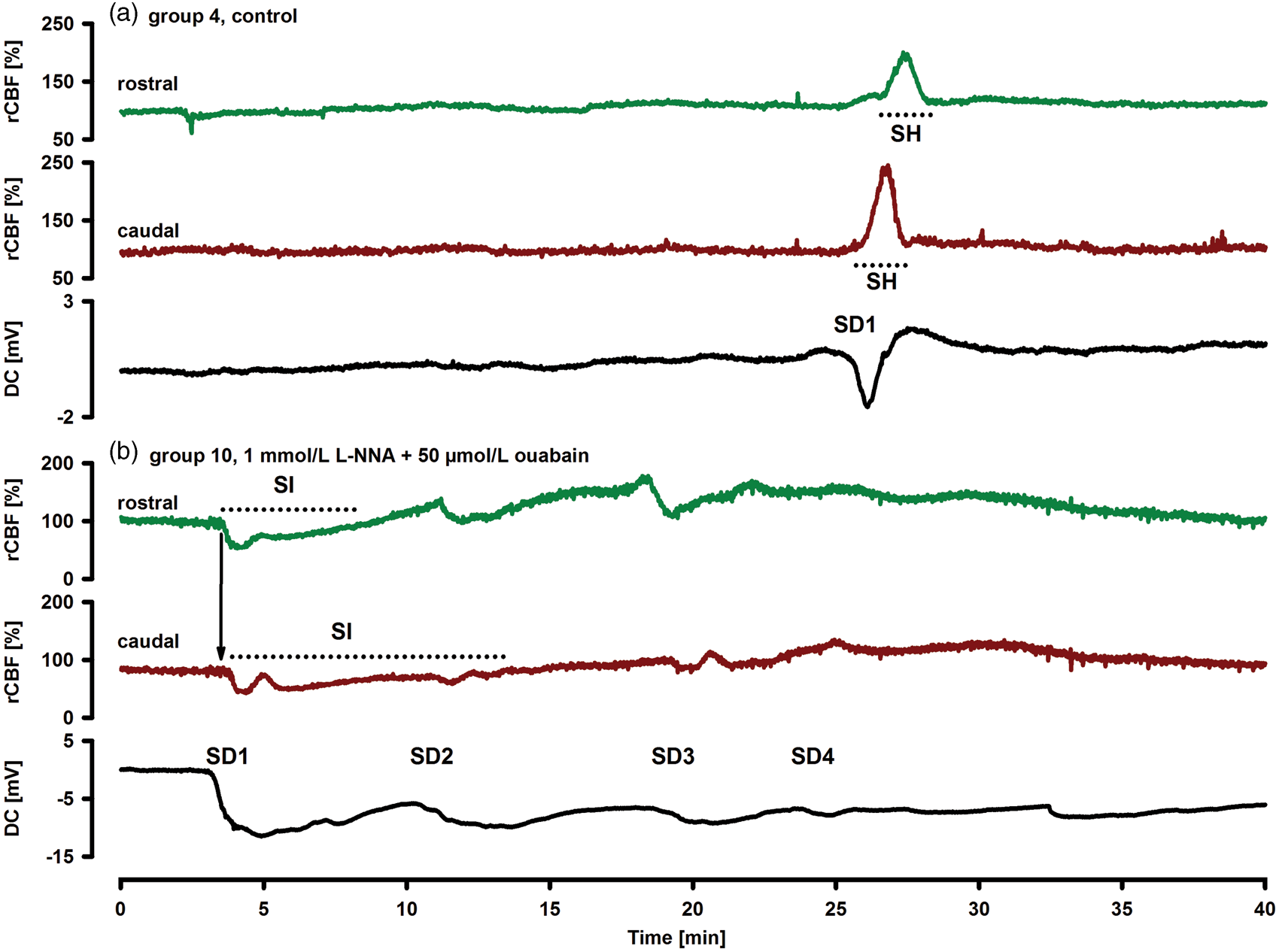
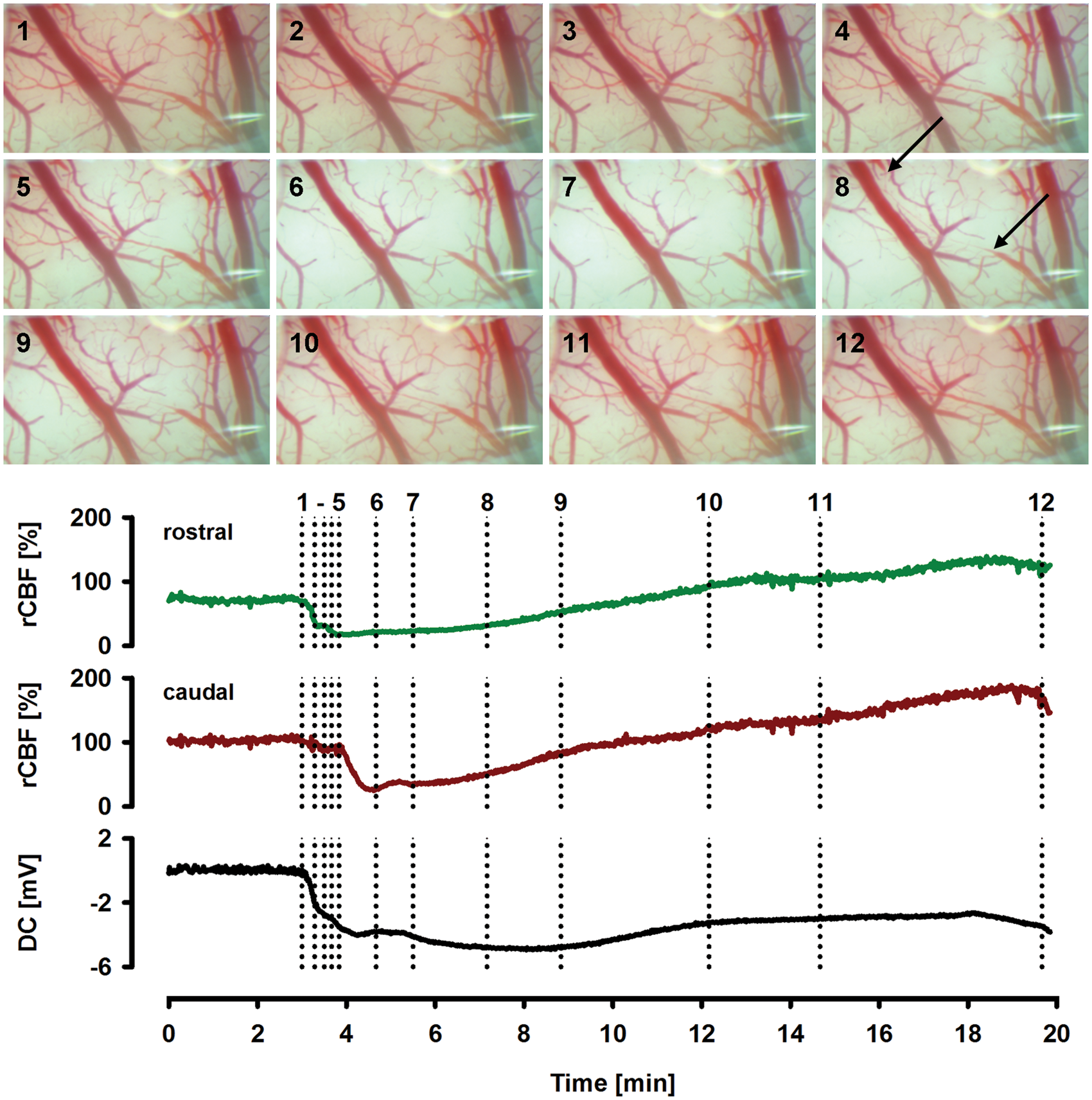
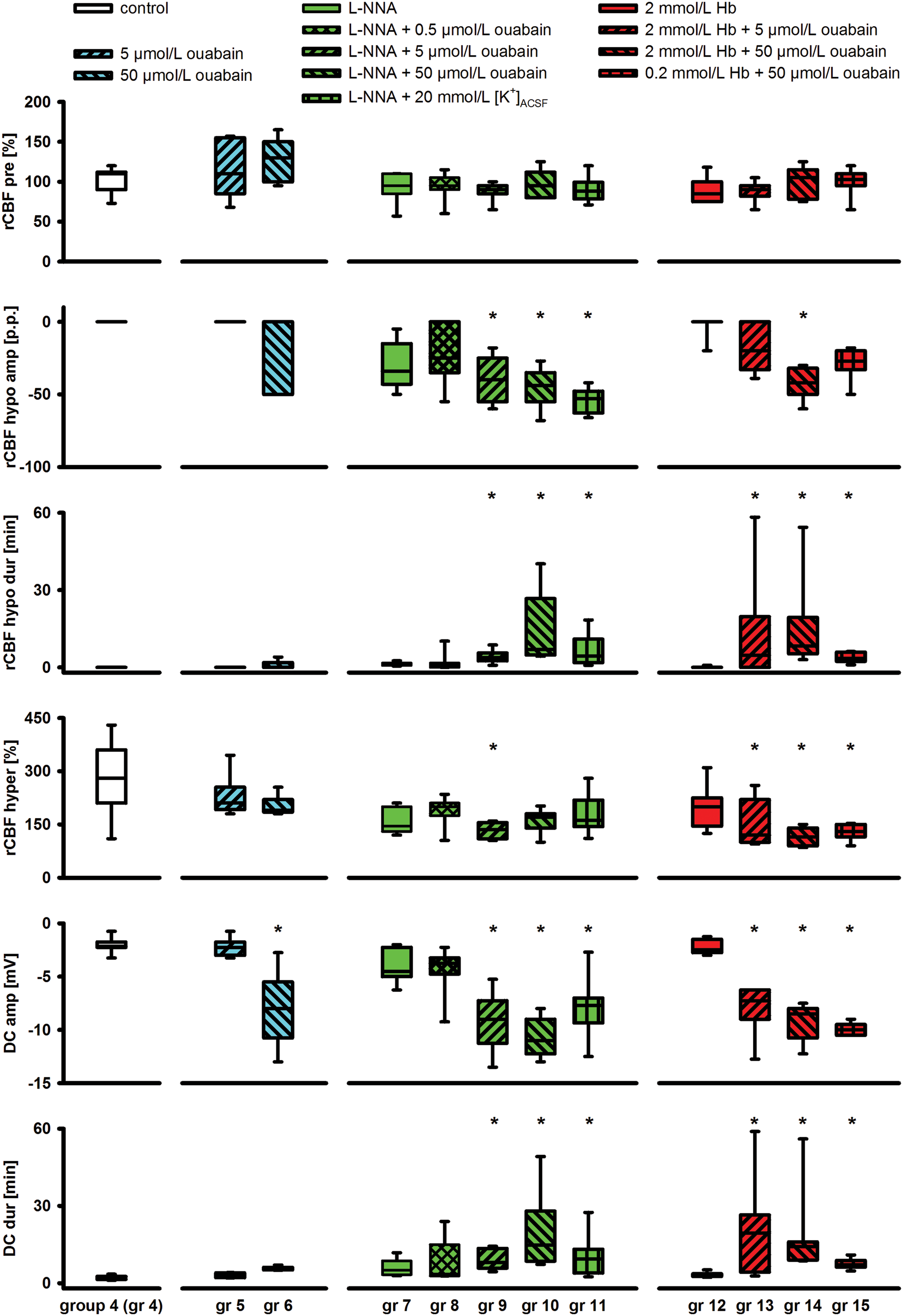
Topical application of ouabain (5 µmol/L) (groups 5, 9, 13) did not entail spontaneous SDs. Therefore, it was artificially triggered by KCl at a small open window. By contrast, ouabain (50 µmol/L) (groups 6, 10, 14, 15) led to one or more spontaneous SDs in practically all experiments similar to 100 µmol/L in rat hippocampal slices. 21 All 11 experimental groups were compared to a control group in which physiological ACSF was applied to the brain throughout the experiment and SD was artificially triggered by KCl at a small open window (group 4; n = 7; ANOVA on Ranks with post-hoc Dunn's tests versus control group; Figure 3).
Similar to previous studies in animals and patients,5,22,23 linear regression analysis found that the durations of initial hypoperfusion (rCBFhypo Dur in Figure 3) and negative DC shift (DC Dur in Figure 3) significantly correlated (a) when ACSF containing both elevated [K+]ACSF (20 mmol/L) and L-NNA (1 mmol/L) was topically applied (group 11, n = 8, adjusted R2 = 0.697, P = 0.006, n = 7), or (b) when either L-NNA (groups 7–10, adjusted R2 = 0.857, P < 0.001, n = 28) or Hb (groups 12–15, adjusted R2 = 0.917, P < 0.001, n = 28) were topically co-applied to the brain with increasing concentrations of ouabain. Notably, baseline rCBF before SD was not reduced in any of the groups which displayed SI (Figure 3).
