Cortical spreading depression (CSD) is a temporary disruption of local ionic homeostasis that propagates slowly across the cerebral cortex. Cortical spreading depression promotes lesion progression in experimental stroke, and may contribute to the initiation of migraine attacks. The purpose of this study was to investigate the roles of the marked increase of nitric oxide (NO) formation that occurs with CSD. Microdialysis electrodes were implanted in the cortex of anesthetized rats to perform the following operations within the same region: (1) elicitation of CSD by perfusion of high K+ medium; (2) recording of CSD elicitation; (3) application of the NO synthase inhibitor, NG-nitro-l-arginine methyl ester (l-NAME); and (4) recording of dialysate pH changes. The primary effect of l-NAME (0.3 to 3.0 mmol/L in the perfusion medium) was a marked widening of individual CSD wave, resulting essentially from a delayed initiation of the repolarization phase. This change was due to NO synthase inhibition because it was not observed with the inactive isomer d-NAME, and was reversed by l-arginine. This effect did not appear to be linked to the suppression of a sustained, NO-mediated vascular change associated with the superposition of NO synthase inhibition on high levels of extracellular K+. The delayed initiation of repolarization with local NO synthase inhibition may reflect the suppression of NO-mediated negative feedback mechanisms acting on neuronal or glial processes involved in CSD genesis. However, the possible abrogation of a very brief, NO-mediated vascular change associated with the early phase of CSD cannot be ruled out.
Cortical spreading depression (CSD) is a transient suppression of electrical activity, resulting from a temporary disruption of local brain ionic homeostasis that propagates slowly across the cerebral cortex (Bureš et al., 1974; Martins-Ferreira et al., 2000). Experimental and clinical data suggest that CSD may be the underlying cause of migraine attacks, even when they are not preceded by aura symptoms (Cao et al., 1999; Lauritzen, 1994). Stroke is another neurologic disorder that may involve CSD, because recurrent periinfarct CSD promotes lesion progression in experimental models of focal ischemia (Hossmann, 1996; Obrenovitch, 1995).
Increased nitric oxide (NO) formation is likely to occur during CSD because NMDA-receptor antagonists suppress CSD (Marrannes et al., 1988; Obrenovitch and Zilkha, 1996), intracellular Ca2+ levels increase markedly during CSD (Hansen and Zeuthen, 1981), and NMDA-receptor activation leads to a large increase in Ca2+/calmodulin-dependent nitric oxide synthase (NOS) activity (Garthwaite and Boulton, 1995). A CSD-induced NO release was confirmed by real-time measurements of NO in the cortex of anesthetized cats, which showed a rapid and sustained increase of NO levels for the duration of K+-induced CSD activity (Read et al., 1997; Read and Parsons, 1998).
Another possible role of NO release during CSD may be rapid feedback inhibition of processes that are involved in CSD genesis (e.g., ion channels, glutamate release), thereby contributing to minimize the duration of this potentially deleterious event. This is a plausible hypothesis because NO is very diffusible, the NO-cyclic GMP pathway is capable of subsecond operations (Bellamy and Garthwaite, 2001), and NMDA-receptors would be likely candidates because there is robust evidence for negative feedback of NMDA-receptors by endogenous NO (Manzoni and Bockaert, 1993; Moller et al., 1995; Tanaka et al., 1993).
The purpose of this study was to investigate the role(s) of NO release during CSD using an innovative experimental strategy that relied on (1) selective application of the NOS inhibitor l-NAME to the site of CSD elicitation; (2) comprehensive, quantitative analysis of both individual and repeated CSD; and (3) analysis of the recovery from acidosis to evaluate the possible involvement of NO-mediated vascular effects. We show that local NOS inhibition delays markedly the initiation of recovery from individual CSD wave. This effect does not appear to be a consequence of the suppression of any local, NO-mediated hyperperfusion associated with CSD.
MATERIALS AND METHODS
Animal preparation and intracerebral microdialysis
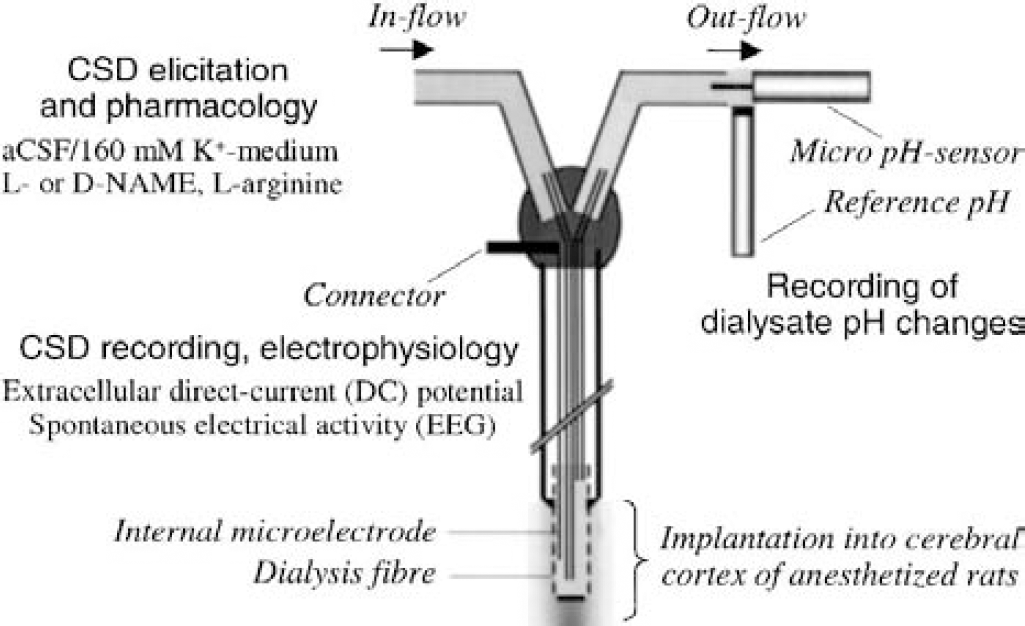
Experiments were performed on adult, male Sprague Dawley rats (weight [mean ± SD], 312 ± 27 g, n = 45; Harlan UK Ltd, Bicester, U.K.) with food and water available ad libitum. They were housed in the animal unit of our institution for at least 1 week before use. All animal procedures complied with the British Home Office guidelines, Animals (Scientific Procedure) Act of 1986. All efforts were made to minimize the number of animals used and their suffering. Animals were anesthetized throughout with halothane (5% for induction, 2.0% to 2.5% during surgery, and 1.5% to 1.75% for maintenance during the rest of the experiment) in 30:70 oxygen to nitrous oxide mixture while the animal breathed spontaneously. Concentric microdialysis probes incorporating a recording electrode (Obrenovitch et al., 1993; AN69 Hospal membrane, 0.25-mm outer diameter x 1-mm fiber length; type ME-H1, Applied Neuroscience Ltd., London, U.K.) were implanted in the frontoparietal cortex (coordinates: 1.3 to 1.5 mm anterior to bregma, 2 mm lateral, and 1.4 to 1.5 mm deep from the cortical surface) (Paxinos and Watson, 1986) and held in place with acrylic dental cement embedding two small stainless-steel skull screws used as anchor. Unless otherwise stated, microdialysis probes were perfused with aCSF (composition in millimoles per liter: 125 sodium chloride, 2.5 potassium chloride, 1.18 magnesium chloride, and 1.26 calcium chloride; pH 7.3 adjusted with 1-mol/L sodium hydroxide) at 1 μL/min with a syringe pump (CMA/100; CMA/Microdialysis, Solna, Sweden). These multifunctional devices allowed the following operations to be carried out simultaneously, within the same region of the cerebral cortex (Fig. 1): (1) reproducible elicitation of CSD by switching from normal aCSF to a medium containing 160-mmol/L K+ (K-medium); (2) recording of the extracellular DC potential to detect CSD initiation precisely at the site of its elicitation; (3) local application of the NOS inhibitor, l-NAME (alone or with l-arginine) or its inactive isomer d-NAME; and (4) recording of dialysate pH changes in one series. Restricting NOS inhibition to the site of CSD elicitation ruled out any possible interference from peripheral NOS inhibition. Indeed, drug-induced changes in arterial blood pressure could have altered the magnitude of hyperperfusion associated with K+-induced increase in brain energy demand, thereby influencing acid-base homeostasis (Taylor et al., 1994), and acidosis is known to inhibit both NMDA-receptor function (Traynelis and Cull-Candy, 1990; Urenjak et al., 1997) and CSD (Gardner-Medwin, 1981; Marrannes et al., 1985).
