
Abstract
We studied whether endothelial nitric oxide synthase (eNOS) is upregulated and uncoupled in large cerebral arteries after subarachnoid hemorrhage (SAH) and also whether this causes cerebral vasospasm in a mouse model of anterior circulation SAH. Control animals underwent injection of saline instead of blood (n=16 SAH and n=16 controls). There was significant vasospasm of the middle cerebral artery 2 days after SAH (lumen radius/wall thickness ratio 4.3±1.3 for SAH, 23.2±2.1 for saline, P<0.001). Subarachnoid hemorrhage was associated with terminal deoxynucleotidyl transferase dUTP nick-end labeling, cleaved caspase-3, and Fluoro-Jade-positive neurons in the cortex and with CA1 and dentate regions in the hippocampus. There were multiple fibrinogen-positive microthromboemboli in the cortex and hippocampus after SAH. Transgenic mice expressing lacZ under control of the eNOS promoter had increased X-gal staining in large arteries after SAH, and this was confirmed by the increased eNOS protein on western blotting. Evidence that eNOS was uncoupled was found in that nitric oxide availability was decreased, and superoxide and peroxynitrite concentrations were increased in the brains of mice with SAH. This study suggests that artery constriction by SAH upregulates eNOS but that it is uncoupled and produces peroxynitrite that may generate microemboli that travel distally and contribute to brain injury.
Introduction
Subarachnoid hemorrhage (SAH) comprises only ∼7% of all strokes worldwide, but is associated with a disproportionate percentage of mortality and morbidity due to the relatively young age of those affected and the overall severity of the disease (Taylor et al, 1996). Subarachnoid hemorrhage is spontaneous bleeding into the subarachnoid space usually due to rupture of an intracranial aneurysm or even more frequently due to trauma. It is associated with a number of secondary pathophysiologic events, such as angiographic cerebral vasospasm, delayed cerebral ischemia, cortical spreading depolarization, microcirculatory dysfunction, and microthromboembolism (Hansen-Schwartz et al, 2007; Macdonald et al, 2007; Pluta et al, 2009).
Several theories have been proposed to explain angiographic vasospasm, delayed cerebral ischemia, and microthromboembolism, among which the involvement of nitric oxide (NO) is prominent. Nitric oxide is produced by endothelial, neuronal, or inducible NO synthases (NOSs). Endothelial NOS (eNOS) in endothelial cells regulates vascular tone and preserves endothelial function and may also be neuroprotective and antiinflammatory (Hashiguchi et al, 2004). Endothelium-derived NO inhibits platelet aggregation and adhesion (Alheid et al, 1987), smooth muscle proliferation, endothelial cell apoptosis, and leukocyte adhesion to the endothelium (Forstermann and Munzel, 2006). Under physiologic conditions, eNOS maintains a tightly regulated connection between its homodimers, so that electron flow generates NO from the ferrous–dioxygen complex. However, under pathologic conditions that can occur owing to oxidative stress or a decrease in NOS cofactors such as L-arginine and tetrahydrobiopterin, the ferrous–dioxygen complex can become uncoupled and generate O−2 instead of NO (Forstermann and Munzel, 2006). O−2 can react with residual NO to form ONOO−, which further oxidizes cofactors such as tetrahydrobiopterin and increases eNOS uncoupling. Specific concentrations of O−2 and NO are required to produce ONOO− (Espey et al, 2002).
It was suggested that subarachnoid blood scavenges NO and stimulates production of endogenous inhibitors of eNOS, leading to decreased NO and transient vasoconstriction (Pluta, 2006). Disruption of endothelial-dependent vasodilation was suggested to contribute to angiographic vasospasm after SAH and to post-SAH reductions in cerebral blood flow (CBF) (Kasuya et al, 1995; Hino et al, 1996; Pluta et al, 1996). Diminished eNOS activity could also aggravate outcome after SAH as it is associated with increased brain injury after ischemia (Atochin et al, 2007). Increased asymmetric dimethylarginine, an endogenous eNOS inhibitor, in the cerebrospinal fluid of patients with SAH may facilitate delayed vasospasm (Jung et al, 2007).
In contrast, upregulating eNOS activity with the HMG-CoA (3-hydroxy-3-methylglutaryl coenzyme A) reductase inhibitors (statins) decreased angiographic vasospasm after experimental SAH and may decrease delayed cerebral ischemia and improve outcome in patients with SAH (McGirt et al, 2006; Tseng et al, 2007). Conversely, there are some inconsistencies in the hypothesis that decreased eNOS/NO causes vasospasm and other complications of SAH. Increased endothelial cell shear stress associated with angiographic vasospasm should upregulate eNOS. Hemoglobin is a potent NO scavenger but how would it affect eNOS-derived NO when it is on the adventitial side of the artery? One study suggested that hemoglobin did penetrate the arterial wall after SAH, but it was not clear from the images provided how close to the internal elastic lamina it got (Foley et al, 1993). Although there are reports that vasospasm can be decreased by NO donors (Afshar et al, 1995; Pluta et al, 1997), administration of NO donors did not decrease vasospasm in one model, despite elevating the target messenger cyclic guanosine monophospate in smooth muscle cells (Aihara et al, 2003).
This study used a mouse model of SAH and mice with lacZ under control of the eNOS promoter to examine the mechanism of eNOS/NO dysfunction after SAH. The hypothesis was that SAH leads to oxidative stress, eNOS uncoupling, and large-artery vasospasm. The loss of NO also contributes to microthrombi forming in large arteries that can embolize distally, and oxidative stress also can form microthrombi in small arterioles. All of these cause neuronal injury. The goal of the study is to determine whether the hypothesis is true.
Materials and methods
Animals
Experimental protocols were approved by the Institutional Animal Care Committee. Endothelial NOS-lacZ transgenic mice weighing 19 to 25 g were anesthetized with ketamine (10 mg/kg) and xylazine (4 mg/kg) intraperitoneally. Body temperature was maintained at 37°C ± 0.5°C with a homeothermic heating pad (Harvard Apparatus, Holliston, MA, USA) and monitored using a rectal probe. Generation of eNOS-lacZ transgenic mice used the murine eNOS promoter with the eNOS-ATG site mutated to prevent translation (Teichert et al, 2000). The promoter was coupled to a lacZ open-reading frame. Mice were genotyped using Southern blot analysis of genomic tail DNA, and both sexes were used at an age of 2 to 6 months.