Effect of elevated [K+]ACSF and ouabain, respectively, on baseline [K+]o in vivo
Effects of increasing concentrations of ouabain (group 16) and [K+]ACSF (group 17/18) on baseline [K+]o in vivo.
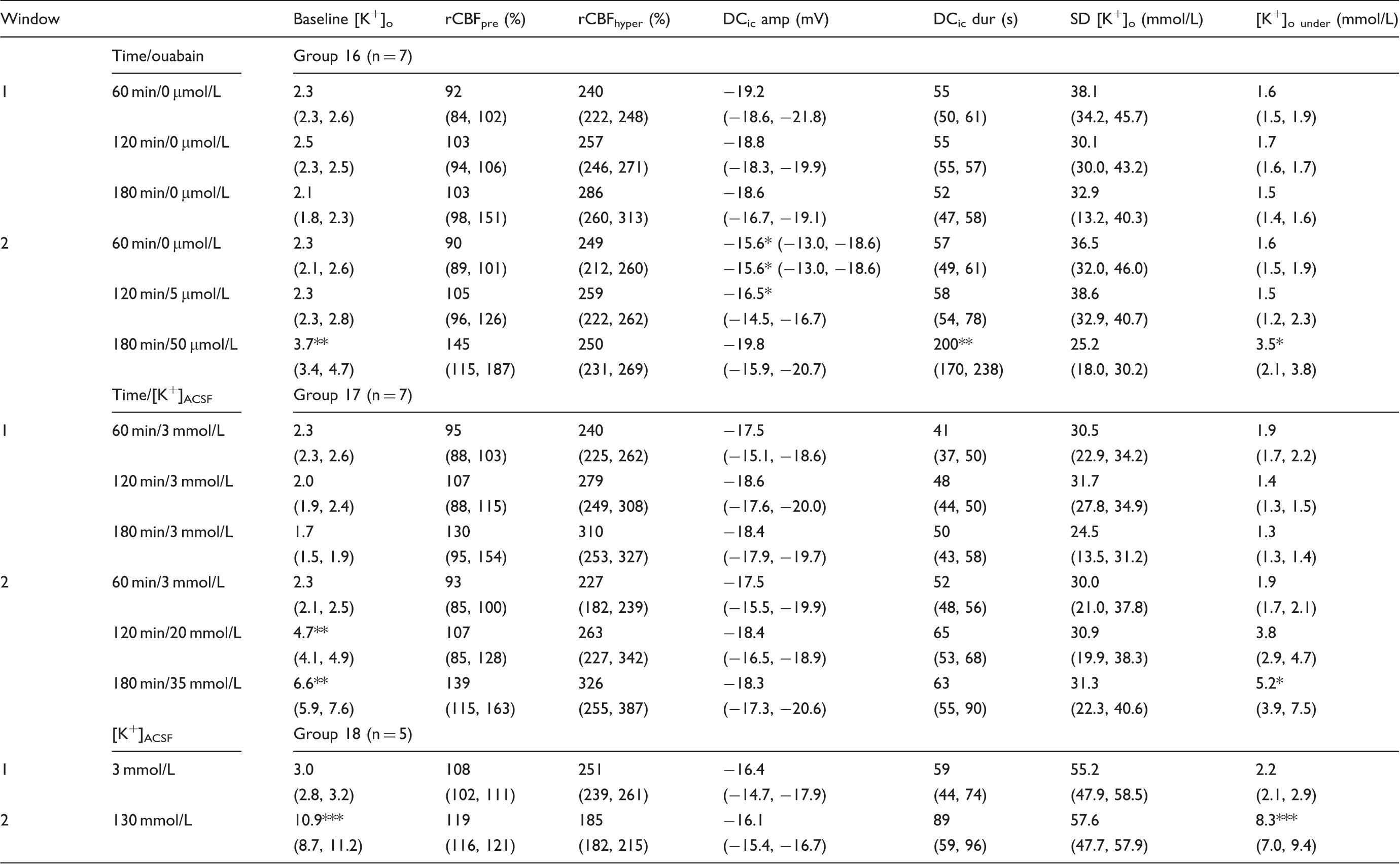
In groups 16 and 17, SD was elicited with KCl and propagated to two cranial windows where rCBF, [K+]o, and the intracortical DC potential were recorded. Group 16: Whereas window 1 was superfused with physiological ACSF throughout the experiments, ouabain was raised in a stepwise manner at 60 min intervals in window 2. Group 17: Similar to group 16, window 1 was superfused with physiological ACSF throughout the experiments. In window 2, [K+]ACSF was added to the ACSF. [K+]ACSF was raised every 60 minutes. Group 18: Window 1 was superfused with physiological ACSF whereas [K+]ACSF was raised to 130 mmol/L at window 2. SD occurred spontaneously in all five animals and propagated from window 2 to window 1. In groups 16 and 17, data were compared between window 1 and 2 at corresponding time periods with either paired T-test or Wilcoxon-signed rank test (*P < 0.05; **P < 0.01; ***P < 0.001). The same tests were used in group 18.