A simplified schematic representation of mitogen- and stress-activated signaling pathways discussed in this article. MAPK, mitogen-activated protein kinase; ERK, extracellular signal-regulated kinase; MEK, MAPK/ERK kinase; SAPK, stress-activated protein kinase; CREB, cyclic-AMP response element binding protein; HSP27, heat shock protein 27.
A femoral artery was catheterized for continuous monitoring of arterial blood pressure, and a vein for rapid induction of cardiac arrest on completion of the experimental procedure. To minimize any possible interference of halothane anesthesia with the processes under study (Carla and Moroni, 1992; Schlame and Hemmings, 1995), once the surgical procedure had been completed, the depth of anesthesia was carefully controlled by monitoring the EEG and arterial blood pressure, and the concentration of halothane in the breathing mixture kept to a minimum (1.5% to 1.75%). It is relevant to note that halothane did not interfere with neuronal NOS-mediated control of CBF in a previous study (Zagvazdin et al., 1998). Body temperature was maintained at 37.0°C throughout the experiment.
Recording of extracellular DC potential and electroencephalogram
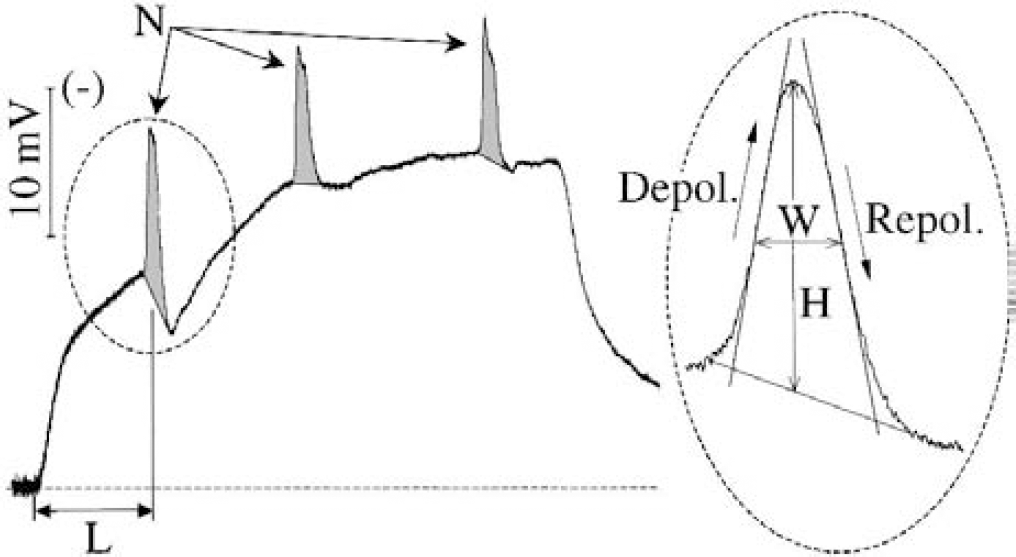
These electrophysiologic variables were derived from the potential between the electrode built into the microdialysis probe and a silver/silver chloride reference electrode placed under the scalp (Obrenovitch et al., 1993). This signal was first amplified with a high-impedance input, AC/DC preamplifier (NL834 or NL102; Neurolog System, Digitimer Ltd., Welwyn Garden City, U.K.). The alternating current component in the 1 to 30 Hz window (x5000–10000 overall amplification) provided the EEG, and the DC component (x250 overall amplification) the DC potential. All the recorded variables were continuously digitized, displayed, and stored using a personal computer equipped with an A/D converter. Cortical spreading depression is recognized as a transient, negative shift of the extracellular DC potential, superimposed on the sustained shift resulting from the imposed high extracellular K+ concentration (Fig. 2).
Representative changes in the DC potential produced by perfusion of high K+-medium through a microdialysis probe, and set of variables used for quantitative determination of the cortex sensitivity to K+-induced cortical spreading depression (CSD; main trace): number of CSD (N); cumulative CSD area (shaded area; in millivolts per minute); and latency for occurrence of first CSD (L, in minutes). The insert to the right shows the variables used to analyze drug effects on individual CSD waves: CSD height (H, in millivolts); width at 1/2 height (W, in seconds); maximum slope of depolarization (Sld, in millivolts per minute); and maximum slope of repolarization (Slr, in milllivolts per minute).
Recording of changes in dialysate pH
The CSD-induced pH changes in the dialysate were recorded as it emerged from the outlet of the implanted microdialysis probe (Fig. 1), by using a micro pH-sensor and separate reference electrode (MINI-Flex NMPH2 and Flex-ref, respectively; World Precision Instruments Ltd., Stevenage, U.K.). The sensitive tip of the pH electrode was inserted within the lumen of the probe outlet tube, and the reference positioned laterally in such a way that suitable contact with the emerging fluid was maintained throughout the recording period (Fig. 1). The potential difference between these two electrodes was amplified (x250 overall amplification; NL106 and NL530, Neurolog System, Digitimer Ltd.), digitized, and acquired together with the other variables for off-line analysis. The sensitivity of the pH sensor was linear within the pH range of 6.7 to 7.3, with a slope of approximately 24 mV per pH unit. The time response of the pH sensor was rapid (seconds) and, therefore, the limiting factor regarding time resolution of changes in dialysate pH was the microdialysis process.
Experimental procedures
Experiments were performed at least 2 hours after probe implantation. Cortical spreading depressions were triggered by switching the perfusion medium from normal aCSF to K-medium (composition in millimoles per liter: 2.5 sodium chloride, 160 K+, 1.18 magnesium chloride, and 1.26 calcium chloride; pH 7.3 adjusted with 1-mol/L sodium hydroxide) using a liquid switch (CMA/110, CMA/Microdialysis AB). Previous studies showed that such a high concentration of K+ is required to evoke consistent, periodic CSD under these experimental conditions (Herreras and Somjen, 1993; Obrenovitch and Zilkha, 1996; Szerb, 1991), possibly because of the efficient uptake of K+ by brain cells (Chen and Nicholson, 2000; Xiong and Stringer, 2000) and because a minimum volume of tissue (approximately 1 mm3) has to be depolarized for successful CSD elicitation (Koroleva and Bureš, 1996). Each K+ stimulus was followed by 40 minutes of recovery (i.e., perfusion of a normal K+ concentration medium). Three series of experiments were performed.
Series 1
Three groups were considered in this series (control, l-NAME, d-NAME), with a single microdialysis electrode implanted in each animal. Five episodes of repetitive CSD where produced by perfusion of K-medium for 20 minutes. L-or d-NAME (dissolved in aCSF and K-medium) was applied at the concentration of 0.3, 1, and 3 mmol/L during the second, third, and fourth CSD episode (in respective order), with 60 minutes of drug application started 20 minutes before the corresponding perfusion of K-medium.