Subarachnoid Hemorrhage
Subarachnoid hemorrhage was created by injection of autologous blood into the prechiasmatic cistern (Sabri et al, 2009). The head was fixed in a stereotactic frame equipped with a mouse adaptor (Stoelting Company, Wood Dale, IL, USA). Relative CBF was measured using a laser Doppler flow probe (BLT21, Transonics Systems, New York, NY, USA). A 0.9-mm hole was drilled in the midline of the skull 4.5 mm anterior to the bregma. The drill was angled 40° caudally, and a 27-G spinal needle was advanced at the same angle through the burr hole to the base of the skull. For the SAH group (n = 16), autologous blood (nonheparinized, 60 μL) was withdrawn from the ventral tail artery using a 25-G needle and injected through the spinal needle over 15 seconds. For the control group, the same volume of saline was injected (n = 16). Allocation to groups was sequential with a SAH and a control animal operated each day. Mice were killed 48 hours after surgery. They were perfused through the left cardiac ventricle with NaCl, 0.9%, 10 mL, followed by 150 mL 4% paraformaldehyde in phosphate-buffered saline (PBS, n = 8 per group). The brains were removed and fixed in 4% paraformaldehyde for 48 hours. Brain blocks were embedded in paraffin. Sections of 7-μm thickness were cut using a microtome (Leica, Wetzlar, Germany) for histologic studies.
Hematoxylin and Eosin Staining and Vasospasm Measurement
Paraffinized sections were incubated in xylene and rehydrated through a decreasing gradient of ethanol solutions for deparaffinization. Slides were stained with hematoxylin and eosin, coverslipped with xylene-based mounting medium (Permount, Sigma-Aldrich, St Louis, MO, USA), and viewed under a light microscope. Slides were scanned using a digital scanner (MIRAX scanner, Carl Zeiss, Göttingen, Germany) and viewed with MIRAX software (Carl Zeiss). The lumen area and thickness of the middle cerebral artery was quantified by a blinded observer at 200 × magnification using Image J (National Institutes of Health (NIH), Bethesda, MD, USA). The sizes of the images were calibrated based on their magnification, and middle cerebral artery lumen was outlined using the free hand tool to obtain lumen area. Artery wall thickness was measured at four equally spaced points along the artery circumference and averaged to obtain artery thickness.
LacZ Staining and Detection
Brain sections obtained from 2-month-old hemizygous eNOS-lacZ mice were excised and dissected into 3-mm sections to maximize the surface area of exposure. Sections were rinsed in PBS and then immersed in fixative solution (containing 0.2% glutaraldehyde, 2% formaldehyde, 5 mmol/L EGTA, 2 mmol/L MgCl2, and 100 mmol/L Na2H2PO4, pH 7.3). Sections were again rinsed in PBS, blotted, and immersed in an aqueous X-gal solution (1 mg/mL 5-bromo-4chloro-3-indolyl-B-D-galactopyranoside, pH 7.3, Boehringer Mannheim, Laval, QC, Canada) for 16 hours in the dark at room temperature. Tissue was embedded in paraffin, sectioned (7 μm), and counterstained with neutral red (Sigma-Aldrich) (Teichert et al, 2000). Localization and expression of β-galactosidase was assessed by light microscopy by two independent observers blinded to the type and group of the mice.
Peroxynitrite Assay and Antinitrotyrosine
Tissue sections were deparaffinized with xylene and dehydrated in graded ethanol solutions. Antigen retrieval was performed as described above and endogenous peroxidase activity was quenched by incubating the sections for 30 minutes in 0.3% H2O2 in water. Sections were blocked with 10% normal goat serum in PBS for 20 minutes to attenuate nonspecific IgG binding. Polyclonal antinitrotyrosine antibody (rabbit polyclonal 1:200) was used as a marker for peroxynitrite (ONOO−) and was added to the sections and incubated for 60 minutes. A secondary biotinylated antibody (goat anti-rabbit) was added to the specimens for 30 minutes. The VIP reaction was then performed using the Vectastain ABC Kit (Vector, Burlingame, CA, USA).
Nitric Oxide, Superoxide Anion Radical Detection
Superoxide anion radical (O−2) and NO were detected in homogenized fresh or deep frozen brain tissue using spectrophotometric methods. Cell-permeable DAF-2DA (fluorophore 4,5-diaminofluorescein-2-diacetate, Alexis Biochemicals, Gruenberg, Germany) was used to detect NO and a chemiluminescence probe, 2-methyl-6-(p-methoxyphenyl)-3,7-dihydroimidazo[1,2-]-pyrazin-3-one (MCLA) for O−2 detection. Homogenized brain tissues were incubated with either 10 μmol/L DAF-2DA for 30 minutes or 4 μmol/L MCLA in transparent 96-well plates (Fisher, Ottawa, ON, Canada) at room temperature in the dark. DAF-2DA was excited at 495 nm and emission read at 515 nm in a spectrofluorometer (SpectraMAX-Gemini, Molecular Devices, Sunnyvale, CA, USA). The luminescence signal of MCLA was read directly at 495 nm. Reactions were followed for 10 minutes for DAF-2DA and 6 minutes for MCLA and repeated three times. To test the specificity of MCLA, increasing concentrations of superoxide dismutase (1 to 10 U/mL) was used to reveal a concentration-dependent MCLA luminescence signal (data not shown). As the MCLA crosses cell membranes, the O−2 detected is intracellular and extracellular.