Baseline [K+]o: baseline [K+]o immediately before SD; rCBFpre: rCBF level immediately before SD; rCBFhyper: highest rCBF level during transient hyperemia; DCic amp: amplitude of intracortical DC shift during SD; DCic dur: duration of negative intracortical DC shift during SD; SD [K+]o: highest extracellular K+ concentration during SD; [K+]o under: extracellular K+ concentration immediately after SD (“undershoot”). [K+]ACSF: K+ concentration of the artificial cerebrospinal fluid; rCBF: regional cerebral blood flow; SD: spreading depolarization; DC: direct current; ACSF: artificial cerebrospinal fluid.
Whereas baseline [K+]o remained unchanged under ouabain (5 µmol/L) in group 16, it significantly increased to 3.7 (3.4–4.7) mmol/L under ouabain (50 µmol/L) (Table 2). Elevation of [K+]ACSF from 2.8 to 20 and to 35 mmol/L significantly increased baseline [K+]o to 4.7 (4.1–4.9) and 6.6 (5.9–7.6) mmol/L while baseline [K+]o remained unchanged at the control window (Table 2). In absence of an NO-lowering agent, [K+]ACSF (35 mmol/L) did not cause spontaneous SDs consistent with previous observations.10,24
In group 18 (n = 5), physiological ACSF was topically applied at the control window, whereas [K+]ACSF was increased to 130 mmol/L at the second window. While baseline [K+]o remained unchanged in the control window, it rose to 10.9 (8.7–11.2) mmol/L in the second window before the first spontaneous SD occurred. This level of baseline [K+]o was significantly higher than that immediately before the first SD under ouabain (50 µmol/L) in group 16 and before the remotely triggered SD under [K+]ACSF at 35 mmol/L in group 17, respectively (P < 0.001 and P < 0.01, one-way ANOVA with Bonferroni post-hoc tests, Table 2).
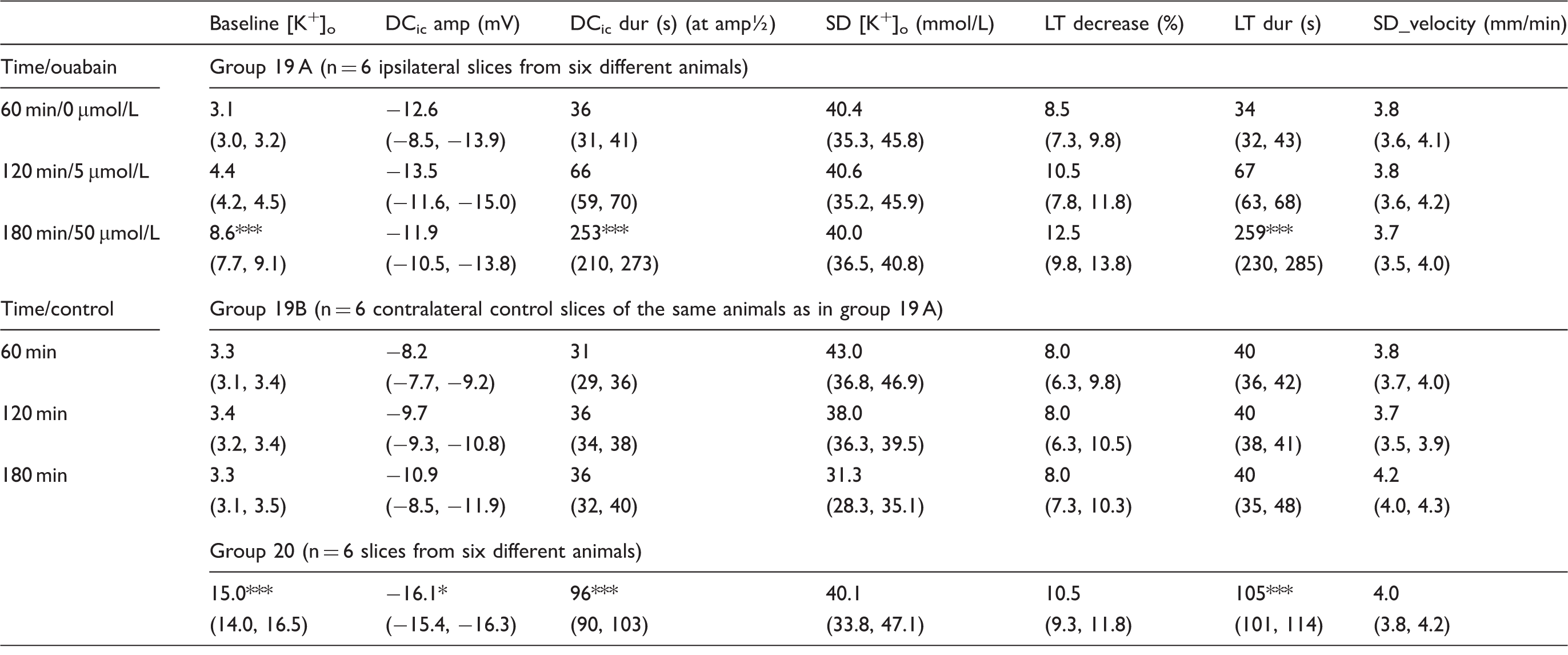
Further, we performed analogous experiments in rat neocortical slices to gauge the potential influence of the circulation on [K+]o in vivo, via, e.g. siphoning of K+ to capillaries. In these experiments, SD was recorded electrographically and optically in 12 slices from six different animals (Figure 4). The first SD was triggered by KCl microinjection during perfusion with physiological ACSF (group 19A, n = 6 slices from six different animals, Table 3). Subsequently, the slices were perfused with ACSF containing ouabain (5 µmol/L). This induced a slow increase of baseline [K+]o to 4.4 (4.2, 4.5) mmol/L. After 60 min, another SD was triggered by KCl microinjection. Then, ouabain was raised to 50 µmol/L, which induced a further gradual rise of baseline [K+]o to 8.6 (7.7, 9.1) mmol/L. SD occurred spontaneously in five slices under ouabain (50 µmol/L) within 60 min, and was triggered by KCl microinjection in the remaining one. Notably, ouabain (50 µmol/L) significantly prolonged the duration of the negative DC shift (Table 3). In the control group 19B, three SDs were triggered in the corresponding slice of the contralateral hemisphere during perfusion with physiological ACSF (n = 6) (Table 3). No differences were observed between the three subsequent SDs.