Series 2
The purpose of this series was to determine whether the effects of NOS inhibition on CSD could be reversed by l-arginine, and to examine whether the same effects could be obtained without application of l-NAME before CSD initiation. Accordingly, l-NAME (with or without l-arginine) was only coapplied with K+ in these experiments. Because series 1 showed that the primary effect of l-NAME was a marked alteration of individual CSD, only a single CSD wave was triggered per K+ application (i.e. K-medium was perfused for 5 minutes only). To reduce the number of animals required, two microdialysis electrodes were implanted symmetrically in each animal of a single group. Five separate CSD were elicited on each side. During the second, third, and fourth CSD elicitation period, the K-medium was supplemented with 1-mmol/L l-NAME on one side, and with 1-mmol/L l-NAME plus l-arginine (10, 3, and 1 mmol/L, respectively) on the other. The range of l-arginine concentrations tested was reversed (i.e., highest concentration applied first) because l-NAME has a much higher affinity for NOS than l-arginine (Alderton et al., 2001) and series 1 showed that the effects of l-NAME on CSD persisted for more than 40 minutes (Fig. 3). Cortical spreading depression was not elicited simultaneously at each probe because we had observed in pilot experiments that CSD on one side of the cortex provoked a small negative shift of the contralateral DC potential. Therefore, one CSD was produced on one side, and 20 minutes within the following recovery period, CSD was then elicited contralaterally.
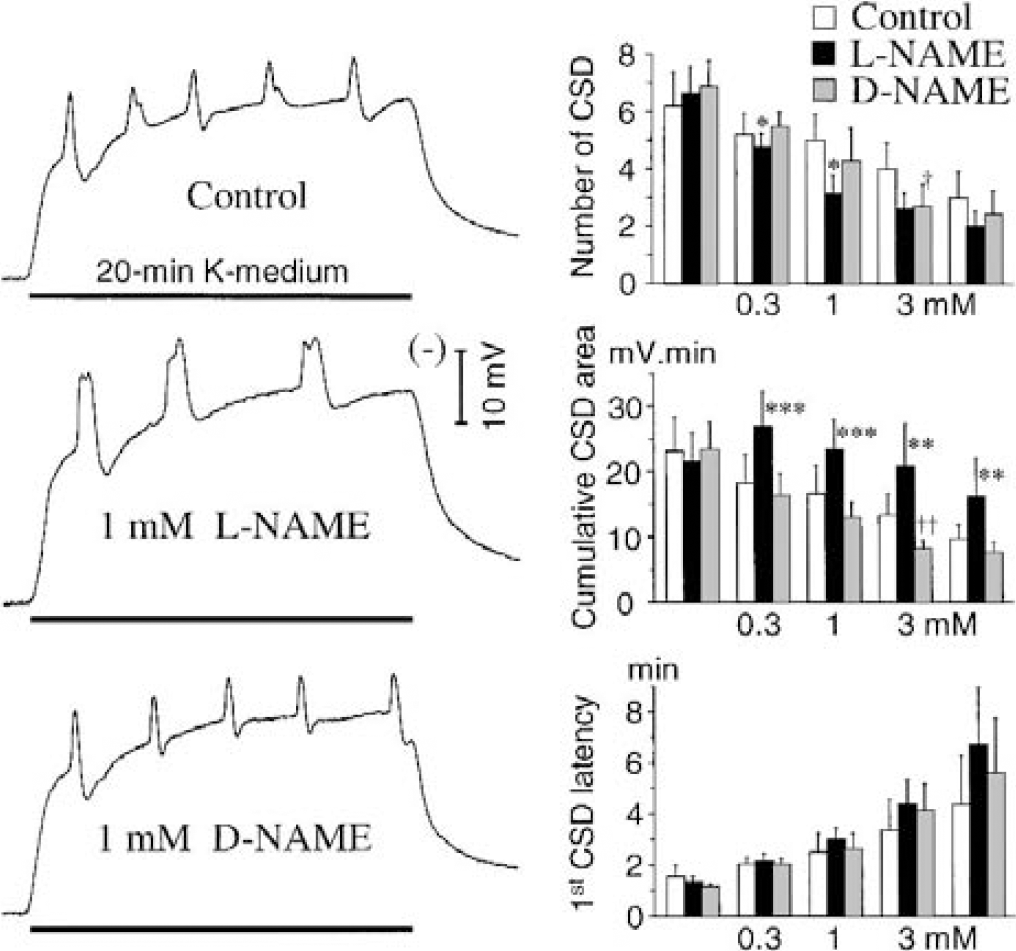
Effects of local nitric oxide synthase inhibition with l-NAME on repetitive cortical spreading depression (CSD) in the cerebral cortex of rats, and comparison with the inactive isomer d-NAME (series 1). Five separate episodes of repetitive CSD were produced by perfusion of 160-mmol/L K+ for 20 minutes through a microdialysis probe/electrode. In the l-NAME and d-NAME groups, 0.3, 1.0, and 3.0 mmol/L of the drug was coper-fused with high K+ during the second, third, and fourth challenge, respectively. The left traces are representative changes of the DC potential recorded with the third K+ stimulus. In agreement with previous reports, the polarity of the DC potential was reversed so that negative shifts (i.e., depolarization) produce an upward deflection. Values are means ± SD in the bar charts. Note the marked increase in the cumulative CSD area under nitric oxide synthase inhibition (l-NAME), and that this effect persisted when l-NAME was omitted from the last K+ stimulus. *P < 0.05, **P<0.01, ***P < 0.001, comparison of the l-and d-NAME groups; †P < 0.05, ††P < 0.01, comparison of the d-NAME and control groups; Student's t-test (n = 6-8).
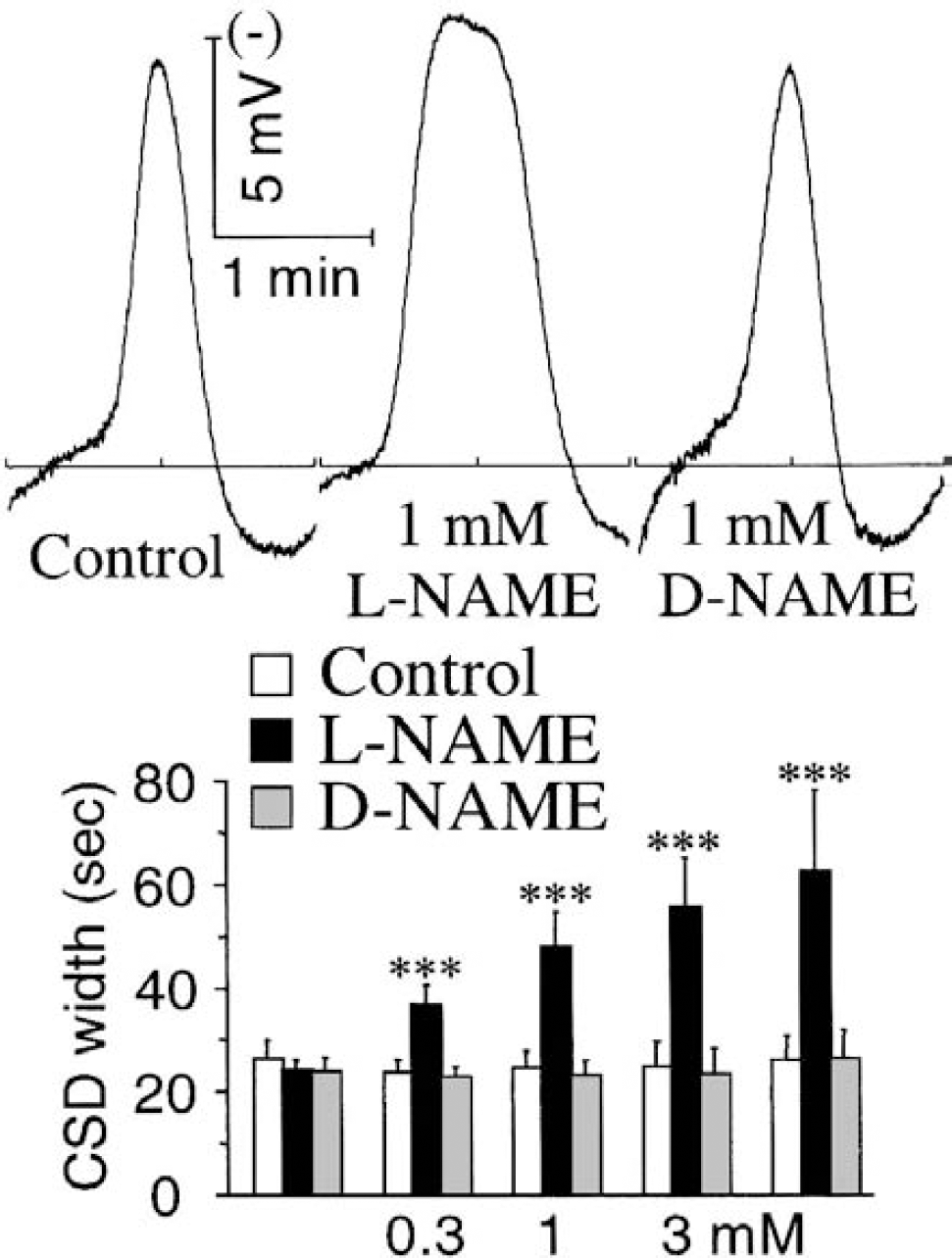
Effects of local nitric oxide synthase (NOS) inhibition with l-NAME on the pattern of the first individual cortical spreading depression (CSD) elicited by the high extracellular K+ concentrations (series 1; see Fig. 3 legend for methodologic details). The left traces are representative recordings of the first CSD wave obtained with the third K+ stimulus. Values are means ± SD in the bar chart. Note the marked increase in the width of individual CSD under NOS inhibition (l-NAME), with this effect persisting when l-NAME was omitted from the last K+ stimulus. ***P < 0.001, comparison of the l- and d-NAME groups; Student's t-test (n = 7 and 8).