Western Blots for Endothelial Nitric Oxide Synthase and Inducible Nitric Oxide Synthase
The brain tissue (n = 8 per group) was excised and stored at −80°C. Tissue was homogenized in 300 μL 1% RIPA buffer with 0.1% protease inhibitor, and centrifuged at 13,000 r.p.m. for 12 minutes at 4°C. Protein was quantified using the Bradford method, in which the RIPA buffer was used as the blank standard. In all, 30 μg protein was loaded and separated by electrophoresis on 8% SDS-polyacrylamide gels and transferred into nitrocellulose membrane. We used Ponceau S and Gel Code to stain the membrane and gel, respectively. Blots were incubated with 5% milk for 60 minutes, followed by incubation with primary monoclonal antibodies (1:1,000 dilution) against phosphorylated S1177-eNOS (BD Biosciences, San Jose, CA, USA) and inducible NOS (iNOS) (Abcam, Cambridge, MA, USA). After washing in PBS, membranes were incubated in horseradish peroxidase-conjugated anti-goat polyclonal antibody (Abcam) at a dilution of 1:1,000 for 50 minutes at room temperature. Reactions were developed with ECL reagent mix (Amersham Biosciences, Baie d'Urfe, Quebec, Canada). Protein intensities were quantified by densitometric analysis using Image J software (National Institutes of Health).
Immunohistological Staining for Fibrinogen, Caspase-3, and Neuronal Nuclei
After deparaffinization and rehydration, antigen was retrieved by heating the sections for 25 minutes in 0.01 mmol/L sodium citrate (pH 6.0) at 96°C. For fibrinogen staining, endogenous peroxidase activity was quenched by incubating the sections for 30 minutes in 0.3% H2O2 in water. Sections were blocked with 10% normal goat blocking serum in PBS for 20 minutes, incubated with fibrinogen first antibody (chicken anti-rat 1:200, Immunology Consultants Laboratory, Newberg, OR, USA) for 60 minutes. They were washed in PBS and incubated with secondary biotinylated anti-body (goat anti-chicken, Millipore, Billerica, MA, USA) for 30 minutes. Staining was visualized with VIP using the Vectastain ABC Kit (Vector) and counterstained with 0.5% methyl green.
For caspase-3 and neuronal nuclei (NeuN) staining, after antigen retrieval, sections were permeabilized with 0.3% Triton X-100 for 15 minutes and then incubated with 10% normal goat serum, 1% bovine serum albumin, and 0.1% sodium azide for 60 minutes. Primary antibodies were goat anti-mouse NeuN (1:400, Invitrogen, Carlsbad, CA, USA) and rabbit anti-human active cleaved caspase-3 (1:400, BD Pharmingen, Franklin Lakes, NJ, USA). Secondary antibodies were Alexa Fluor 568-conjugated goat anti-mouse for NeuN (1:1,000, Invitrogen) and Alexa Fluor 488 conjugated goat anti-rabbit for caspase-3 (1:1,000, Invitrogen). After incubation, sections were coverslipped with antifading mounting medium and sealed with nail polish. Slides were viewed with a confocal microscope and images were taken using constant parameters.
Terminal Deoxynucleotidyl Transferase dUTP Nick-End Labeling and Fluoro-Jade Staining
Apoptosis was assessed using terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL, DeadEnd Flurometric kit, Promega, Fitchburg, WI, USA). Fluoro-Jade B (Histo-Chem, Jefferson, AR, USA) staining was used as a neuronal injury marker, and as a possible indicator of neurodegeneration. Staining was performed according to a previously published protocol (Sabri et al, 2009). In brief, after deparaffinization and subsequent rehydration, the slides were incubated in 0.06% potassium permanganate (VWR International, Strasbourg, France) for 15 minutes. Slides were then rinsed in deionized water and immersed in 0.001% Fluoro-Jade B working solution (0.1% acetic acid) for 30 minutes. They were then washed and dried in an incubator (60°C) for 15 minutes. Sections were cleared in xylene and coverslipped with a nonaqueous, low-fluorescence, styrene-based mounting medium (Sigma-Aldrich). Slides were viewed under a confocal microscope and images were taken using constant parameters (such as laser power, exposure time, and pin hole size).
Statistical Analysis
All data are presented as means ± s.d. Data were compared within groups over time and between groups at each time by ANOVA (analysis of variance) or Student's t-test for continuous variables and by χ2 test for categorical variables. P < 0.05 was considered significant.
Results
Cerebral Blood Flow and Vasospasm After Subarachnoid Hemorrhage
Relative CBF after injection of autologous blood decreased compared with baseline (10% ± 6% of baseline). The decrease was greater than in the saline group (34% ± 11% of baseline, Figure 1A, P > 0.05). The duration of decrease in CBF was also longer in the SAH group. After 40 minutes, CBF of saline-injected mice recovered to near baseline levels (95% ± 3%). In contrast, CBF in SAH mice stabilized at 79% ± 14%, which was significantly lower than baseline (Student's t-test, P < 0.05).