(a) Induction of SD by stepwise increases in [K+]ACSF. Arrows indicate increases in [K+]ACSF. (b) SD propagation in rat brain slice: Changes in IOS depicting depolarized tissue are marked with blue color. The SD was induced by microinjection of K+. The K+ threshold concentration for SD induced by ouabain (group 19 A) is lower than the K+ threshold concentration for SD induced by [K+]ACSF (group 20) in neocortical rat brain slices. In group 19, SDs were elicited with KCl microinjection unless they spontaneously occurred under ouabain at 50 µmol/L (n = 5 of 6). In group 19 A, only the first SD occurred under control conditions. Thereafter, ouabain was added to the ACSF as indicated in the table. In group 19B, all SDs occurred under control conditions. Data within groups 19A and 19B were compared with one-way repeated measures ANOVA and Bonferroni-test or Friedman repeated measures ANOVA on ranks and Tukey test. In group 20, [K+]ACSF was raised in steps of 2.5 mmol/L at 60 min intervals until SD was recorded. Data of group 20 were compared with those of spontaneous SD caused by ouabain at 50 µmol/L in group 19A using T-tests as described in the body text (*P < 0.05; **P < 0.01; ***P < 0.001). Baseline [K+]o: baseline [K+]o immediately before SD; DCic amp: amplitude of intracortical DC shift during SD; DCic dur: duration of negative intracortical DC shift at half-maximal amplitude during SD; SD [K+]o: highest extracellular K+ concentration during SD; LT decrease: decrease of light transmission during SD; LT dur: duration of light transmission change at half maximal amplitude during SD; SD velocity: velocity of SD propagation as measured at different regions of interest; [K+]ACSF: K+ concentration of the artificial cerebrospinal fluid; SD: spreading depolarization; DC: direct current; LT: light transmittance; ACSF: artificial cerebrospinal fluid.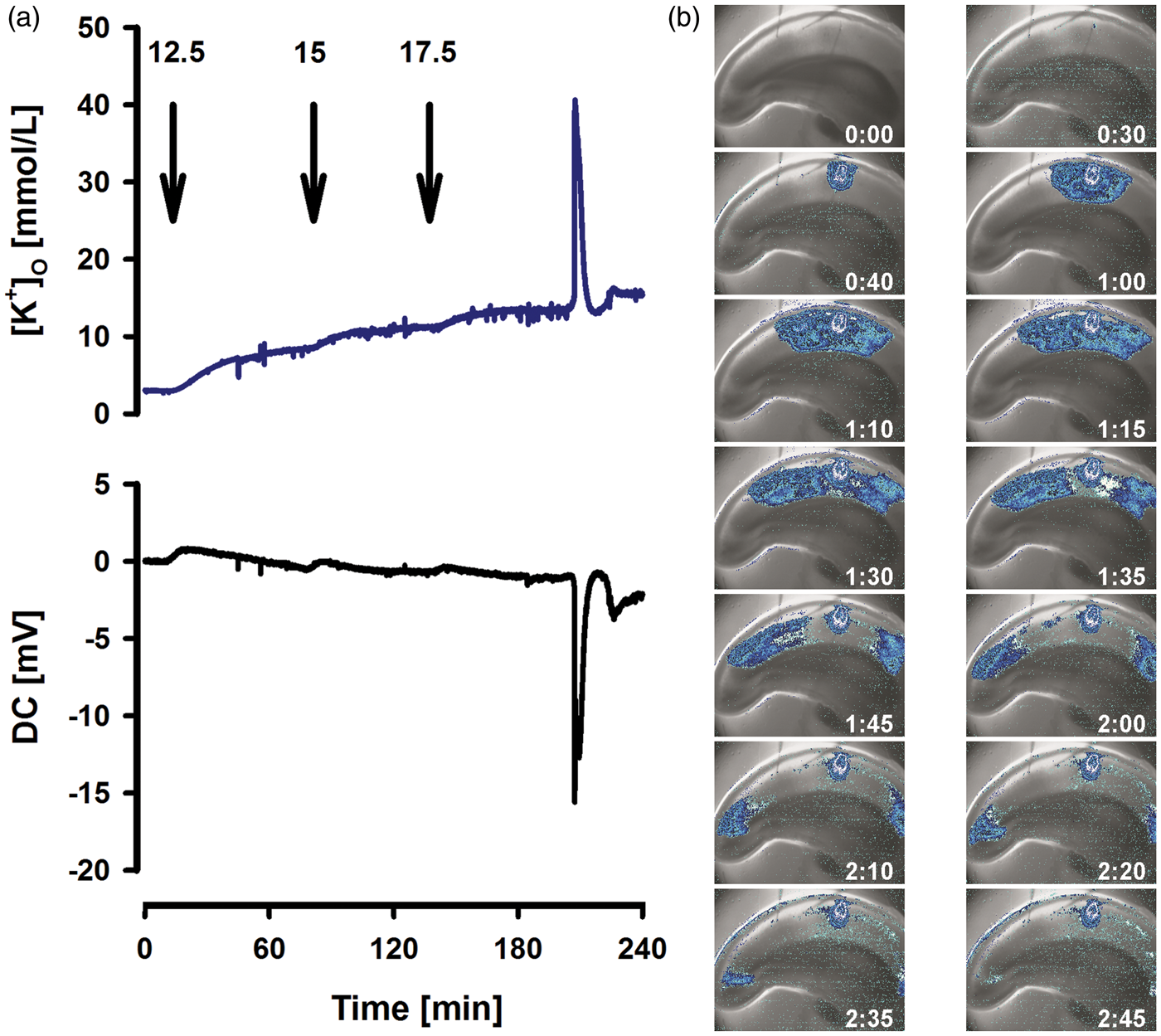
Dose-response curve of [K+]ACSF inducing SD in rat neocortical slices
In group 20 (n = 6), [K+]ACSF was raised in rat neocortical slices from six different rats in steps of 2.5 mmol/L at 60 min intervals until SD occurred (Figure 4). It was not necessary to raise [K+]ACSF beyond 17.5 mmol/L for this purpose. A baseline [K+]o level of 15.0 (14.0, 16.5) mmol/L was measured immediately before the first SD occurred. Similar to the in vivo findings, baseline [K+]o immediately before the first spontaneous SD in vitro was significantly higher under high [K+]ACSF (group 20) than under ouabain (50 µmol/L) (group 19A) (T-test, P < 0.001). SD under high [K+]ACSF was characterized by a negative intracortical DC shift of −16.1 (−15.4, −16.3) mV, lasting for 96 (90, 103) s at half-maximal amplitude (T½max). The duration of the negative DC shift was significantly longer than that under physiological conditions in group 19B, suggesting that high baseline [K+]o interfered with the repolarization after SD (P < 0.001, T-test) (Table 3).
Co-application of NOS inhibitor and ouabain inverts the hemodynamic response to SD but not the hemodynamic response to electrical stimulation
There may be mechanistic overlaps between the hemodynamic response to SD and the hemodynamic responses to functional neuronal activation, electrical stimulation or ictal epileptic activity. Therefore, we tested if simultaneous inhibition of NaKA and NOS inverts the rCBF response to direct electrical stimulation of the cortex in an open window model (group 21, n = 6).
Under physiological conditions, a 20 Hz stimulus train for 10 s resulted in a negative intracortical DC shift of –3.1 (−2.2, −4.0) mV associated with a rise of [K+]o from 2.7 (2.6, 2.9) to 6.5 (6.0, 8.3) mmol/L and a local rise of rCBF from 101% (97%, 113%) to 193% (170%, 240%). Topical application of 1 mmol/L L-NNA for 90 min significantly prolonged the time needed for rCBF to reach 90% of its maximum value from 6 (5, 9) to 16 (12, 18) s, suggesting a role of NO in the initial phase of the hemodynamic response to electrical stimulation (one-way repeated measures ANOVA and Tukey post-hoc tests, P < 0.01). There was no significant difference between L-NNA alone and the combination of L-NNA and ouabain (5 µmol/L) when stimulated 60 min later. The last stimulation under continued topical application of L-NNA and ouabain (50 µmol/L) to the brain for 40 min resulted in an initial rise of rCBF at the rostral laser probe from 94% (75%, 129%) to 114% (80%, 178%) in parallel with a stimulus-induced negative intracortical DC shift of –3.4 (−3.2, −3,6) mV and rise in [K+]o from 3.5 (2.5, 4.3) to 8.9 (6.5, 4.2) mmol/L. Superimposed on these changes, stimulation triggered SD to which SI was coupled (Figure 5(a), Table 4). This implies that, despite the presence of L-NNA (1 mmol/L) and ouabain (50 µmol/L), no inverse but rather a hyperemic rCBF response was triggered by the electrical stimulus-induced rise of [K+]o which remained below the ceiling level of ∼12 mmol/L. Only during the subsequent SD, the hemodynamic response became inverted.