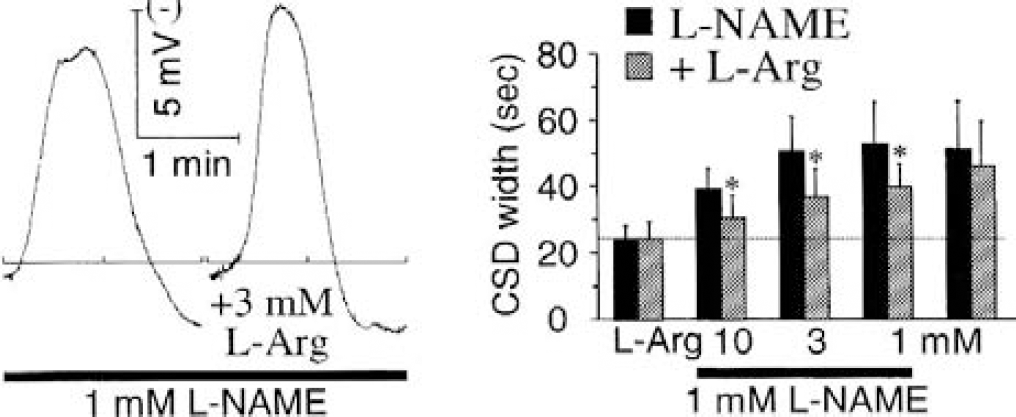
Effect of local nitric oxide synthase (NOS) inhibition with l-NAME on individual K+-induced cortical spreading depression (CSD; series 2), and its reversal with l-arginine. Five single CSD were elicited, each by 5-minute perfusion of 160-mmol/L K+ with 40 minutes of subsequent recovery (i.e., perfusion of aCSF). On the l-NAME side, 1 mmol/L of the drug was dissolved in the K-medium for the second, third, and fourth challenge (no pretreatment and posttreatment in this series). On the contralateral side (+L-Arg), l-arginine was coapplied with l-NAME, with the highest concentration of l-arginine applied first (10, 3, and 1 mmol/L) because series 1 showed that the effects of l-NAME persisted for more than 40 minutes (Figs. 3 and 4). The left traces are representative CSD recorded with the third K+ stimulus. Values are means ± SD in the bar charts. *P < 0.05, comparison of the l-NAME and l-NAME plus l-arginine sides (n = 9 and 7); Student's t-test.
Series 3
The aim of this series was to help to determine whether the effects of NOS inhibition on CSD, identified through series 1 and 2, could result from the suppression of any local, NO-mediated vascular change induced by CSD. Two groups were considered (control and l-NAME), with implantation of a single microdialysis electrode because we did not have the necessary equipment for dual pH monitoring. Five separate, individual CSD were elicited, each by perfusion of K-medium for approximately 5 min. In these experiments, perfusion of K-medium was interrupted as soon as CSD was elicited, to standardize the state of the CSD plus K challenge when recovery was initiated, and thereby the period during which dialysates pH normalization was studied (see Data presentation and analysis). In the control group, standard K-medium was used throughout for CSD elicitation. In the l-NAME group, 1 mmol/L of the drug was added to the K-medium used to trigger the second, third, and fourth CSD wave.
Drugs
Both l-NAME and d-NAME were purchased from Tocris Cookson Ltd. (Bristol, U.K.), and l-arginine was purchased from Sigma Chemicals (Poole, U.K.). All other chemicals were of analytical grade. Drug solutions were prepared on the day of the experiment, and ultimately dissolved in aCSF or K-medium with pH adjusted to 7.3.
Estimation of the microdialysis probe delivery ratios
In vitro experiments were performed to evaluate the percentage of l-NAME and l-arginine delivered through the probe. Microdialysis probes identical to those used in vivo were perfused with l-NAME (1 mmol/L) or l-arginine (10 mmol/L) at 1 μL/min, and immersed in normal aCSF maintained at 37.0°C. The remaining concentration of each compound in the dialysate was measured in three consecutive 20-minute dialysate samples by ultraviolet detection (270 and 202 nm for l-NAME and l-arginine, respectively), and the delivery ratio (%) was calculated as
Data presentation and analysis
As in previous studies (Obrenovitch and Zilkha, 1996; Urenjak and Obrenovitch, 2000), drug effects on the cortex susceptibility to repeated CSD elicited by K+ (series 1) were assessed through the analysis of changes in the following variables (Fig. 2): number of CSD elicited (N), cumulative CSD area (A, in millivolts x minute), and latency for occurrence of first CSD (L, in minutes). Because NOS inhibition altered the pattern of individual CSD waves, the following variables were also examined (Fig. 2): CSD height (H, in millivolts), width at 1/2 CSD height (W, in seconds), and maximum slope of depolarization (Sld, in millivolts per minute) and repolarization (Slr, in millivolts per minute). For series 1, this second set of variables was considered only for the first CSD of each K-medium application to avoid any possible interference from previous CSD within the same K stimulus. Comprehensive in vitro evaluation of the pH sensor under our experimental conditions revealed an interference of large changes in the K+-concentration of the solution tested (i.e., switch from aCSF to K-medium produced a steady shift in the recording potential of 0.78 mV, equivalent to a change of approximately 0.03 pH units). In addition, the time resolution of the microdialysis/pH-sensor combination could not resolve any possible change specific to CSD alone. Therefore, the quantitative analysis of the pH signal in series 3 was restricted to the recovery phase, and consisted in the determination of the rate of recovery from acidosis resulting from the combined CSD/K challenge, when any interference of high K+ with the pH recording could be ruled out (Fig. 6, bottom trace). All values in Results are means ± SD. The Student's t-test was used for statistical analysis, and a probability less than 5% was considered statistically significant.