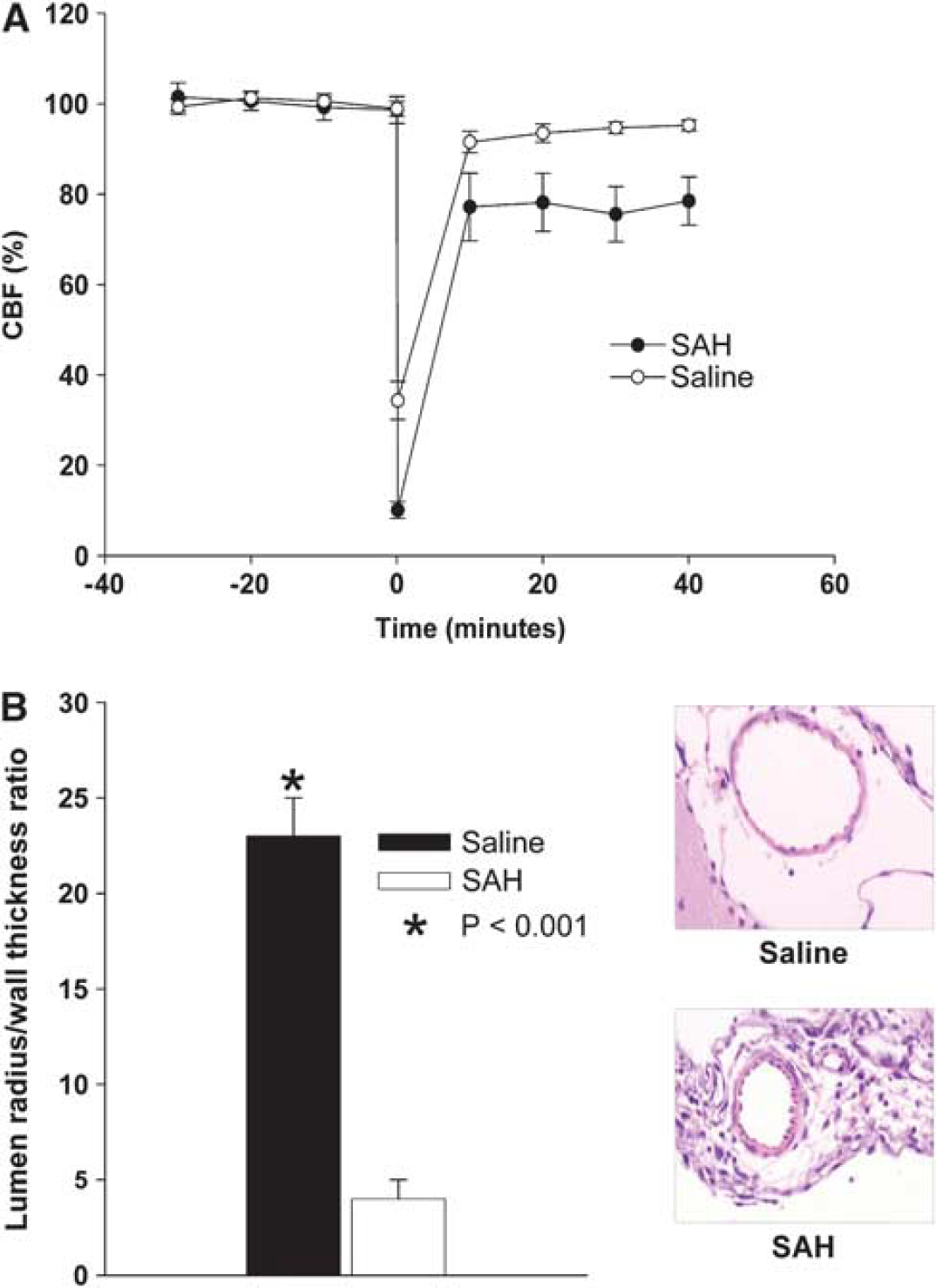
CBF and vasospasm. (
Mice with SAH developed large-artery vasospasm as shown by the significantly decreased lumen radius/wall thickness ratio in comparison with control mice (4.3 ± 1.3 for SAH, 23.2 ± 2.1 for saline, P < 0.001, Figure 1B).
X-Gal and Peroxynitrite Staining
We hypothesize that large-artery vasospasm could increase eNOS expression but that oxidative stress due to SAH uncoupled eNOS, resulting in ONOO− production, endothelial thrombogenicity, and formation of microemboli. lacZ/eNOS transgenic mice were subjected to SAH (n = 8), and X-gal staining was performed to localize the activity of eNOS in the brains and cerebral arteries of saline-injected control and SAH mice. X-gal staining was localized to the endothelium of large arteries. After SAH, there was an increase in staining in the endothelium of the middle cerebral artery in comparison with that of control mice (Figure 2A). This was consistent with an upregulation of eNOS due to increased shear stress but not with NO deficiency contributing to vasospasm, although these results do not exclude the possibility that decoupling and competitive inhibition of eNOS produce the observed results.
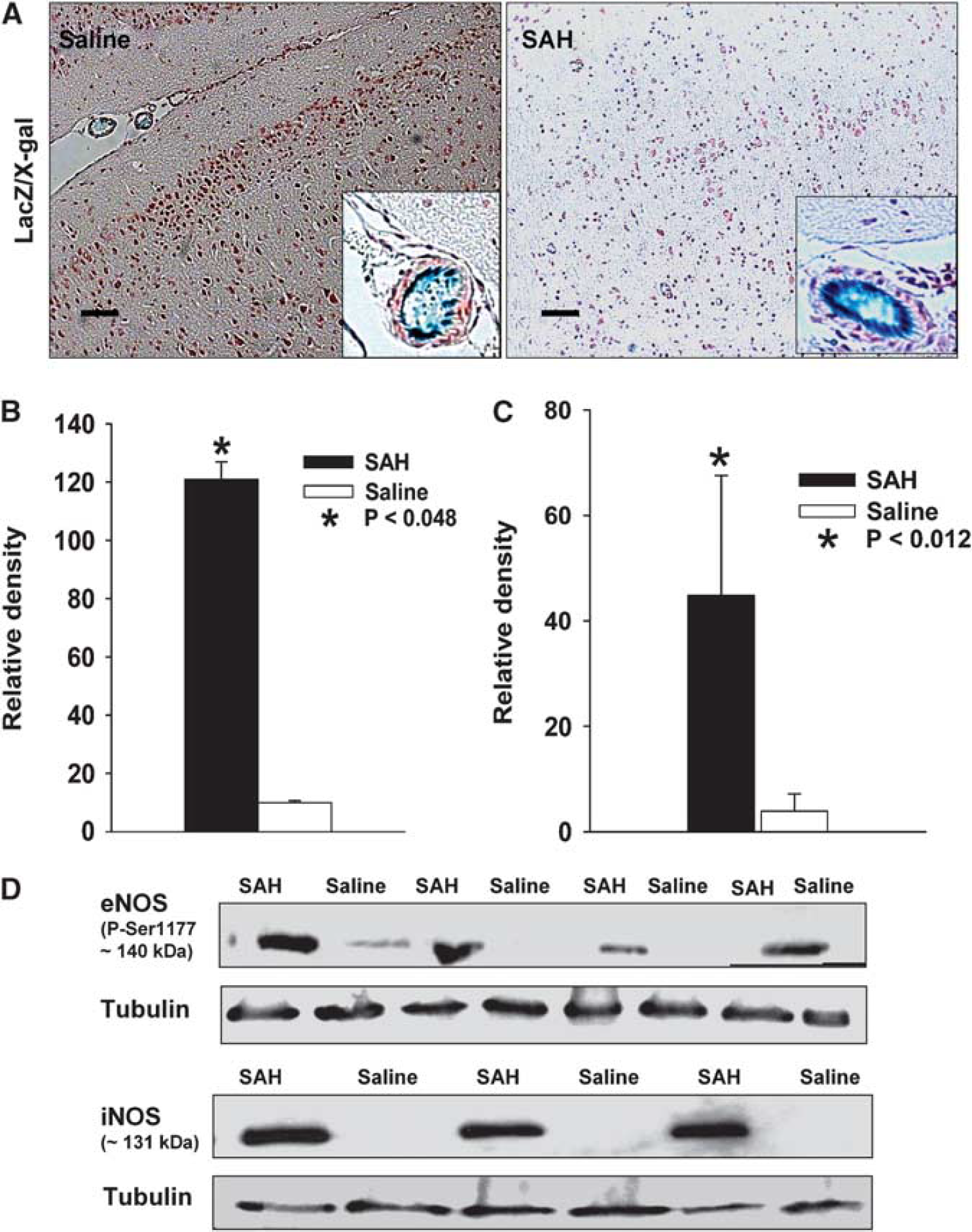
X-gal staining and western blot analysis. X-gal staining was used as a reporter for eNOS expression. (
Western blotting was also used to assess eNOS and iNOS protein expression. There was a significant increase in the eNOS protein in the brains of SAH mice compared with controls (120 ± 2 relative units for SAH, 5.3 ± 1.2 relative units for saline control, P = 0.048, Figures 2B and 2D). Similarly, the iNOS protein was significantly increased in the SAH group in comparison with the control group (42 ± 2 for SAH, 3.5 ± 1.0 relative units for saline control, P = 0.012, Figures 2C and 2D).