(a,b) Comparison of the hemodynamic responses to electrical stimulation and to SD in vivo. Stimulation with a 20 Hz pulse for 10 s induced an increase in rCBF under physiological conditions. Following treatment with ACSF containing ouabain (50 µmol/L), electrical stimulation induced first an increase in rCBF before it triggered SD both in absence of NO under treatment with L-NNA (1 mmol/L) (a) and in presence of NO (b). During the electrically triggered SD, rCBF then showed a marked and prolonged decrease when NO was absent (=SI) (a) whereas it further increased in presence of NO (=spreading hyperemia) (b Co-application of the NOS inhibitor L-NNA with ouabain (50 µmol/L) only inverts the coupling between neuronal activation and rCBF if SD occurs. Data are given for spreading ischemia (SD/SI, group 21) and spreading hyperemia (SD/SH, group 22). A K+-sensitive microelectrode, a bipolar stimulation electrode and a laser-Doppler flow probe were positioned rostrally and a laser-Doppler probe caudally. SD/SI and SD/SH were triggered electrically. Group 21 was compared with group 22 using Student’s t-test or Mann–Whitney rank-sum test (*P < 0.05; **P < 0.01). Baseline [K+]o: value immediately before stimulation in presence of ouabain at 50 µmol/L; rCBFpre: rCBF level immediately before stimulation; rCBFstim: rCBF level in response to stimulation before onset of SD; rCBFhypo: lowest rCBF level during initial hypoperfusion in response to SD; rCBFhypo dur: duration of rCBFhypo; rCBFhyper: highest rCBF level during transient hyperemia following SD; rCBFhyper dur: duration of rCBFhyper; rCBF delay: delay between onsets of rCBF responses to SD at the two laser-Doppler probes; DCic amp: amplitude of the intracortical DC shift during SD; DCic dur: duration of negative intracortical DC shift during SD; SD [K+]o: highest extracellular K+ concentration during SD; [K+]o under: extracellular K+ concentration immediately after SD (“undershoot”); NOS: nitric oxide synthase; L-NNA: NG-nitro-L-arginine; rCBF: regional cerebral blood flow; SD: spreading depolarization; DC: direct current; SI: spreading ischemia; SH: spreading hyperemia.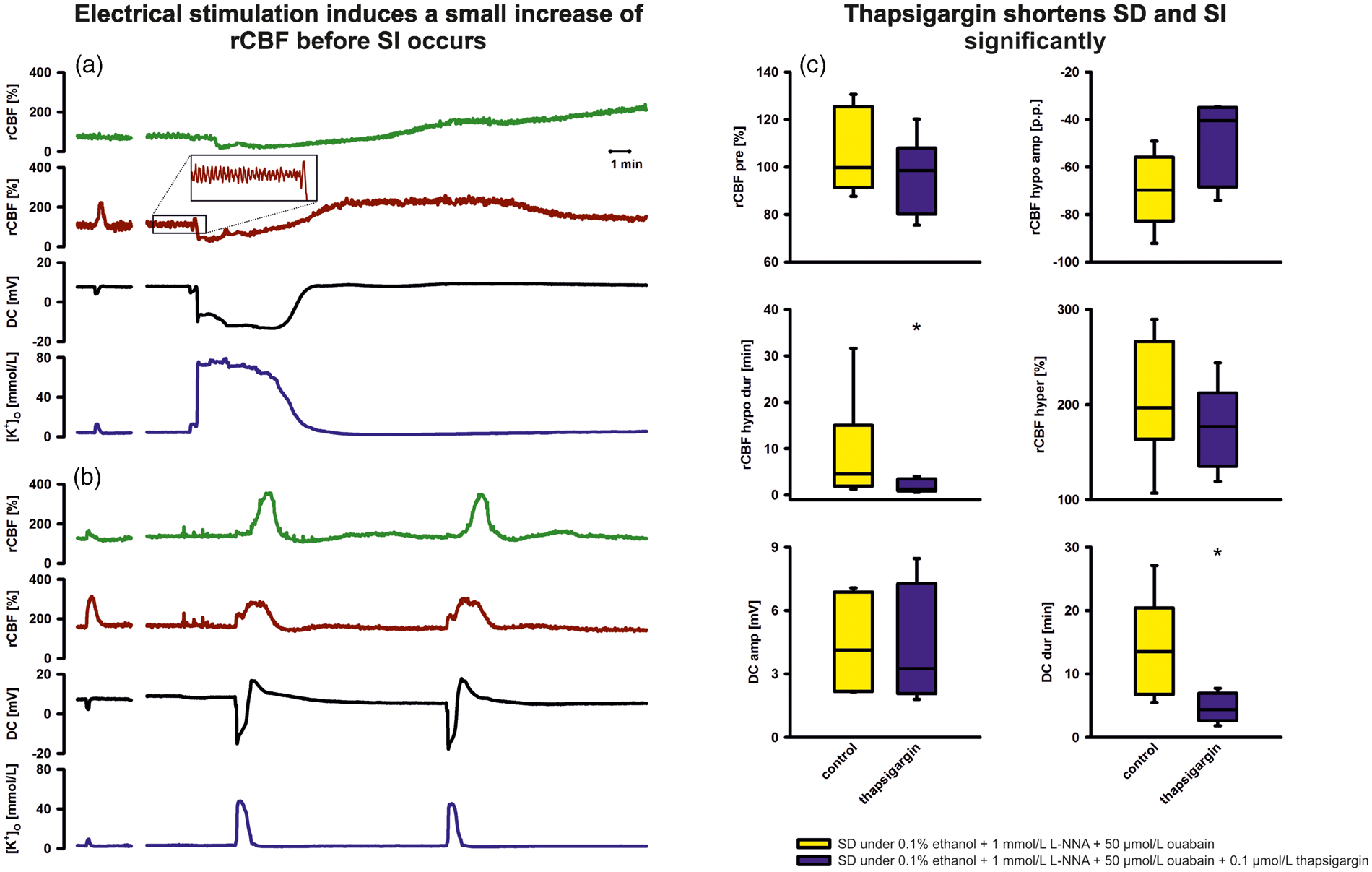
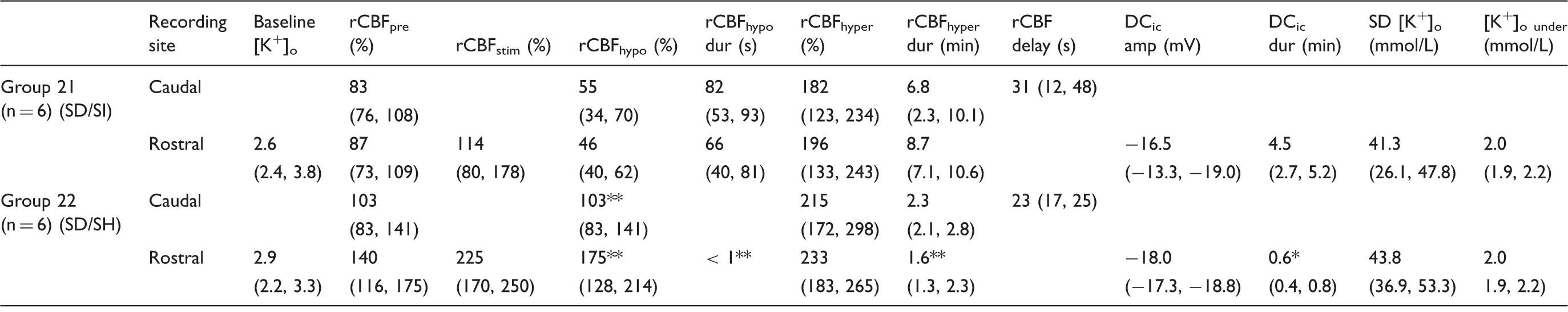
Without L-NNA, there was no significant change between first and second stimulation under physiological ACSF and the third stimulation after wash-in of ouabain (5 µmol/L) (group 22, n = 6). Stimulation 40 min after wash-in of ouabain (50 µmol/L) resulted in a negative intracortical DC shift of −4.3 (−2.1, −4,5) mV associated with a rise of [K+]o from 3.2 (2.1, 3.3) to 8.0 (6.8, 10.0) mmol/L and rCBF increase from 140% (116%, 175%) to 225% (170%, 250%). Superimposed on these changes, SD occurred to which a spreading hyperemia was coupled (Figure 5(b), Table 4).