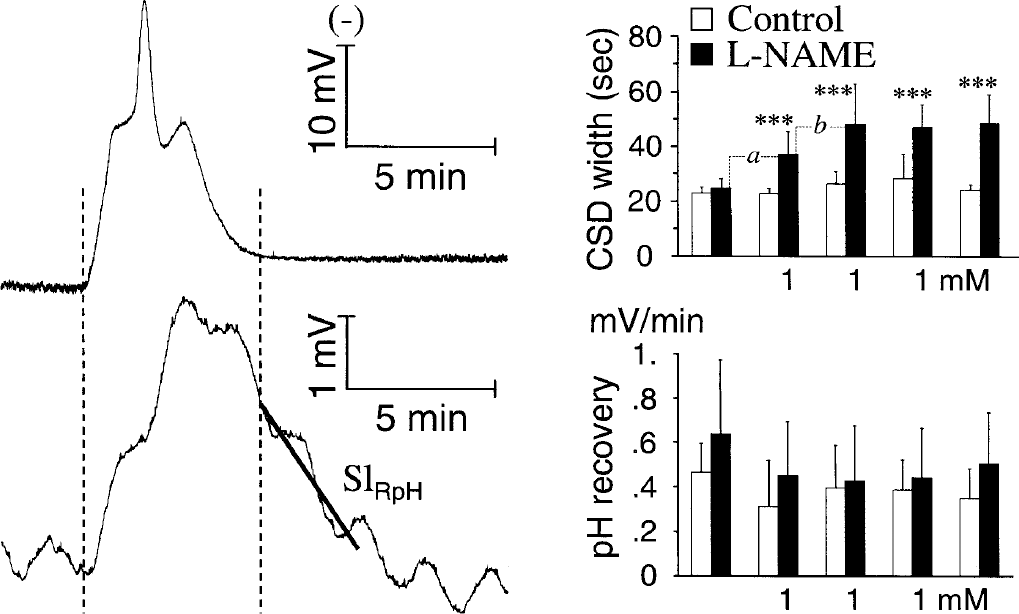
Effects of local nitric oxide synthase (NOS) inhibition with 1-mmol/L l-NAME on cortical spreading depression (CSD) width (upper bar chart), and rate of pH recovery (SlRpH) from the CSD plus K-application challenge (lower bar chart) observed in series 3. Five single CSD were elicited by perfusion of 160-mmol/L K+, with each subsequent 40 minutes of recovery (i.e., perfusion of aCSF) initiated as soon as CSD elicitation was detected. In the l-NAME group, 1 mmol/L of the drug was dissolved in the K-medium for the second, third, and fourth challenge (i.e. no pre-CSD and post-CSD drug treatment in this series). The left traces are representative recordings of the DC potential (upper trace) and pH changes (lower trace) during CSD and K-application. The latter shows the data sequence used for computation of the rate of pH recovery (SlRpH). In agreement with previous reports, the polarity of the DC potential was reversed so that negative shifts (i.e., depolarization) produce an upward deflection. In the pH trace, an upward trend of the signal indicates an acidification of the dialysate; the regular oscillations of the pH signal were due to cyclic irregularities in the rate of perfusion of the “high-precision” syringe pump. Values are means ± SD in the bar charts (n = 7–9). ***P < 0.001, comparison of the l-NAME and control groups with Student's t-test. Comparisons within the l-NAME group (paired t-test): aP< 0.001, comparison of the second versus the first CSD (i.e., first CSD with l-NAME to CSD without drug, respectively); bP< 0.01, comparison of the second versus the third CSD (i.e., first and second CSD with coapplication of l-NAME, respectively).
RESULTS
The delivery ratio was 37.7 ± 5.6% (n = 7) for l-NAME (i.e., the extracellular concentration achieved with 1 mmol/L of the drug in the perfusion medium was estimated to be approximately 0.4 mmol/L). A similar delivery ratio was found for l-arginine (32.9 ± 4.2%, n = 7).
Series 1
In the control group, the first 20-minute perfusion of 160-mmol/L K+ elicited 6.2 ± 1.2 CSD waves, with a corresponding cumulative CSD area of 23.2 ± 5.1 mV/min, and 1.6 ± 0.4 minutes as first CSD latency. As in previous studies based on the same experimental protocol (Obrenovitch and Zilkha, 1996; Urenjak and Obrenovitch, 2000), the subsequent K+ stimuli were progressively less effective in triggering CSD, as shown by the reduced number of CSD elicited, reduced cumulative area, and prolonged latency (Fig. 3, bar charts). l-NAME reduced significantly the number of CSD, but this change was not associated with any prolongation of first CSD latency and, paradoxically, the cumulative CSD area was markedly increased (Fig. 3). These sets of changes suggested strongly that the primary effect of local NOS inhibition was a marked “widening” of individual CSD (Fig. 3, l-NAME trace). In contrast, only the highest concentration of d-NAME tested (i.e., 3 mmol/L) altered CSD, reducing slightly (but significantly) both the number of CSD and corresponding area (Fig. 3, bar charts).
In the control group (n = 6), and for the first CSD elicited by the first K+ stimulus, the peak height and width (at ½ peak) were 11.7 ± 2.4 mV and 26.1 ± 4.0 seconds, respectively. There was no change in these variables with the successive K+ in control and d-NAME groups (see Fig. 4 for CSD width, data not shown for height). l-NAME increased slightly the height of the first CSD, but this change was only significant with the 3-mmol/L concentration (9.0 ± 0.9 and 10.3 ± 1.3 mV for d- and l-NAME groups, respectively; P < 0.05, n = 7 and 8). The most striking change with NOS inhibition was a marked, concentration-dependent increase in CSD width (Fig. 4). This effect was consistent within each K+-stimulus (Fig. 3, l-NAME trace), very significant throughout the range or l-NAME concentration tested, and it persisted when l-NAME was omitted for the last challenge (Fig. 4, bar chart).
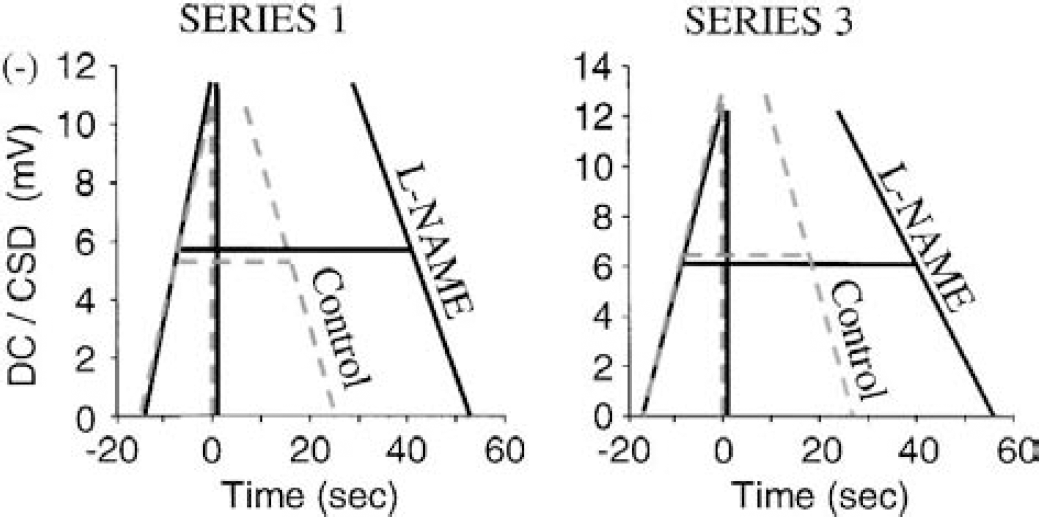
To determine whether this effect involved a change in the rate of depolarization (Sld) and/or repolarization (Slr), these two variables were determined for the first CSD of the third K+-stimulus (i.e., test of 1-mmol/L l-NAME and d-NAME). Sld was 43.6 ± 11.8, 50.4 ± 9.3 and 43.3 ± 9.8 mV/min in the control, l-NAME, and d-NAME group, respectively (n = 6–9). The slight increase of Sld with l-NAME was not significant. Slr was 34.6 ± 1.7, 28.4 ± 5.6 and 39.1 ± 6.3 mV/min in the control, l-NAME, and d-NAME group, respectively. Slr was significantly reduced by 1-mmol/L l-NAME (i.e., repolarization was slower; P < 0.01, comparison between d-NAME and l-NAME, n = 7 and 9). Therefore, a slower recovery of the DC-potential negative shift indicative of CSD did contribute to the increased width of CSD under NOS-inhibition, but the major contributor was actually a delay in the initiation of the repolarization phase (Fig. 7).
Diagrams of changes in the pattern of individual cortical spreading depression (CSD) wave produced by 1-mmol/L l-NAME, which show that the major contributor to wider CSD under nitric oxide synthase inhibition was a marked delay in the initiation of the repolarization phase. These diagrams were constructed from the averaged CSD height, width at ½ height, and maximum rates of depolarization and repolarization, computed for the third K+ challenge in the control (dashed lines) and l-NAME group (solid lines) in series 1 and 3.