To investigate whether eNOS could be uncoupled after SAH, resulting in the production of ONOO− rather than NO, antinitrotyrosine staining was performed. This showed an increase in nitrotyrosine in the endothelium of large arteries of SAH mice in comparison with those of control mice (Figure 3A). The pattern of increased oxidative reaction (nitrotyrosine staining for ONOO−) in the same region where eNOS is upregulated (X-gal staining for eNOS) supports the hypothesis of eNOS uncoupling.
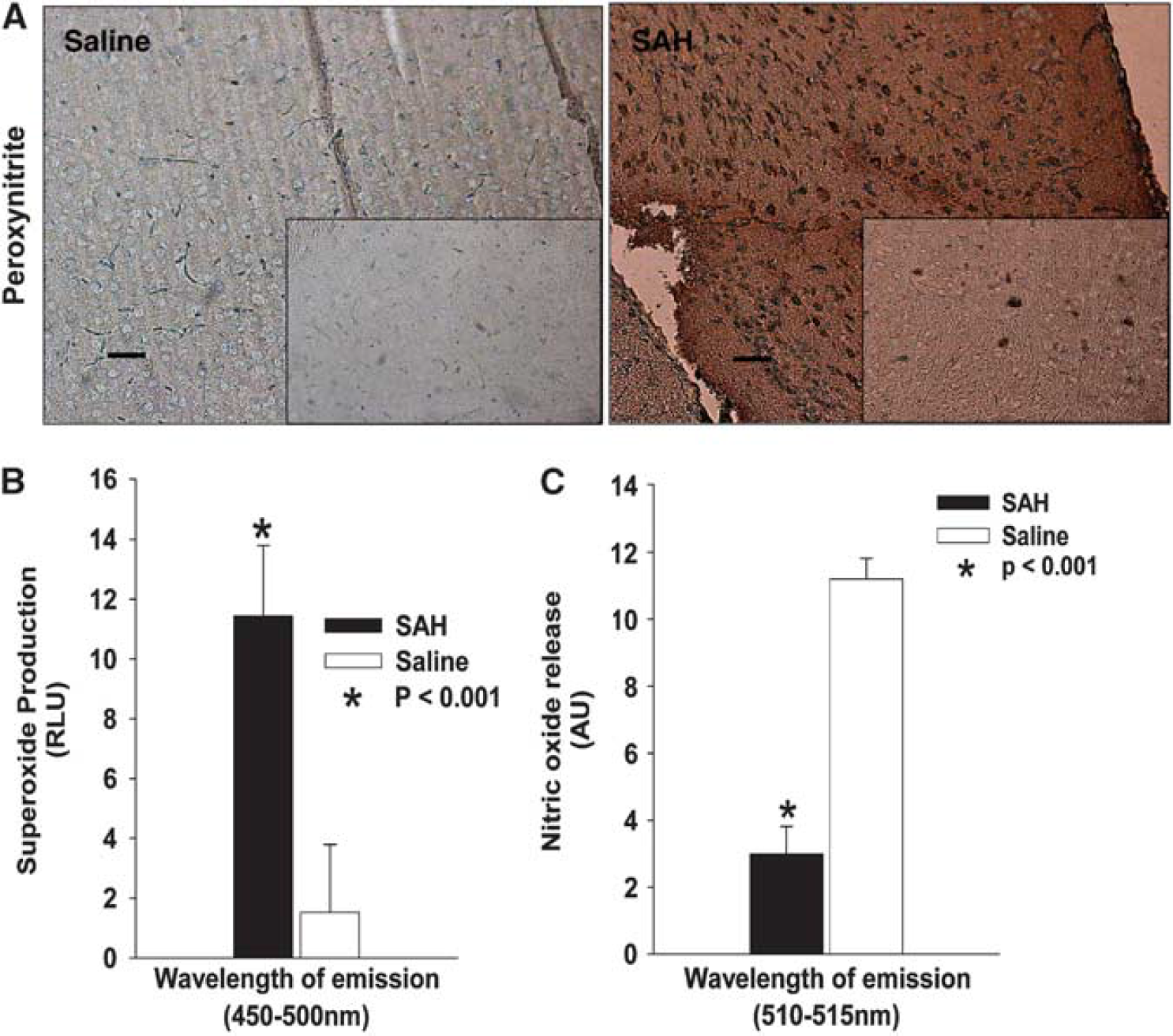
Nitrotyrosine staining and chemiluminescent detection of O−2 and NO. Nitrotyrosine staining was used as a marker for ONOO−. (
O−2 Production, Endothelial Nitric Oxide Synthase and Inducible Nitric Oxide Synthase Expression, and Decreased Nitric Oxide
Subarachnoid hemorrhage is associated with perivascular oxidative reactions due to oxidation of the extravascular hemoglobin due to hemoglobin itself, bilirubin oxidation products, or some associated oxidative reaction (Clark et al, 2008; Macdonald and Weir, 1994). This is one possible mechanism for the uncoupling of eNOS. To test this, we measured O−2 in brain homogenates by fluorescence assay and detected a significant increase in O−2 in the brain tissue of mice with SAH compared with saline-injected control animals (11.2 ± 2.8 and 1.8 ± 0.8 activity units for SAH and control mice, respectively, P < 0.001, Figure 3B). In contrast to the significantly increased phosphorylated S1177-eNOS and iNOS expression and increased O−2, there was a significant reduction in NO in the brain tissue obtained from SAH mice as compared with control mice (2.8 ± 1.2 and 10.6 ± 2.5 activity units for SAH and control mice, respectively, P < 0.001, Figure 3C).