In addition, group 21 confirms the conclusion from the experiments in groups 16–20 that ouabain did not substantially increase baseline [K+]o at concentrations triggering the switch from spreading hyperemia to SI. Indeed, it can be seen in Table 4 that baseline [K+]o even remained within the physiological range until the electrical stimulus triggered the SD to which SI was coupled. Further, [K+]o fully returned to the physiological range after the SD. This suggests that L-NNA did not modify the effect of ouabain on baseline [K+]o.
SI and intracellular Ca2+ stores
The α2/α3 isoforms of NaKA are important regulators of intracellular Ca2+ signaling. 25 Decreased α2 activity in vascular smooth muscle cells (VSMC) and astrocytes thus augments Ca2+ signaling secondary to enhanced Ca2+ storage in the junctional sarco(endo)plasmic reticulum (jSER). Therefore, we investigated the role of intracellular Ca2+ stores for SI using thapsigargin. Thapsigargin is an inhibitor of the sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA), 26 which leads to depletion of intracellular Ca2+ stores by inhibition of refilling. 27 Thapsigargin was topically co-applied to the brain with L-NNA (1 mmol/L) and ouabain (50 µmol/L) in a closed cranial window preparation (group 23, n = 6). Sixty minutes after application of ACSF containing L-NNA, thapsigargin (100 nmol/L) was added to the ACSF. Another 30 min later, ouabain (50 µmol/L) was added on top. Application of thapsigargin did not prohibit SI, but significantly shortened the durations of both hypoperfusion and negative DC shift compared with a vehicle control (group 24, n = 6), in which ACSF was applied containing ethanol (0.1%), L-NNA (1 mmol/L), and ouabain (50 µmol/L) (Figure 5(b) and Supplemental Table 1).
Effects of ouabain on the isolated MCA
It was previously described that extracellular ionic changes during SD (=cocktailSD) cause dilation of the isolated, pressurized MCA in absence of NOS inhibition but constriction in its presence corresponding with the shift from spreading hyperemia to SI in vivo. 16 In a similar fashion, cocktailSD was extraluminally applied in group 25 (n = 5) of the present study first in absence and then in presence of L-NNA (10 µmol/L). This produced the same responses as observed previously (Supplemental Figure 1). By contrast, cocktailSD was extraluminally applied first in absence and then in presence of L-NNA (10 µmol/L) plus ouabain (5 µmol/L) in group 26 (n = 5). Overall, the additional effect of ouabain was small (upper graph in Supplemental Figure 1). Ouabain only augmented the vasoconstrictor response to cocktailSD under NOS inhibition when the response was analyzed in relation to the dilator response to cocktailSD in absence of NOS inhibition, which corrects for the differences in vascular reactivity between individual MCAs (P = 0.003, n = 5 in both groups, Two-Way Repeated Measures analysis of variance (ANOVA) [One Factor Repetition] with Bonferroni post-hoc test, lower graph in Supplemental Figure 1).
Discussion
The protective role of NO against SI
NO depletion alone was not sufficient to evoke full-blown SI in previous studies.9,28,29 Yet, no experimental protocol has been found so far, which resulted in SI but did not cause NO depletion at the same time.8,30 Existing experimental protocols resulting in SI caused NO depletion either by NOS inhibition, NO scavenging, 9 or lack of molecular oxygen.11–13 The latter causes NO depletion because molecular oxygen is needed as a substrate in the synthesis of NO by all three isoforms of NOS to donate one oxygen atom to form L-citrulline and the other one to form NO from L-arginine. Accordingly, hypoxia has been shown to decrease both (a) NO synthesis by attenuation of NOS activity 15 and (b) acetylcholine-induced endothelium-dependent relaxation. 31 The present data support the notion that NO depletion is a necessary condition for SI because ouabain merely triggered the switch from spreading hyperemia to SI under simultaneous NO depletion. Admittedly, Hb also shows a plethora of other actions than NO depletion.32–34 Yet, the good agreement with the results observed under the otherwise very different drug L-NNA suggests that NO scavenging is the relevant action of Hb concerning SI.
NO is ideally situated to fight vasoconstrictor effects of increases in cytosolic free Ca2+ ([Ca2+]cyt) since in both neuronal NOS, expressed by neurons, astrocytes, and vascular myocytes, and endothelial NOS, expressed by endothelial cells and astrocytes, calmodulin binding is brought about by an increase in [Ca2+]cyt with half-maximal activity between 200 and 400 nmol/L. When calmodulin affinity to NOS rises by the Ca2+/calmodulin interaction, it facilitates the flow of electrons from NADPH in the reductase domain to the heme in the oxygenase domain, thereby NO synthesis increases. 35 SD causes a large increase in [Ca2+]cyt in various cell types. 36 Accordingly, it was found using NO-sensitive dyes that SD induces NO synthesis in neurons and endothelial cells. 24
Relaxing actions of NO on vascular myocytes include: (a) cGMP-mediated desensitization of the contractile machinery to Ca2+ by activation of myosin phosphatase, 37 (b) activation of Ca2+-activated K+ channels,38,39 (c) voltage-independent inhibition of L-type Ca2+ channels via cGMP-dependent protein kinase,40,41 and (d) inhibition of Ca2+ release from internal stores elicited by inositol trisphosphate, 42 cADP-ribose, 43 or caffeine. 44 However, the antagonistic effect of NO on SI not only involves direct vasodilatior but also permissive actions. The conceptual term permissive describes that a certain level of NO is necessary (a) to support other vasodilators such as protons or prostaglandin E2 to unfold their effects45,46 and/or (b) to blunt the action of vasoconstrictors such as high [K+]o. 16 Consistent with a permissive effect, it was previously found that the NO donor S-nitroso-N-acetylpenicillamine (SNAP) caused SI to revert to an almost normal hyperemic response to SD in presence of NOS inhibition. 18
Beyond being a vasodilator, NO is also a signaling molecule in neural networks. It is interesting in this respect that NO depletion caused marked reduction in the K+ threshold of SD both in vivo and in neocortical slices.10,24,47 Further, it increased the propagation velocity of SD in the isolated chicken retina. 48 We here investigated whether this could be due to NO depletion-induced decline in NaKA activity because (a) reduced NaKA activity led to a lower electrical threshold of SD in genetically engineered mice carrying the human W887R mutation in the ATP1A2 orthologous gene, resulting in loss of function of the mutant α2 isoform, 49 and (b) physiological in contrast to toxic levels of NO seem to activate NaKA via increase of its apparent Na+ affinity. 50 However, our coupled enzyme assay did not support this hypothesis since reduced NaKA activity by elevated [K+]ACSF was similar in presence and absence of NOS inhibition. This rather supported previous observations that loss of cGMP-independent modulatory effects of NO on P/Q-type voltage-gated Ca2+ channels and N-methyl-D-aspartate receptor (NMDAR)-controlled channels are responsible for the reduced K+-threshold. 47 Our finding may also imply that the physiological regulation of NaKA by NO is canceled when elevated [K+]ACSF down-regulates α2/α3 activity.