Series 2
On the l-NAME side, the width (at ½ peak) of the first CSD (i.e., K-medium without any drug) was 24.2 ± 3.7 seconds (n = 9). This variable was consistently and significantly increased in all the subsequent CSD (Fig. 5), and this change was very similar qualitatively and quantitatively to that observed in series 1 (Fig. 4). This effect was reversed significantly by l-arginine at all the concentrations tested (Fig. 5). These data confirm that the effect of l-NAME on CSD was due to effective NOS inhibition in the cell populations surrounding the microdialysis fiber. These findings also show that this is readily achieved when l-NAME is added to the perfusion medium, because l-NAME was not applied before the K+ stimuli in these experiments. However, note on the l-NAME side that the drug effect on CSD width was less marked with the first drug application than with the subsequent ones (Fig. 5, bar chart).
Series 3
In the l-NAME group, the width (at ½ peak) of the first CSD (i.e., K-medium without any drug) was 24.6 ± 3.5 seconds (n = 8), and this variable increased significantly with all the subsequent challenges (Fig. 6). In agreement with series 2 data (1-mmol/L l-NAME without l-arginine), the magnitude of the increase obtained with the second K+ challenge (i.e., first CSD with 1-mmol/L l-NAME) was one half that observed with the subsequent CSD, also with 1 mmol/L l-NAME; however, the effect of l-NAME persisted at the fifth K+ challenge when the drug was no longer present (Figs. 5 and 6, CSD width bar charts). Regarding pH recovery (Fig. 6, lower trace and bar chart), the average SlRpH for the first CSD plus K challenge in the control group was 0.47 ± 0.13 mV/min (n = 9), approximately 0.02 pH units/min), and there was no significant change at any time between the control and l-NAME groups.
DISCUSSION
Local nitric oxide synthase inhibition and cortical spreading depression: primary effect
In series 1, l-NAME reduced significantly the frequency of repetitive CSD during 20-minute perfusion of K-medium, but this effect did not reflect inhibition of CSD elicitation because the first CSD latency was not prolonged (Fig. 3). Measures of cumulative CSD area and individual CSD waves showed that the primary effect of l-NAME was a marked widening of each CSD (i.e., increased CSD duration), resulting essentially from a delayed initiation of the repolarization phase (Figs. 4, 5 and 7). This action of l-NAME was effectively due to NOS inhibition, because it was not observed with d-NAME and was reversed by l-arginine (Figs. 3–5). The reduced CSD frequency under l-NAME was presumably a consequence of this primary effect, through a prolongation of the refractory period. Indeed, each CSD is followed by a refractory period during which the elicitation of subsequent CSD is inhibited (Brand et al. 1998), and the refractory period is likely to be prolonged by longer-lasting CSD as a result of more pronounced disruption of cellular ionic homeostasis and metabolism (Koroleva and Bureš 1980, 1996). When the same concentration of l-NAME (1 mmol/L) was coapplied with several consecutive K+-challenges (series 2 and 3), the drug effect was consistently less with the first K challenge. This also occurred when l-NAME was applied 20 minutes before CSD elicitation (Wang M, Urenjak J, Obrenovitch TP, unpublished data, 2001). In other terms, the full effect of the concentration of l-NAME tested on CSD required its application in association with a previous CSD, thus suggesting activation-dependent inhibition of NOS or the presence of preformed pools of NO-containing factors (Davisson et al., 1996).
Comparison of the effects of l-NAME on cortical spreading depression with those of 7-nitroindazole
In a previous study with the purported in vivo neuronal selective NOS inhibitor 7-nitroindazole (Moore and Handy 1997; Reiner and Zagvazdin, 1998; Alderton et al., 2001), very different effects were observed (Urenjak and Obrenovitch, 2000). First, perfusion of 1-mmol/L 7-nitroindazole through the probe produced a small but detectable negative shift of the DC potential, which was not observed with l-NAME. Second, 7-nitroindazole inhibited significantly CSD elicitation as indicated by reduced frequency, decreased cumulative CSD area, and increased first CSD latency. These changes were not reversed by l-arginine. One possible origin for these different effects is the monoamine oxidase B-inhibitory action of 7-nitroindazole, which was not found with l-NAME (Desvignes et al., 1999; Di Monte et al., 1997), because CSD implies synchronous neurotransmitter release, including that of dopamine (Moghaddam et al., 1987), and the dopamine-receptor D2 agonist, Quinpirole inhibited spreading depression in the retina model (de Azeredo and Ribeiro, 1992). One common action of 7-nitroindazole and l-NAME was the widening of CSD, but it was much smaller with 1-mmol/L 7-nitroindazole (1.2-fold vs. twofold with 1-mmol/L l-NAME in this study). This difference in magnitude may be linked to the different potency of these two NOS inhibitors; 7-nitroindazole is approximately 30-times less potent than l-NAME (Alderton et al., 2001).
Delayed initiation of repolarization during nitric oxide synthase inhibition: direct neuronal or indirect vascular origin?
It is likely that perfusion of l-NAME through the microdialysis probe resulted in an equivalent inhibition of both neuronal and endothelial NOS in the surrounding cell populations because in vitro tests showed that the IC50 for l-NAME inhibition of NOS is approximately 0.3 μmol/L for both isoforms (Alderton et al., 2001). In addition, in the brain tissue, NO formation subsequent to activation of either endothelial or neuronal NOS can contribute to the coupling of rCBF to metabolism (Ayata et al., 1996; Wiencken and Casagrande, 1999), and CSD does imply increased energy demand with associated hyperperfusion (Lauritzen, 1994). Therefore, the delayed initiation of repolarization demonstrated during NOS inhibition could reflect either a direct action at neuronal level (e.g., suppression of NO-mediated negative feedback control of processes involved in CSD) or an indirect action at vascular level (i.e., suppression of NO-mediated hyperperfusion associated with CSD, with secondary effect on CSD recovery). Two elements are especially relevant when considering the possibility of a vascular origin: (1) the precise kinetic of rCBF changes in relation to CSD; and (2) the role of NO in the coupling of rCBF to CSD. The point-by-point analysis provided next is centered on these two elements.
Studies of rCBF changes with laser Doppler flowmetry showed that the blood flow response to CSD consisted essentially in a sharp increase in rCBF, lasting around 2 minutes in untreated cortex, and starting well within the repolarization phase (Gold et al., 1998; Hashimoto et al., 2000). This finding suggests that the hyperperfusion (even if NO mediated) actually occurred after the primary effect of NOS inhibition on CSD that we observed (i.e., delayed initiation of repolarization; Figs. 4, 5 and 7).
When the hyperperfusion subsequent to CSD was markedly attenuated by the serotonin 2A/2C receptor antagonist ritanserin (Gold et al., 1998), the DC-potential recovery was significantly prolonged, apparently because of a slower rate of recovery. When NOS was inhibited locally in our study, the rate of recovery of the DC potential was also slower, but the primary contributor to wider CSD was a delayed initiation of repolarization (Fig. 7).
Four studies from different laboratories showed that administration of NOS inhibitors in rats did not alter the marked hyperperfusion that is associated with CSD, despite independent measures proving that brain NOS activity was effectively inhibited (Fabricius et al., 1995; Read and Parsons, 1998; Wolf et al., 1996; Zhang et al., 1994). In two studies, NOS inhibition revealed or enhanced a brief initial hypoperfusion that preceded CSD (Duckrow, 1993; Fabricius et al., 1995), suggesting that an initial vasoconstrictor component may be present as CSD is elicited. Only in one study did NOS inhibition reduce the hyperperfusion associated with CSD (Duckrow, 1993). Overall, there is little evidence supporting the notion that NO is the primary mediator of hyperemia induced by CSD in rats.
Delayed initiation of repolarization during nitric oxide synthase inhibition: a consequence of altered energy status in the brain region under study?