Terminal Deoxynucleotidyl Transferase dUTP Nick-End Labeling, Caspase-3, and Fluoro-Jade
To determine whether SAH was associated with brain injury, we assessed the brains of mice with SAH or saline injection by TUNEL, caspase-3, and NeuN staining. Immunofluorescence staining showed a significant increase in the number of TUNEL-positive neurons in the SAH group compared with saline-injected controls. The TUNEL-positive cells were localized in the outer two cortical layers and hippocampal CA1 to CA3 and dentate gyrus regions (Figures 4A to 4F). To determine whether apoptosis was occurring in neurons, we performed double staining for caspase-3 and NeuN. There was a similar pattern of localization of double positively stained cells to that of TUNEL staining with neurons expressing cleaved caspase-3 after SAH but not saline injection (Figures 4G to 4L). Fluoro-Jade staining was also performed to assess neuronal degeneration. There was a similar pattern of increase in Fluoro-Jade-positive neurons in the brains of SAH mice, suggesting that SAH is associated with neuronal apoptotic and degenerative death (Figures 4M and 4N).
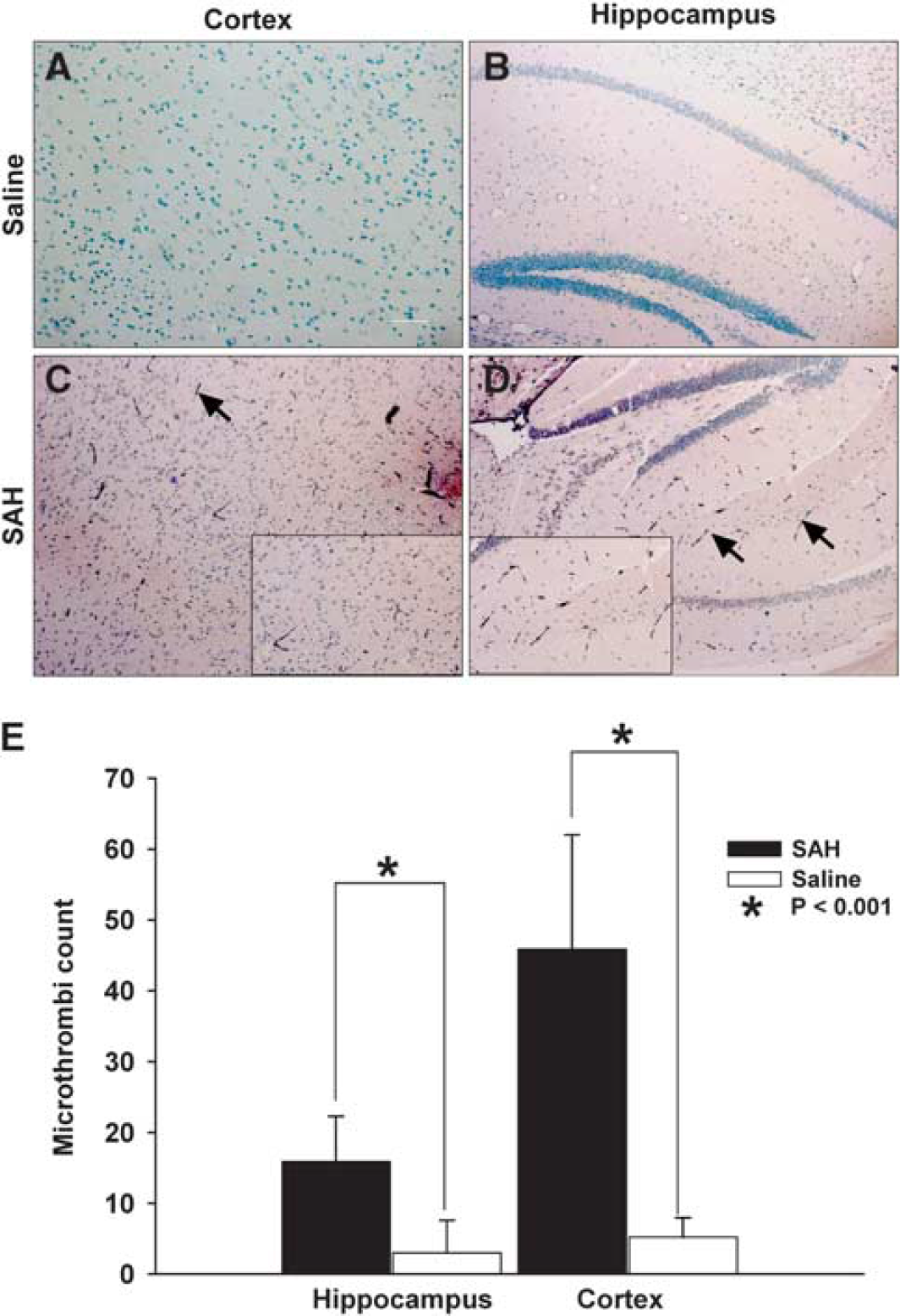
Fibrinogen staining and microthromboembolism quantification. Upper panels show representative images of cortical (
Microthromboemboli Formation After Subarachnoid Hemorrhage
Having observed neuronal injury due to SAH, it was noted that the degree of vasospasm may not be sufficient to reduce CBF and cause injury by ischemia. Laser Doppler recording of CBF, at least acutely after the SAH, also showed a return of CBF to probable nonischemic levels. Thus, we examined mice for other mechanisms of neuronal injury, such as microthromboemboli that have been postulated to contribute to brain injury after SAH (Stein et al, 2006). Antifibrinogen staining of cortical and hippocampal sections of saline and SAH mice at 2 days after the injections showed that sections obtained from mice with SAH had many fibrinogen-positive-stained microvessels scattered throughout the multiple layers of the cerebral cortex and in the dentate gyrus and CA3 regions of the hippocampus (Figures 5C and 5D). In comparison, sections obtained from saline-injected mice showed no fibrinogen staining in the microvessels or parenchyma (Figures 5A and 5B). Counts of microthromboemboli in SAH animals were significantly increased in both cortical and hippocampal sections in comparison with saline animals, wherein there was negligible thrombosis in microvessels (Figure 5E).