Elevated [K+]ACSF causes decline in α2/α3 NaKA activity
Apart from NO depletion, the shift from spreading hyperemia to SI requires that a second condition is fulfilled, such as presence of the NaKA inhibitor ouabain in the current paper or elevated baseline [K+]o in previous ones.8,9 NaKA is an αβ heterodimeric transmembrane complex expressed in virtually all animal cells. The catalytic subunit, α, contains the Na+, K+, ATP, and ouabain binding sites. In rodent brain, three different α isoforms (α1-α3) have been described. Whereas α1 represents the housekeeping enzyme expressed by all cells, most cell types additionally express NaKA with another α isoform: astrocytes, endothelial cells, and VSMCs express α2 whereas most neurons express α3. 25
Interestingly, Hajek and colleagues reported that prolonged increase of [K+]o significantly reduced NaKA activity in astrocytic culture. Therefore, we investigated whether NaKA activity is reduced by increase of [K+]ACSF to a level that was previously found to induce SI. 9 We found a selective reduction of α2/α3 NaKA activity by ∼50%. The reduction in total NaKA activity by ∼33% was only slightly above the value of 20% reported in astrocytic culture. Our result also corresponded well to a previous study, in which decline of total NaKA activity by 39% was observed 1h after focal cerebral ischemia in mice. 51 Similar to reduced NaKA activity by elevated [K+]ACSF, the one by ischemia (a) did not result from loss of high-energy phosphate levels (evidenced by the fact that the enzyme assay was performed under optimal biochemical conditions, including an excess of ATP concentration), and (b) was entirely due to reduced activity of the α2/α3 fraction. No modifications in protein and mRNA expression of any isoforms were observed, but suppression of enzyme activity rather resulted from modifications of specific active sites, trapping the enzyme in an inactive phosphorylated state. 52
Possibly, the mechanistic basis of reduced NaKA activity by ischemia and elevated baseline [K+]o is of similar nature because ischemia entails first a rise of [K+]o in core and penumbra from 3 to a value lower than the ceiling level of ∼12 mmol/L. This is due to activation of neuronal ATP-sensitive or G protein–dependent Ca2+-sensitive K+ channels. 14 Only 1–5 min later, the first SD occurs which is accompanied by further rise in [K+]o to ∼50 mmol/L. Depolarization and rise in [K+]o are often persistent in the core while they are progressively shorter-lasting from the penumbra toward surrounding well-perfused tissue. However, even if tissue recovers from the first SD, [K+]o typically remains elevated in the penumbra in a range between 3 and ∼12 mmol/L. During further SDs, [K+]o increases again to ∼50 mmol/L in both penumbra and surrounding well-perfused tissue. The whole cerebral hemisphere rather than the ischemic zone alone thus undergoes repeated marked increases in [K+]o during ischemia, which could potentially lead to down-regulation of α2/α3 NaKA activity.
Evidence that elevated [K+]ACSF contributes to SI via reduced α2/α3 NaKA activity
Ouabain directly inhibits plasmalemmal NaKA in a variety of cell types. In rats, the half maximal inhibitory concentration is 48 µmol/L for α1, 58 nmol/L for α2, and 6.7 nmol/L for α3. 53 α2 and α3 can thus be distinguished from the α1 isoform by their higher affinity to ouabain. 54 To determine whether reduced α2/α3 activity is a candidate mechanism by which elevated [K+]ACSF contributes to SI, we depleted NO similar to previous studies,9,16 but replaced [K+]ACSF (20 mmol/L) with ouabain (5 µmol/L), and found that it led to SI in similar fashion.
In additional in vivo experiments, we investigated the effect of ouabain (5 µmol/L) on baseline [K+]o and did not find any increase. Even the higher concentration of 50 µmol/L increased baseline [K+]o to only ∼3.7 mmol/L in absence of NOS inhibition and did not increase baseline [K+]o in presence of NOS inhibition. By contrast, [K+]ACSF (20 mmol/L) increased baseline [K+]o to 4.6 mmol/L although ouabain (50 µmol/L) induced spontaneous SDs in contrast to [K+]ACSF (20 mmol/L). In the neocortical slice experiments, ouabain increased baseline [K+]o to somewhat higher levels than in vivo. This may result from (a) a shorter distance between ACSF flooded surface and electrode tip in the tissue and (b) less efficient buffering of K+ in brain slices because they have a smaller volume and lack an intact circulation. Nonetheless, also in slices, the ouabain-induced rise was markedly smaller than the [K+]ACSF-induced rise in baseline [K+]o. Further, the level of baseline [K+]o from which spontaneous SDs arose under ouabain (50 µmol/L) was markedly lower than the one from which they arose under elevated [K+]ACSF. These findings suggest that elevated baseline [K+]o is not the primary mechanism of ouabain by which it triggers the switch from spreading hyperemia to SI.
Intracellular Ca2+ stores are necessary to maintain SI
An important cause for the roles of the different α subunit isoforms of NaKA has been given by their subcellular localization. Whereas α1 NaKA is ubiquitously distributed in the plasma membrane, excluding microdomains adjacent to the jSER, α2/α3 NaKAs are restricted to exactly these microdomains. 55 The plasmalemmal Na+/Ca2+-exchanger (NCX) is confined to the same microdomains similar to different transient receptor protein canonical (TRPC) proteins as components of receptor- and store-operated cation-selective channels. 56 In mesenteric artery VSMCs, the functional interrelationship of these transport proteins was supported by the observation that α2 (but not α1) NaKA, NCX1, TRPC1, and TRPC6, were all upregulated by prolonged ouabain application both in vivo and in vitro. 56 Altogether, these structures form the plasmerosome as a functional unit, in which the plasma membrane and the jSER are separated by only 12 to 20 nm. As a consequence, diffusion of Na+ and Ca2+ between plasmerosome and bulk cytosol is restricted. Standing Na+ and Ca2+ concentration gradients between plasmerosome and bulk cytosol can thus be maintained. 25
The ubiquitous distribution of α1 NaKA in the plasma membrane led to the hypothesis that it controls, primarily, the intracellular Na+ concentration in the bulk cytosol ([Na+]cyt) whereas α2/α3 NaKAs regulate the local Na+ concentration in the plasmerosome. The differential regulation of the Na+ concentration in plasmerosome and bulk cytosol is determined by the lower affinity of α2/α3 NaKAs than α1 NaKA for Na+, which favors the relative increase of the local Na+ concentration in the plasmerosome. In this concept, selective inhibition of α2/α3 activity by exogenous or endogenous ouabain should cause only limited rise in [Na+]cyt and a comparably small depolarization. 57 The local decline in the electrochemical gradient for Na+ in the plasmerosome, however, would affect the Ca2+ transport by the neighboring NCX. The NCX would be either deactivated or even switched into reverse mode so that Ca2+ is taken up instead of released. 58 This organizational arrangement in the plasmerosome would link cellular Na+ and Ca2+ regulation. Notably, the secondary, NCX-mediated redistribution of Ca2+ may only slightly increase [Ca2+]cyt but SERCA is activated, which pumps Ca2+ into the jSER. 59 As a result of Ca2+ sequestration in the jSER, a new steady state would be achieved with a substantially augmented Ca2+ store. The additionally stored Ca2+ would then be available for mobilization whenever the cell is activated. The jSER would thus effectively act as a Ca2+ amplifier when, for example, α2/α3 NaKAs are selectively inhibited by ouabain. 60
Augmented Ca2+ stores may shift vasodilator to vasoconstrictor responses
[Ca2+]cyt is a key signaling mechanism in virtually all cells. For example, VSMCs and pericytes are electrically coupled, transmitting Ca2+ signals between arterioles and venules dependent on gap junctional communication and Ca2+ influx through L-type Ca2+ channels. Decrease in [Ca2+]cyt of myocytes causes vasorelaxation and increase vasoconstriction. In response to constrictor stimuli, increase in [Ca2+]cyt results from (a) opening of L-type Ca2+ channels in the plasma membrane and/or (b) the release of Ca2+ to the cytosol by internal stores such as the jSER. Therefore, it is plausible that augmented Ca2+ stores by reduced α2 activity could contribute to SI because more Ca2+ is released to the cytosol of VSMCs in response to SD. The same may apply to pericytes because they also contract in response to Ca2+ release from the jSER and seem to express α2 NaKA.46,61 Accordingly, the SERCA inhibitor thapsigargin, emptying the jSER Ca2+ store, significantly shortened SI under ouabain and NOS inhibition in the present study.
[Ca2+]cyt is moreover a key signaling mechanism of astrocytes; astrocytic processes encase more than 90% of the surface area of intracerebral arterioles. 62 Under physiological conditions, astrocytes cause vasodilation via Ca2+-dependent activation of BK channels 63 and K+ release from astrocytic endfeet. Inward rectifier VSMC K+ channels are in turn activated, VSMC membrane hyperpolarizes and voltage-dependent L-type Ca2+ channels close if the local rise in [K+]o remains below ∼20 mmol/L. At this point, it should be noted that microelectrode recordings of [K+]o may not accurately reflect perivascular [K+]o because their tip diameter is ∼3 µm and the distance between astrocytic endfeet and VSMC is merely on the order of 100 nm. Perivascular [K+]o during SD could thus be lower than the values measured in the sample volume around the electrode tip.