Using a 3 × 6-mm perfused cortical window in anesthetized rats, Dreier et al., (1998, 2001) have observed that superimposition of high K+ (35 mmol/L) on NOS inhibition (1-mmol/L NG-nitro-l-arginine) produced clear signs of imbalance between local energy demand and blood supply that were similar to those observed with ischemia. Therefore, one could speculate that the altered CSD waves observed under NOS inhibition were a consequence of their elicitation in an energy-compromised region (i.e., the cellular population surrounding the implanted microdialysis probe/electrode). The following elements observed during NOS inhibition do not support this hypothesis: (1) the latency for CSD elicitation was not prolonged, in contrast to what would be expected from a region with disrupted cellular ionic homeostasis and metabolism (Koroleva and Bureš, 1980, 1996); (2) a much slower rate of repolarization should have been observed, especially when K+ application and NOS inhibitor were applied for 20 and 60 minutes, respectively (series 1; Fig. 7, left diagram); (3) there was no detectable change in the rate of recovery from acidosis (Fig. 6); (4) the rate of early repolarization after switching from K-medium to aCSF was not altered by l-NAME, even when the drug application persisted within the recovery period (series 1; data not shown); (5) a comprehensive examination of the recorded EEG signals (series 1; control and l-NAME experiments) did not reveal any sign of ischemia when l-NAME was superimposed on 160-mmol/L K+, and there was no significant difference in the rate of EEG recovery from individual CSD wave (203 ± 48 and 185 ± 119%/min in the control and l-NAME group, respectively). It is likely that, in our study, ischemia was not produced because of the very small region that was exposed to the combination of high K+ concentration and NOS inhibition.
From this analysis of our and previously published data, we speculate that the delayed initiation of repolarization, observed when CSD was elicited under local NOS inhibition, reflected the suppression of NO-mediated negative feedback mechanism(s) controlling the duration of CSD. By matching the mechanisms involved in CSD with possibilities for NO-mediated modulation, three different molecular targets can be identified for such a negative feedback: (1) NMDA-receptors (Choi and Lipton, 2000), though ongoing experiments do not support this hypothesis (Urenjak J, Wang M, Obrenovitch TP, unpublished data, 2001); (2) glutamate exocytosis (Sistiaga et al., 1997); and (3) gap junctions, which are necessary for the propagation of CSD (Nedergaard et al., 1995) and can be modulated by NO (O'Donnell and Grace, 1997; Rorig and Sutor, 1996).
An alternative explanation for the effect of l-NAME that we observed, which cannot be ruled out by the data presented herein, is the suppression by l-NAME of a very brief, NO-mediated vasodilation associated with the early phase of CSD, such as that reported by Duckrow (1993) and Fabricius et al. (1995). Should such a transient NO-mediated vasodilation and its impact on the initiation of repolarization be confirmed, than an important role of NO-mediated vasodilatation in the brain may be to respond rapidly to changes in neuronal activity and/or depolarization before other less responsive cerebrovascular control mechanisms.
Selective pharmacologic and genetic manipulations have shown that the pathogenesis of neuronal ischemic injury can be altered by NO, either positively or negatively, depending on the stage of evolution of ischemic injury and on the cellular source of NO (Dawson and Dawson, 1998; Iadecola, 1997; Shimizu-Sasamata et al., 1998). The current consensus is that NO produced by endothelial NOS is beneficial by favoring blood supply to vulnerable regions (e.g., the penumbra), whereas post-ischemic production of NO by neuronal NOS may be neurotoxic. Our finding that NO production associated with CSD promotes the initiation of the recovery phase from this potentially deleterious event adds a new degree of complexity to this already complicated picture.
Footnotes
Acknowledgements
The authors thank Alexandra Egan and Jack Chan (Biomedical Sciences, University of Bradford) for their technical help.
Abbreviations used
References
1.
AldertonWKCooperCEKnowlesRG (2001) Nitric oxide synthases: structure, function and inhibition. Biochem J357:593–615
2.
AyataCMaJMengWHuangPMoskowitzMA (1996) L-NA-sensitive rCBF augmentation during vibrissal stimulation in type III nitric oxide synthase mutant mice. J Cereb Blood Flow Metab16:539–541
3.
BellamyTCGarthwaiteJ (2001) Sub-second kinetics of the nitric oxide receptor, soluble guanylyl cyclase, in intact cerebellar cells. J Biol Chem276:4287–4292
4.
BrandSFernandes de LimaVMHankeW (1998) Pharmacological modulation of the refractory period of retinal spreading depression. Naunyn Schmiedebergs Arch Pharmacol357:419–425
5.
BurešJBuresováOKrivánekJ (1974) The mechanisms and applications of Leao's spreading depression of electroencephalographic activity.New York: Academic Press
6.
CaoYWelchKMAuroraSVikingstadEM (1999) Functional MRI-BOLD of visually triggered headache in patients with migraine. Arch Neurol56:548–554
7.
CarlaVMoroniF (1992) General anesthetics inhibit the responses induced by glutamate receptor agonists in the mouse cortex. Neurosci Lett146:21–24
8.
ChenKCNicholsonC (2000) Spatial buffering of potassium ions in brain extracellular space. Biophys J78:2776–2797
9.
ChoiYBLiptonSA (2000) Redox modulation of the NMDA receptor. Cell Mol Life Sci57:1535–1541
10.
DavissonRLBatesJNJohnsonAKLewisSJ (1996) Use-dependent loss of acetylcholine- and bradykinin-mediated vasodilation after nitric oxide synthase inhibition: evidence for preformed stores of nitric oxide-containing factors in vascular endothelial cells. Hypertension28:354–360
11.
DawsonVLDawsonTM (1998) Nitric oxide in neurodegeneration. Prog Brain Res118:215–229
12.
de AzeredoFARibeiroMF (1992) A simple biological way to screen dopaminergic agonists. Metab Brain Dis7:211–221
13.
DesvignesCBertLVinetLDenoroyLRenaudBLambas-SenasL (1999) Evidence that the neuronal nitric oxide synthase inhibitor 7-nitroindazole inhibits monoamine oxidase in the rat: in vivo effects on extracellular striatal dopamine and 3,4-dihydroxyphenylacetic acid. Neurosci Lett261:175–178
14.
Di MonteDARoylandJEAndersonACastagnoliKCastagnoliNJrLangstonJW (1997) Inhibition of monoamine oxidase contributes to the protective effect of 7-nitroindazole against MPTP neurotoxicity. J Neurochem69:1771–1773
15.
DreierJPKörnerKEbertNGornerARubinIBackTLindauerUWolfTVillringerAEinhauplKMLauritzenMDirnaglU (1998) Nitric oxide scavenging by hemoglobin or nitric oxide synthase inhibition by N-nitro-l-arginine induces cortical spreading ischemia when K+ is increased in the subarachnoid space. J Cereb Blood Flow Metab18:978–990
16.
DreierJPPetzoldGTilleKLindauerUArnoldGHeinemannUEinhäuplKMDirnaglU (2001) Ischemia triggered by spreading neuronal activation is inhibited by vasodilators in rats. J Physiol (Lond)531:515–526
17.
DuckrowRB (1993) A brief hypoperfusion precedes spreading depression if nitric oxide synthesis is inhibited. Brain Res618:190–195
18.
FabriciusMLauritzenM (1996) Laser-Doppler evaluation of rat brain microcirculation: comparison with the [14C]-iodoantipyrine method suggests discordance during cerebral blood flow increases. J Cereb Blood Flow Metab16:156–161
19.
FabriciusMAkgörenNLauritzenM (1995) Arginine-nitric oxide pathway and cerebrovascular regulation in cortical spreading depression. Am J Physiol269:H23–H29
20.
FaraciFMBrianJEJr (1994) Nitric oxide and the cerebral circulation. Stroke25:692–703
21.
Gardner-MedwinAR (1981) Possible roles of vertebrate neuroglia in potassium dynamics, spreading depression and migraine. J Exp Biol95:111–127
22.