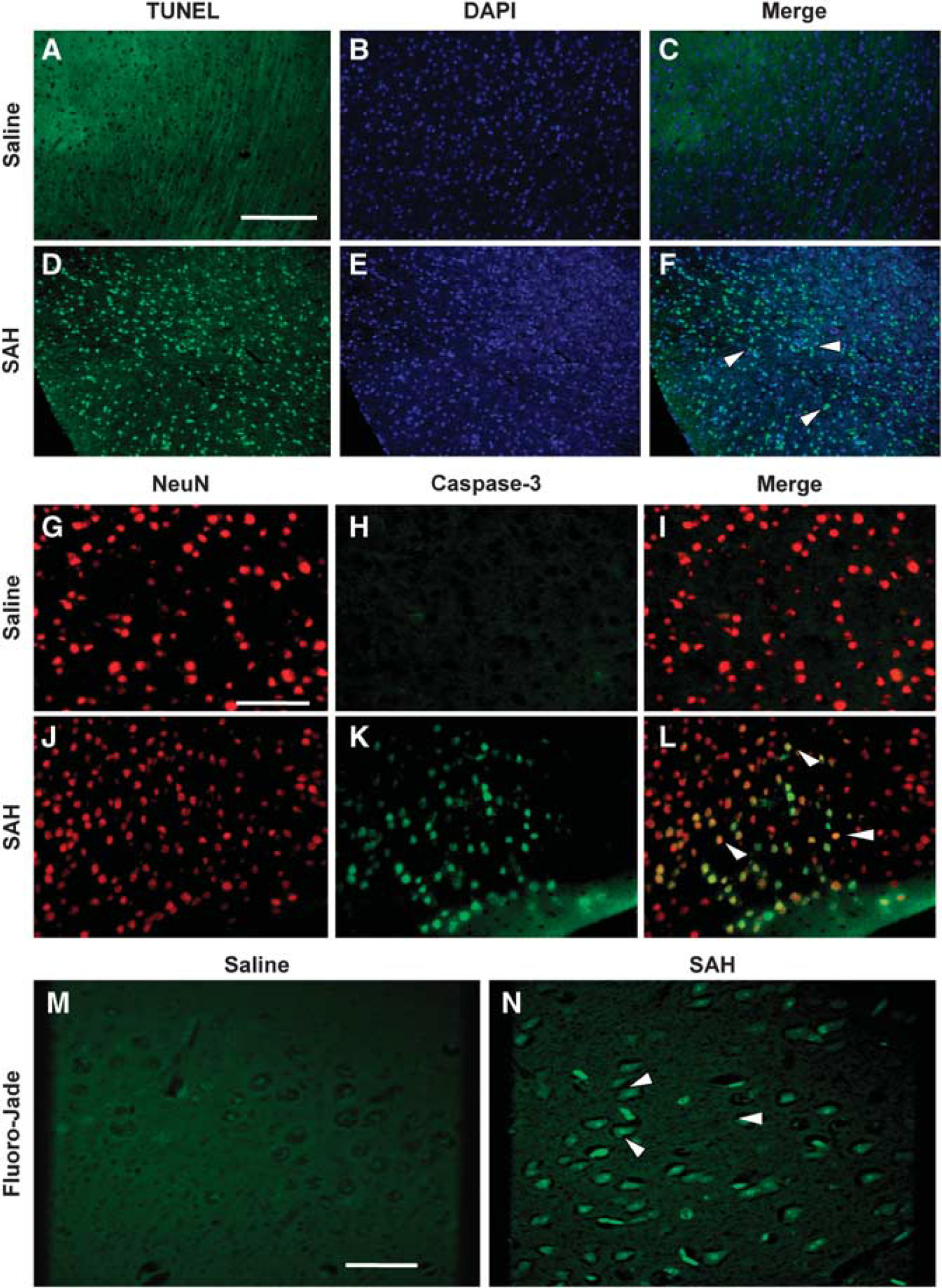
TUNEL, caspase-3/NeuN, and Fluoro-Jade staining. (
Discussion
Endothelial NOS uncoupling and production of injurious nitrosative stress has been suggested to contribute to diseases characterized by vascular injury, such as hypertension, atherosclerosis, hypercholesterolemia, and diabetes (Forstermann and Munzel, 2006). Under these conditions, oxidative stress may be generated by nicotinamide adenine dinucleotide phosphate oxidases. However, after SAH, there is already an abundant perivascular source of oxygen-free radicals, namely ferrous oxyhemoglobin (Pluta, 2006). Although NO deficiency seems to be important in several vascular diseases, these conditions may actually be associated with the upregulation of eNOS (Forstermann and Munzel, 2006). The present experiments are consistent with this. Increased phosphorylation of eNOS on Ser1177 was accompanied by a reduction in available NO in SAH animals, suggesting an uncoupling of eNOS. Furthermore, there was an increase in O−2 and nitrosylation in the brain parenchyma, also suggesting eNOS uncoupling, which could exacerbate the oxidative stress already present after SAH (Macdonald and Weir, 1994). Our data are consistent with studies in other vascular diseases in which endothelial dysfunction was characterized by decreased NO synthesis coupled with increased O−2 formation and upregulation of eNOS messenger RNA, such as hypertension (Bouloumie et al, 1997) and hyperglycemia (Cosentino et al, 1997). Conversely, it is recognized that the present experiments assess the available NO and whether NO production is decreased was not specifically determined. The brain level of NO is not necessarily a marker of eNOS dysfunction because neuronal NOS and iNOS, as well as nonenzymatic sources of NO could also contribute. In addition, the relative concentrations of O−2 and NO can determine what chemicals are produced (Espey et al, 2002).