However, transition between astrocyte-evoked vasodilation and vasoconstriction occurs when the intraastrocytic Ca2+ response approximately doubles from normally 300–400 nmol/L to 700–800 nmol/L at the endfeet. 64 Under this condition, BK channel activation is strongly enhanced because BK channels show a 16-fold increase in open probability when [Ca2+]cyt doubles. 65 As a consequence, local [K+]o in the restricted perivascular space may exceed 20 mmol/L. This will (a) depolarize vascular myocytes, (b) activate L-type Ca2+ channels, and (c) cause contraction. 64 In relationship with aSAH and the inverse hemodynamic response, this mechanism is interesting because enhanced BK channel activity in response to increased activity-induced astrocytic [Ca2+]cyt oscillations converted vasodilation in control neocortical slices to vasoconstriction in slices from rats that had been previously subjected to SAH. 66
Augmented Ca2+ signaling in astrocytes may also enhance release of arachidonic acid from astrocytes, which is metabolized to the vasoconstrictor 20-HETE in VSMCs. 67 Correspondingly, 20-HETE was shown to increase during SD in animals. 68 Also, 20-HETE is interesting in relationship with aSAH and the inverse hemodynamic response, because it was associated with delayed cerebral ischemia (DCI) in aSAH patients, 69 clusters of recurrent SDs with prolonged depression of spontaneous activity indicated the occurrence of DCI in human electrocorticographic recordings, 70 and such clusters displayed the inverse hemodynamic response to SD more often than isolated SDs.5,7,71
Ouabain and endothelial-derived hyperpolarizing factor (EDHF)
Endothelium expresses α2 NaKA as well, but augmented endothelial Ca2+ signaling should antagonize VSMC and pericyte contraction because endothelial Ca2+ signaling inhibits VSMC and pericyte Ca2+ signaling via NO and EDHF. 72 Interestingly, however, ouabain (1–10 µmol/L) was previously reported to antagonize the endothelium-dependent hyperpolarizing effect of vasodilators such as acetylcholine or protease-activated receptor 2 ligand on VSMCs of rat and cat MCAs.73,74 This would favor VSMC depolarization, activation of L-type Ca2+ channels and contraction. In other words, the above claim suggests that both important dilator responses evoked through endothelial Ca2+ mobilization are abolished when reduced activity of high-affinity NaKAs meets NO depletion.
In our experiments in the isolated, pressurized MCA, the overall effect of ouabain under simultaneous NOS inhibition was small. This corresponds well with earlier work in absence of NOS inhibition in which ouabain showed no significant effects on membrane potential and diameter in isolated, pressurized MCAs unless they were stimulated by endothelium-dependent vasodilators. 74 We also observed in correspondence with earlier work that extraluminal application of ionic cocktail, matching previously measured ion changes during SD, led to marked dilation in absence and constriction in presence of NOS inhibition. 16 [K+]ACSF (50 mmol/L) had been previously identified as predominant constrictor of the ionic cocktail in this in vitro model of SI, acting primarily through direct depolarization of VSMCs by reduced K+ equilibrium potential and activation of L-type Ca2+ channels. 16 However, in contrast to SD in vivo, high [K+]ACSF rises only slowly in this model and may thus cause simultaneous downregulation of high affinity NaKAs. Accordingly, ouabain merely augmented the vasoconstrictor response to the ionic cocktail under NOS inhibition when the response was analyzed in relation with the dilator response to the cocktail in absence of NOS inhibition. However, even though this effect of ouabain was relatively small, it was still significant, and could possibly result from both augmented internal Ca2+ mobilization and blockade of EDHF.
Clinical implications
DCI is the most prominent in-hospital complication following aSAH, and is presumably caused by products of erythrocytes in the subarachnoid space. Clinical signs of DCI are global and focal neurological deficits, which lead to significant deterioration in functional outcome. 75 The delayed ischemic lesions predominantly affect brain cortex.
Aneurysmal SAH is a prototypical disease in which a prolonged state of reduced NO availability is measured in the patient cortex before DCI occurs. 76 NO scavenging by Hb in the subarachnoid blood clot, endogenous NOS inhibitors, degeneration of perivascular nitrergic nerves, uncoupling of endothelial NOS, reactive oxygen species, or rho kinase activation have all been accused to contribute to NO depletion following aSAH.8,77,78 Because SI was discovered in animals under NO depletion,9,79 it was investigated and found in patients with aSAH. 5 This calls for better characterization of this phenomenon in animals with the ultimate goal to improve therapeutic options.
Notably, prophylaxis with the L-type Ca2+ antagonist nimodipine reduced frequency and severity of DCI in patients and is the only approved medication for this indication. Experimentally, nimodipine antagonized SI similar to an NO donor,9,16,80 whereas it had no effect on angiographic vasospasm in aSAH patients. 81 This suggests that SI is a potential pharmacological target by which nimodipine improves the clinical outcome after aSAH. 8 Unfortunately, the clinically applicable dose of nimodipine is limited because it is a potent antihypertensive.
ACSF containing elevated [K+]ACSF in combination with NO depletion has been the original protocol in animals resulting in SI.9,79 In view of the current concepts on NaKA and Ca2+ regulation, the present findings support the hypothesis that (a) elevated [K+]ACSF decreases α2/α3 NaKA activity; (b) decreased α2/α3 NaKA activity causes augmented Ca2+ release from the jSER during SD; (c) augmented Ca2+ release from the jSER produces severe contraction of vascular myocytes and/or pericytes under the condition that the basal level of NO is low and [Ca2+]cyt-evoked increases of [NO] are suppressed. Further, ouabain may contribute to VSMC depolarization and activation of L-type Ca2+ channels because it counteracts EDHF.73,74
Conclusion
SI is clinically important in different conditions including aSAH5–7,82 but exceedingly complex, involving dysregulation in all cell types of the neurovascular unit8,30 and all segments of the vascular tree from large arteries 83 up to capillaries. 84 The physiological level of NO shows preventive effect, pursuing a defense-in-depth strategy that fights the occurrence of SD in the first place, and second, aims at maintaining the normal hyperemic response in the case that SD occurs. This supports the therapeutic approach in patients with aSAH to sustain the basal level of NO.76,78,85,86 Beyond NO depletion and plasmalemmal Ca2+ entry via L-type Ca2+ channels also Ca2+ release from internal stores seems to contribute to SI, providing a new therapeutic target which could potentially complement nimodipine prophylaxis against DCI. The caveat is added, though, that thapsigargin is not suitable for use as a drug since SERCA inhibition eventually leads to apoptosis in any mammalian cell. 87 Another limitation is that the concept of intracellular Ca2+ regulation by high affinity NaKAs has been partially based on other vascular beds than the cerebral circulation.25,56 More research is thus needed to investigate these mechanisms in the brain.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Deutsche Forschungsgemeinschaft (DFG DR 323/6-1), the Bundesministerium für Bildung und Forschung (Center for Stroke Research Berlin, 01 EO 0801) and Era-Net Neuron 01EW1212 to Dr. Dreier and DFG PE1193/2-1, Else-Kröner Fresenius Foundation 2010_A16 and German Center for Neurodegenerative Diseases (DZNE) to Dr. Petzold.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors' contributions
Sebastian Major: designed and performed experiments, analyzed data, drafted and finalized the manuscript and approved the manuscript before submission. Gabor C. Petzold: designed and performed experiments, analyzed data, contributed to the manuscript and approved the manuscript before submission. Clemens Reiffurth: performed experiments, analyzed data, contributed to the manuscript and approved the manuscript before submission. Olaf Windmüller: designed and performed experiments, analyzed data, contributed to the manuscript and approved the manuscript before submission. Ute Lindauer: designed experiments, analyzed data, contributed to the manuscript and approved the manuscript before submission. Marco Foddis: performed experiments, analyzed data, contributed to the manuscript and approved the manuscript before submission. Eun-Jeung Kang: analyzed data, contributed to the manuscript and approved the manuscript before submission. Jens P. Dreier: designed and performed experiments, analyzed data, drafted and finalized the manuscript and approved the manuscript before submission.
Supplementary material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.