GarthwaiteJBoultonCL (1995) Nitric oxide signalling in the central nervous system. Annu Rev Physiol57:683–706
23.
GoldLBackTArnoldGDreierJEinhäuplKMReuterUDirnaglU (1998) Cortical spreading depression-associated hyperemia in rats: involvement of serotonin. Brain Res783:188–193
24.
HansenAJZeuthenT (1981) Extracellular ion concentrations during spreading depression and ischemia in the rat brain cortex. Acta Physiol Scand113:437–445
25.
HashimotoMTakedaYSatoTKawaharaHNaganoOHirakawaM (2000) Dynamic changes of NADH fluorescence images and NADH content during spreading depression in the cerebral cortex of gerbils. Brain Res872:294–300
26.
HerrerasOSomjenGG (1993) Analysis of potential shifts associated with recurrent spreading depression and prolonged unstable spreading depression induced by microdialysis of elevated K+ in hippocampus of anesthetized rats. Brain Res610:283–294
IadecolaC (1997) Bright and dark sides of nitric oxide in ischemic brain injury. Trends Neurosci20:132–139
29.
KorolevaVIBurešJ (1980) Blockade of cortical spreading depression in electrically and chemically stimulated areas of cerebral cortex in rats. Electroencephalogr Clin Neurophysiol48:1–15
30.
KorolevaVIBurešJ (1996) The use of spreading depression waves for acute and long-term monitoring of the penumbra zone of focal ischemic damage in rats. Proc Natl Acad Sci U S A93:3710–3714
31.
LauritzenM (1994) Pathophysiology of the migraine aura: the spreading depression theory. Brain117:199–210
MarrannesRDe PrinsEWauquierA (1985) Influence of CO2 and hyperventilation on spreading depression in the rat. Arch Int Physiol Biochim Biophys94:P19–P20
34.
MarrannesRWillemsRDe PrinsEWauquierA (1988) Evidence for a role of the N-methyl-d-aspartate (NMDA) receptor in cortical spreading depression in the rat. Brain Res457:226–240
35.
Martins-FerreiraHNedergaardMNicholsonC (2000) Perspectives on spreading depression. Brain Res Brain Res Rev32:215–234
36.
MoghaddamBSchenkJOStewartWBHansenAJ (1987) Temporal relationship between neurotransmitter release and ion flux during spreading depression and anoxia. Can J Physiol Pharmacol65:1105–1110
37.
MollerMJonesNMBeartPM (1995) Complex involvement of nitric oxide and cGMP at N-methyl-d-aspartic acid receptors regulating gamma-[3H]aminobutyric acid release from striatal slices. Neurosci Lett190:195–198
38.
MoorePKHandyRL (1997) Selective inhibitors of neuronal nitric oxide synthase—is no NOS really good NOS for the nervous system?Trends Pharmacol Sci18:204–211
39.
NedergaardMCooperAJGoldmanSA (1995) Gap junctions are required for the propagation of spreading depression. J Neurobiol28:433–444
40.
ObrenovitchTP (1995) The ischaemic penumbra: twenty years on. Cerebrovasc Brain Metab Rev7:297–323
41.
ObrenovitchTPZilkhaE (1996) Inhibition of cortical spreading depression by L-701,324, a novel antagonist at the glycine site of the N-methyl-d-aspartate receptor complex. Br J Pharmacol117:931–937
42.
ObrenovitchTPRichardsDASarnaGSSymonL (1993) Combined intracerebral microdialysis and electrophysiological recording: methodology and applications. J Neurosci Methods47:139–145
43.
O'DonnellPGraceAA (1997) Cortical afferents modulate striatal gap junction permeability via nitric oxide. Neuroscience76:1–5
44.
PaxinosGWatsonC (1986) The rat brain in stereotaxic coordinates.London: Academic Press
45.
ReadSJParsonsAA (1998) Nitric oxide does not mediate cerebral blood flow changes during cortical spreading depression in the anaesthetised rat. Neurosci Lett250:115–118
46.
ReadSJSmithMIHunterAJParsonsAA (1997) The dynamics of nitric oxide release measured directly and in real time following repeated waves of cortical spreading depression in the anaesthetised cat. Neurosci Lett232:127–130
47.
ReinerAZagvazdinY (1998) On the selectivity of 7-nitroindazole as an inhibitor of neuronal nitric oxide synthase. Trends Pharmacol Sci19:348–350
48.
RorigBSutorB (1996) Nitric oxide-stimulated increase in intracellular cGMP modulates gap junction coupling in rat neocortex. Neuroreport7:569–572
49.
SchlameMHemmingsHC (1995) Inhibition by volatile anesthetics of endogenous glutamate release from synaptosomes by a presynaptic mechanism. Anesthesiology82:1406–1416
50.
Shimizu-SasamataMBosque-HamiltonPHuangPLMoskowitzMALoEH (1998) Attenuated neurotransmitter release and spreading depression-like depolarizations after focal ischemia in mutant mice with disrupted type I nitric oxide synthase gene. J Neurosci18:9564–9571.
51.
SilverIAErecinskaM (1994) Extracellular glucose concentration in mammalian brain: continuous monitoring of changes during increased neuronal activity and upon limitation in oxygen supply in normo-, hypo-, and hyperglycemic animals. J Neurosci14:5068–5076
52.
SistiagaAMiras-PortugalMTSanchez-PrietoJ (1997) Modulation of glutamate release by a nitric oxide/cyclic GMP-dependent pathway. Eur J Pharmacol321:247–257
53.
SoehleMHeimannAKempskiO (2001) On the number of measurement sites required to assess regional cerebral blood flow by laser-Doppler scanning during cerebral ischemia and reperfusion. J Neurosci Methods110:91–94
54.
SzerbJC (1991) Glutamate release and spreading depression in the fascia dentata in response to microdialysis with high K+: role of glia. Brain Res542:259–265
55.
TanakaTSaitoHMatsukiN (1993) Endogenous nitric oxide inhibits NMDA- and kainate- responses by a negative feedback system in rat hippocampal neurons. Brain Res631:72–76
56.
TaylorDLRichardsDAObrenovitchTPSymonL (1994) Time course of changes in extracellular lactate evoked by transient K+-induced depolarisation in the rat striatum. J Neurochem62:2368–2374
57.
TraynelisSFCull-CandySG (1990) Proton inhibition of N-methyl-d-aspartate receptors in cerebellar neurons. Nature345:347–350
58.
UrenjakJObrenovitchTP (2000) Pharmacological investigation into the involvement of nitric oxide in K+-induced cortical spreading depression. Naunyn Schmiedebergs Arch Pharmacol362:137–144
59.
UrenjakJZilkhaEGotohMObrenovitchTP (1997) Effect of acidotic challenges on local depolarizations evoked by N-methyl-d-aspartate (NMDA) in the rat striatum. Life Sci61:523–535
60.
WienckenAECasagrandeVA (1999) Endothelial nitric oxide synthetase (eNOS) in astrocytes: another source of nitric oxide in neocortex. Glia26:280–290
61.
WolfTLindauerUObrigHDreierJBackTVillringerADirnaglU (1996) Systemic nitric oxide synthase inhibition does not affect brain oxygenation during cortical spreading depression in rats: a non-invasive near-infrared spectroscopy and laser-Doppler flowmetry study. J Cereb Blood Flow Metab16:1100–1107
62.
XiongZQStringerJL (2000) Sodium pump activity, not glial spatial buffering, clears potassium after epileptiform activity induced in the dentate gyrus. J Neurophysiol83:1443–1451
63.
ZagvazdinYSancesarioGFitzgeraldMECReinerA (1998) Effects of halothane and urethane-chloralose anaesthesia on the pressor and cerebrovascular responses to 7-nitroindazole, an inhibitor of nitric oxide synthase. Pharmacol Res38:339–346
64.
ZhangZGChoppMMaynardKIMoskowitzMA (1994) Cerebral blood flow changes during cortical spreading depression are not altered by inhibition of nitric oxide synthesis. J Cereb Blood Flow Metab14:939–943