The prevailing opinion is that perivascular hemoglobin after SAH inhibits physiologic NO-mediated vasorelaxation by binding to NO produced by eNOS and neuronal NOS (Macdonald and Weir, 1991; Pluta, 2006). Moreover, decreased eNOS messenger RNA was found 7 days after SAH in monkeys (Hino et al, 1996) and unchanged or reduced eNOS protein 2 and 7 days after SAH in dogs (Kasuya et al, 1995). In addition, the loss of neuronal NOS associated with SAH was shown by immunoreactivity to rat cerebellar NOS (Pluta et al, 1996). Asymmetric dimethyl-L-arginine, an endogenous inhibitor of eNOS that is produced by protein arginine N-methyltransferase and degraded by dimethylarginine dimethylaminohydrolase, may be increased after SAH (Jung et al, 2007).
In addition to enzymatic sources, nonenzymatic sources of NO could contribute to secondary complications of SAH. For example, nitrite can be reduced to NO during ischemia (van Faassen et al, 2009). The contribution of this nonenzymatic source of NO to the pathophysiology of SAH is not clear. However, nitrite infusion was effective at reducing vasospasm and increasing CBF in nonhuman primates after SAH (Pluta et al, 2005) and at decreasing mean arterial pressure in rats (Rifkind et al, 2007). Therefore, nonenzymatic sources of NO may be useful for treating complications of SAH and other central nervous system disorders (Pluta, 2006).
In contrast to these studies, we observed eNOS activation, as shown by increased phosphorylated S1177-eNOS, 2 days after SAH in mice. There are reports of increased phosphorylated S1177-eNOS in vasospastic basilar arteries 2 days after SAH in rats (Osuka et al, 2009) and rabbits (Santhanam et al, 2005), and reports that total eNOS protein increased, normalized, and decreased 3, 5, and 7 days, respectively, in the femoral arteries of rats that were encased in blood clot (Moon et al, 2001). In these studies and in ours, the increase in eNOS and in some studies, phosphorylated S1177-eNOS, coincided with vasospasm and decreased NO level in brain tissue lysates, suggesting dysfunction in NO synthesis. Previous studies did not examine eNOS in the brain parenchyma, which we also show is increased. In addition, we observed enhanced production of O2− and nitrotyrosine in the brain parenchyma and endothelial layers of pial vessels. These findings are features of eNOS uncoupling (Forstermann and Munzel, 2006).
The limitations of the findings need to be mentioned. The subarachnoid space of mice is small and whether enough blood can accumulate and break down and produce delayed cerebral ischemia similar to that occurring in humans is uncertain. The time course of vasospasm is different among species. We also observed increased iNOS, which could contribute to ONOO− formation, oxidative stress, eNOS uncoupling, vasospasm, microthromboembolism, and neuronal injury (Samdani et al, 1997).
The cause for possible activation of eNOS after SAH remains to be elucidated, but candidate mechanisms would be increased shear stress due to vasospasm (Pluta, 2006) and increased oxidative stress (Forstermann and Munzel, 2006), both of which occur after SAH (Macdonald and Weir, 1994). Activation of eNOS and its associated uncoupling could aggravate vasospasm and oxidative stress, as well as contribute to other complications of SAH such as microthromboembolism, which was observed in this study and in others (Stein et al, 2006).
A prolonged decrease in CBF was observed in both SAH and saline-injected animals. The cause of this was not proven herein. Prolonged decreases in CBF after acute SAH are well documented in animal models and may be due to acute large- and/or small-artery vasoconstriction (Bederson et al, 1998). It is a matter of speculation as to whether microthromboemboli may form acutely, although microvascular perfusion defects were documented in some experimental models of SAH (Asano and Sano, 1977). Subarachnoid saline injection reduced CBF in rats, although the mechanism was not investigated (Ansar and Edvinsson, 2009).
These findings may bear some relevance to the theory that cortical spreading ischemia contributes to brain injury after SAH (Dreier et al, 2009). Dreier et al (1998) produced cortical spreading ischemia in rats by a combination of increased potassium and a NOS inhibitor. They hypothesized that the source of potassium was energy depletion due to activation of adenosine triphosphate-sensitive potassium channels and decreased sodium pump activity and possibly the subarachnoid clot itself. How NO availability was decreased after SAH was less clear, but these results suggest a reason.
In conclusion, these results suggest that SAH is associated with activation of eNOS in the brain tissue. This is accompanied by increased O2− and ONOO− but decreased NO. This suggests that there is eNOS uncoupling, which theoretically could contribute to secondary complications of SAH such as vasospasm and microthromboembolism and eventually, neuronal cell death.
Footnotes
The authors declare no conflict of interest.